12.1
Introduction
In what constituted a major advance in our understanding of bone biology and disease, several groups working on the genetics of rare human skeletal disorders reported a decade ago that mutations in components of the Wnt signaling pathway resulted in abnormal regulation of bone mass homeostasis. These mutations were either in the gene encoding the Wnt coreceptor LRP5 or in Sost, which encodes for sclerostin, a secreted antagonist to Wnt signaling .
There is now compelling evidence that the Wnt signaling pathway affects both skeletal development and postnatal bone mass homeostasis . Indeed, confirming the strong link between Wnt signaling and skeletal homeostasis, multiple genome-wide association studies (GWAS) have demonstrated association between bone mineral density (BMD) at multiple sites and several loci cluster within the Wnt signaling pathway, including AXIN1, CTNNB1, DKK1, LRP4, LRP5, MEF2C, RSPO3, SFRP4, SOST, WLS, WNT3, WNT4, WNT5B , and WNT16 . Mouse genetic studies have confirmed that activation of the canonical Wnt pathway, in which β-catenin is the master regulator (see Section 12.2 ), in the osteoblast cell lineage stimulates proliferation, differentiation, and bone formation. Activation of this signaling in mesenchymal precursor cells favors osteoblast commitment over adipocyte and chondrocyte differentiation, supporting a role for canonical Wnt signaling in cell fate determination . Wnt signaling has also a strong antiapoptotic function, as shown by a significant reduction in the number of apoptotic osteoblasts and osteocytes in mice with activating mutations in LRP5 . In addition to being a central player at early stages of osteoblast differentiation, Wnt signaling is important at later stages of osteoblast differentiation. Mice lacking β-catenin in mature osteoblasts and osteocytes display a low bone mass phenotype. Importantly, these studies have demonstrated that the reduced bone mass observed in these mice is due to increased osteoclastogenesis, while osteoblast function is unaffected. The opposite is true in mice expressing constitutively active β-catenin in mature osteoblasts and osteocytes . Thus, in addition to its effect on osteoblastogenesis, Wnt signaling also regulates osteoclastogenesis by osteoblasts for the most part through the regulation of RANKL (receptor activator of nuclear-factor kappa-B ligand) and osteoprotegerin (OPG) expression. A direct effect of canonical Wnt signaling on osteoclastogenesis has also been proposed as mice lacking β-catenin in osteoclast precursors develop osteopetrosis because of reduced osteoclast number and activity . Most importantly, a link between canonical Wnt signaling in osteocytes and the response of bone to mechanical loading has also been demonstrated , suggesting that osteocytes transduce, via Wnt signaling, mechanical stress into biological responses of osteoblasts and osteoclasts through bone modeling and remodeling.
While the role of canonical Wnt signaling in bone mass regulation has been extensively investigated, our understanding of noncanonical Wnt signaling in skeletal development and postnatal bone mass homeostasis is much less complete. However, evidence indicates that β-catenin-independent pathways are also central in skeletal development and bone mass regulation . Both the Wnt/Gα q/11 –Pkcδ and the Wnt/Ca 2+ noncanonical cascades are involved in osteoblastogenesis and bone formation and the Wnt/Ror2/JNK axis regulates osteoclastogenesis .
Altogether these studies have established the very prominent role of Wnt signaling in skeletal development and bone mass regulation, notwithstanding the fact that not all aspects of this regulation are clear. Even more important is the fact that Wnt signaling can be targeted for drug development, opening the door for novel approaches to treat osteoporosis and other skeletal diseases. Understanding how we can manipulate the different players within the Wnt signaling pathways is a major focus for developing new anabolic agents for treating bone diseases such as osteoporosis.
This chapter provides an overview of the progress made in understanding the role of Wnt signaling pathways in bone and the resulting possibilities for therapeutic intervention.
12.2
WNT signaling
The name “Wnt” derives from the acronym between wingless (wg) in Drosophila and Int1 (currently known as Wnt1) in mouse. Wg and Int1 genes were identified more than two decades ago and found to belong to an evolutionarily conserved family of extracellular signaling molecules . After these initial findings, several studies in Drosophila , Xenopus , and mouse have identified multiple secreted molecules, intracellular effectors and receptors converging in the same signaling cascade, the Wnt signaling pathway.
In mammals, at least 19 secreted Wnt proteins, several families of secreted antagonists and coactivators, and multiple receptors and coreceptors have been identified . Two Wnt signaling pathways, the canonical pathway (in which β-catenin is the master regulator) and the noncanonical pathway, have been described. In the canonical Wnt pathway, Wnt proteins bind to the Frizzled (Fzd) family of receptors and to the low-density lipoprotein receptor (LDLR)–related protein (LRP5/6) leading to stabilization of cytosolic β-catenin and to its translocation into the nucleus where it activates transcription of downstream target genes. The noncanonical Wnt signaling acts independently of β-catenin and triggers its effects through binding to Fzd receptors alone or together with coreceptors such as Ror2 or RYK, activating alternative pathways, including the Wnt/planar cell polarity (PCP), the Wnt/Ca 2+ , the Wnt–JNK signaling, and the Wnt/Ror pathways ( Fig. 12.1 ) . Although the β-catenin-dependent and β-catenin-independent pathways activate distinct downstream signaling, it appears that it is the balance and integration of these different signals that ultimately determines a specific cellular response.
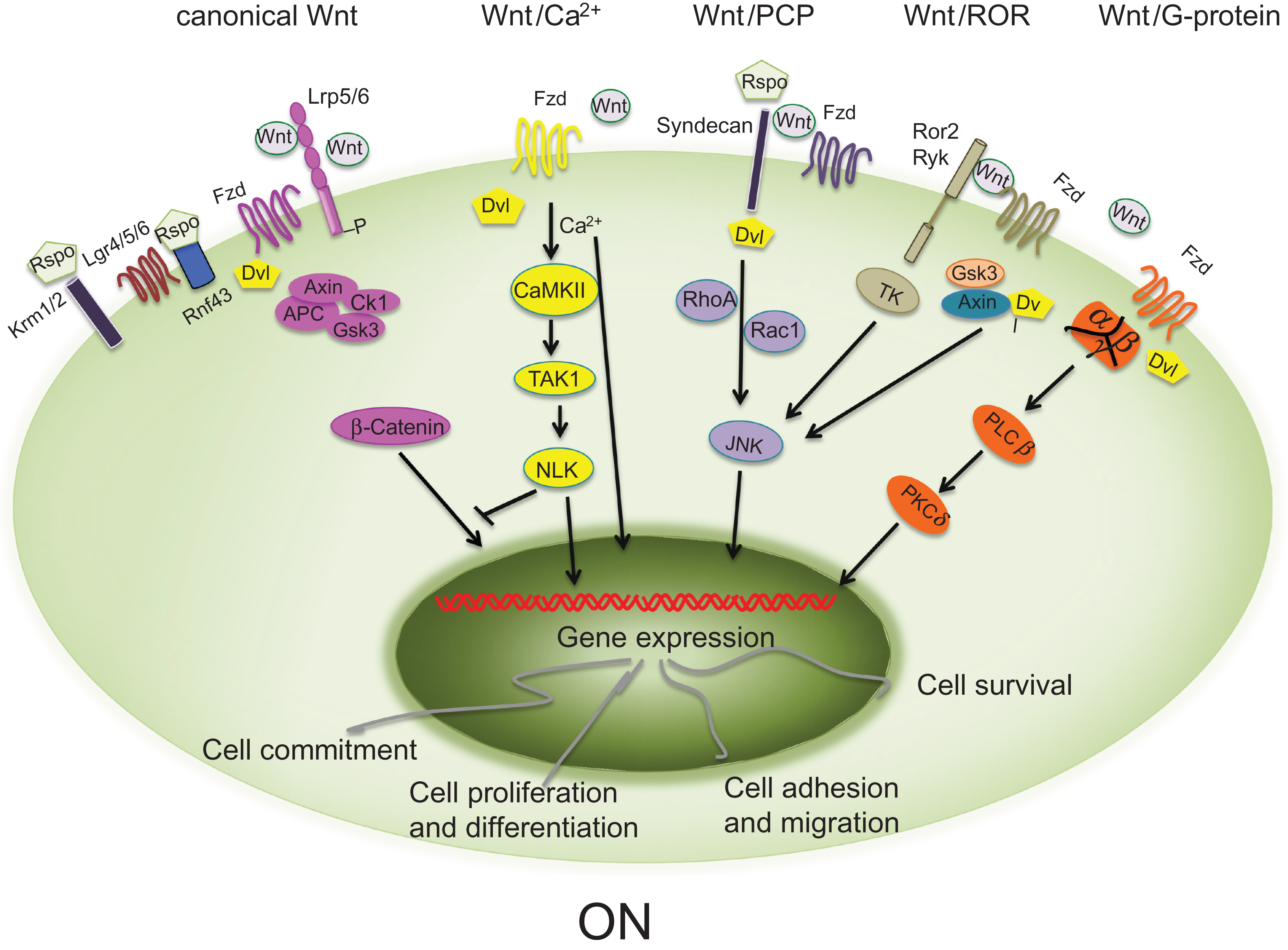
12.2.1
Canonical and noncanonical Wnt signaling pathways
In the canonical Wnt pathway ( Fig. 12.1 ), in the absence of Wnts or if the binding to their receptors is repressed, β-catenin is associated with an intracellular complex (the destruction complex) containing Axin, adenomatous polyposis coli (APC), casein kinase I (CKI), and glycogen synthase kinase 3 (GSK3), and is degraded by ubiquitin-mediated proteolysis. Tcf/Lef transcription factors in the nucleus are bound to transcriptional corepressors (Groucho) thereby preventing gene transcription. Binding of the appropriate Wnt proteins to the Fzd-LRP5/6 coreceptor complex recruits and activates the cytoplasmic signaling protein Dishevelled (Dvl) to the cytoplasmic tail of Fzd. Dvl in turn recruits the axin–GSK3 complex, leading to LRP5/6 cytoplasmic tail phosphorylation and inhibition of the β-catenin destruction complex (axin–GSK3–Ck1–APC), stabilizing cytosolic β-catenin; β-catenin then accumulates in the cytoplasm and translocates to the nucleus where it interacts with Tcf/Lef transcription factors to activate transcription of Wnt downstream target genes .
Noncanonical Wnt signaling triggers its effects via alternative pathways through binding to Fzd or coreceptor complexes of Fzd/Ror2 or Ryk . Noncanonical pathways are complex and include Wnt/Ca 2+ , Wnt/PCP, Wnt/Rho-Rac, Wnt/G-protein-coupled receptors, Wnt/Ror, Wnt–RYK, and Wnt–mTOR pathways ( Fig. 12.1 ). These pathways are involved in developmental processes such as PCP and cell migration and recently some of them have been associated with the regulation of osteoblast differentiation and postnatal bone homeostasis . In the noncanonical Wnt signaling pathways, Wnt proteins signal through the Fzd receptors and activate Dvl leading to the activation of multiple distinct downstream effectors. Thus in these pathways, Dvl functions as a central player. On the other hand, noncanonical Wnt proteins can also signal through membrane receptors such Ror2 and Ryk, thereby activating downstream Wnt signals independently of Dvl .
12.2.2
The ligands: Wnt family of secreted proteins
The findings that Drosophila, Caenorhabditis elegans, Xenopus , and higher vertebrates express multiple Wnt genes indicate that these proteins have only partially redundant biological functions. Wnts are secreted cysteine-rich glycoproteins historically classified as either “canonical” or “noncanonical” depending on their ability/preference to activate β-catenin or β-catenin-independent signaling events, respectively. Although useful, this classification has been challenged by the evidence of cross talk between downstream effectors of the canonical and noncanonical cascades and by the findings that some Wnts can stimulate both pathways depending on the cellular context, the nature of receptors and coreceptors as well the presence of specific coactivators. For example, the “noncanonical” Wnt5a can induce Wnt/β-catenin signaling in the human 293 embryonic kidney cell line in the presence of overexpressed Fzd4 and LRP5 , while Wnt3a, the classical canonical Wnt ligand, can promote bone formation via Wnt/G-protein-coupled receptors . However, the molecular mechanism(s) by which Wnt proteins can activate one or the other signaling pathways remain to be elucidated.
12.2.3
Receptors and coreceptors
There are currently 10 known Fzd receptors. All are seven-transmembrane domain receptors with an extracellular N-terminus domain and an intracellular C-terminus domain. Fzd proteins contain a signal peptide, a cysteine-rich domain (CRD) in the N-terminal extracellular region (which interacts with both Wnts and other Wnt receptors), seven-transmembrane spanning domains, and a C-terminal PDZ domain-binding motif. Different Fzds regulate different intracellular signaling cascades . Importantly, the CDR domain is also found in tyrosine kinase receptors such as Ror2 (see later). Indeed, because Wnts bind the extracellular CRD of these receptors, Fzd receptors activate both canonical and noncanonical signaling, depending upon the nature of the coreceptors that are recruited by the ligand to the complex. Thus while the Wnt/Fzd/LRP complex activates β-catenin-dependent signaling, the Wnt/Fzd/Ror or Wnt/Fzd/RhoA–Rac1 activate noncanonical signaling cascades .
12.2.3.1
Canonical Wnt receptors
LRP5, together with LRP6, LRP4, and LRP7, is a member of a family of the LDLRs . The closely related proteins LRP5 and LRP6, and their homologue in Drosophila (arrow), are required for Wnt-induced β-catenin signaling . Structure–function analysis of LRP5/6 showed that these single-pass transmembrane proteins have an extracellular domain containing four β-propeller domains each followed by an EGF-like domain and three LDLR ligand-binding motifs. The intracellular domain of LRP5/6, responsible for the signaling, is proline- and serine-rich and contains five PPP(S/T)P phosphorylation sites important for the recruitment of axin and GSK3 upon Wnt binding to the receptor complex. LRP5 and LRP6 are normally inactive and exist as oligomers, homodimers, or heterodimers thanks to interactions between their extracellular domains. Although partially redundant, LRP5 and LRP6 display clearly distinct functions. Thus while LRP6-deficient mice die at birth, LRP5-deficient mice are viable, suggesting that these receptors have unique functions that cannot be compensated for by other LRPs .
12.2.3.2
Noncanonical Wnt receptors
Not all the transmembrane Wnt receptors activate the canonical Wnt signaling pathway. Fzd receptors can also activate noncanonical Wnt signaling, by binding to the single-pass transmembrane receptor tyrosine kinases (RTKs) Ryk, Ror1 and Ror2 (RTK-like orphan receptor) . Ror2 has an intracellular tyrosine kinase domain and an extracellular domain containing a Frizzled-like CRD and a kringle domain implicated in protein–protein interaction. Importantly, these receptors function also as autonomous Wnt receptors, with Wnt interactions mediated by Wnt inhibitory factor (Wif) domains in the case of Ryk receptors and by CRDs for Ror receptors ( Fig. 12.1 ). Ror2 binds to Fzd2 and Fzd5 through the CRDs and Wnt5 has been identified as a ligand for this receptor, resulting in activation of the PCP and the JNK noncanonical pathways .
12.2.3.3
Coreceptors for enhancers
Recent work has identified a novel group of Wnt coreceptors, the leucine-rich repeat containing G-protein-coupled receptor (Lgr) 4, 5, 6, which encode for seven-transmembrane receptors . These receptors have been found in stem cells within the small intestine, stomach, and hair follicle (Lgr4 and Lgr5), and in stem cells in the epidermis (Lgr6) and are overexpressed in several types of cancer . Recent studies have demonstrated that in the small intestine Lgr4 and Lgr5 associate with Fzd and LRP5/6 and function as receptors by mediating the effect of R-spondins (Wnt enhancers, see later) on canonical and noncanonical Wnt signaling transduction . Knockout (KO) of Lgr5 in mice leads to neonatal lethality accompanied by ankyloglossia and gastrointestinal distension and loss of Lgr4 results in reduced viability due to developmental defects in many organs, including the kidney, testis, and bone among others. In regard to bone, it has been shown that Lgr4 null mice display delayed osteoblast differentiation, reduced trabecular bone and cortical thickness, and increased osteoclast number and activity and that Lgr4 promotes mesenchymal stem cell proliferation .
12.2.3.4
Coreceptors for antagonists
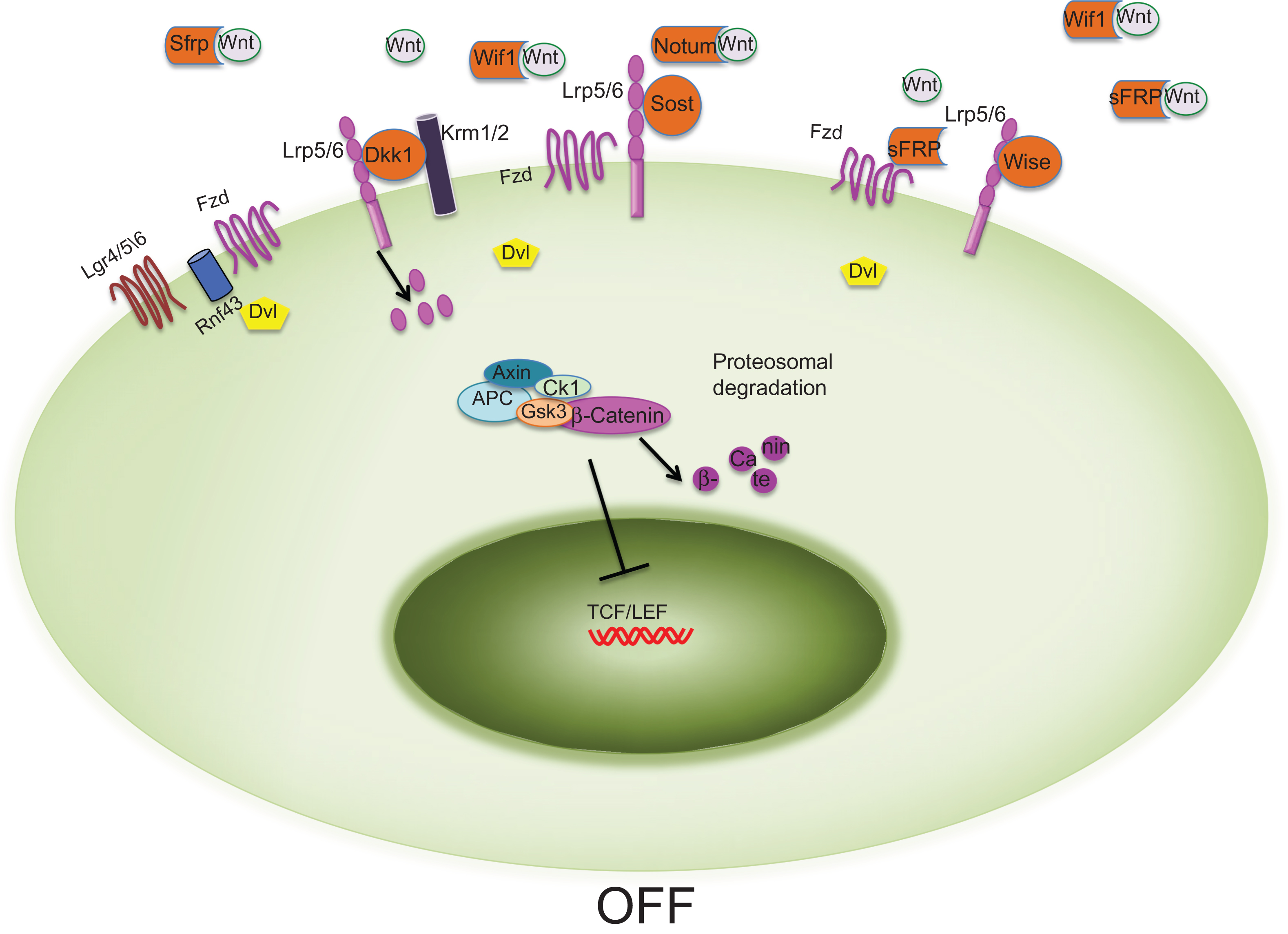
Two coreceptors are central to the downregulation of the canonical Wnt signaling pathway, Kremen1 and Kremen2 (Krm1 and Krm2). These coreceptors bind with high affinity the Wnt antagonist dickkopfs (Dkks) . When canonical Wnt signaling is activated, Wnts interact with Fzd (which activates Dvl) and LRP5/6 (which activate Axin). Dkks bind to kremen1 and 2 and together they form a ternary complex with LRP5/6, which is rapidly endocytosed to inhibit Wnt/β-catenin signaling.
12.2.4
Other ligands: extracellular enhancers and antagonists
12.2.4.1
Wnt enhancers: the R-spondins
In addition to Wnt proteins, vertebrates secrete endogenous enhancers of Wnt signaling. The best known of these are the R-spondins (roof plate–specific spondins), cysteine-rich secreted glycoproteins, which like Wnt proteins are essential for embryonic development and homeostasis . Four R-spondins (Rspo1 to 4) have been identified. These proteins display high structural similarity and 60% sequence homology to each other. They all contain four distinct domains: a putative signal peptide domain, a cysteine-rich furin-like domain, a thrombospondin type I repeat domain, and a basic amino acid–rich domain. Human and mouse genetic studies have demonstrated a biological function for Rspos in vivo. Mutations in the human Rspo1 result in female-to-male sex reversal, a phenotype that has been confirmed in Rspo1 null mice . In addition, a role for Rspo1 in skeletal homeostasis has also been reported . Rspo2 null mice die shortly after birth and display limb patterning defects and craniofacial malformations . Global deletion of Rspo3 results in impaired placental development leading to embryonic lethal phenotype around embryonic day 10 and mutations in the gene encoding Rspo4 lead to anonychia . Several studies have demonstrated that R-spondin synergize with Wnt ligands to activate β-catenin canonical and noncanonical signaling . While some studies reported that Rspos bind to LRP6 and to the coreceptors Krm1 and Krm2, others failed to find interaction between Rspos and these receptors . As mentioned previously, studies suggest that Lgr4, Lgr5, and Lgr6 function as receptors for the Rspos and proposed that Rspos synergize the canonical Wnt signaling via the Lrg4/5 receptors ( Fig. 12.1 ). However, a recent report demonstrated that Rspo2 and Rspo3 can enhance Wnt signaling independently of the presence of these receptors . Adding another layer of complexity, it has been recently suggested that the transmembrane E3 ubiquitin ligase Znrf3 and its homologue ring finger 43 (Rnf43) are the molecular target of Rspos. Indeed, it has been proposed that Znrf3 and Rnf43 inhibit Wnt signaling by promoting the turnover of frizzled and LRP5/6, and this activity is inhibited by Rspos . Interestingly, it has been reported in Zebrafish that Rspo3 functions as a negative regulator of canonical Wnt signaling during dorsoventral and anteroposterior patterning, and that Rspo2 exerts a negative effect on canonical Wnt signaling in colorectal cancer cells , therefore suggesting that the role of the Rspos/Wnt signaling axis in vivo may be more complex. Recently, in vitro studies demonstrated that Rspo3 function as a negative regulator of osteoblast differentiation via a mechanism involving activation of the ERK downstream signaling cascade .
12.2.4.2
Wnt antagonists
Given its critical role in multiple aspects of development, growth, and organ homeostasis, it is not surprising that several endogenous secreted antagonists to Wnt signaling have evolved to dampen the impact of Wnt activation through negative feedback loops. Secreted Wnt inhibitors can antagonize Wnt signaling either by binding directly to the receptors or by functioning as decoy receptors for Wnt proteins ( Fig. 12.1 ).
Antagonists that bind to receptors
Members of the dickkopf family, sclerostin and Wise, interfere with Wnt signaling by interacting with the LRP5/6 coreceptor thus preventing formation of the Wnt/Fzd/LRP5/6 complex. In mammals, the family of Dkk genes is composed of Dkk1, Dkk2, Dkk3, Dkk4, and the Dkk3 related protein termed “Soggy” (Sgy). The Dkk proteins are four structurally related cysteine-rich secreted proteins. Dkk1 was the first protein to be identified as crucial for head formation in Xenopus and in mice . Dkk1 to 4 contain two distinct CRDs, an N-terminal CRD and a C-terminal CRD (which is involved in the binding to both LRP5/6 and Krm1/2), and a potential C-terminal N -glycosylation site, while Sgy lacks the CRD. Dkk proteins do not all function in the same manner: Dkk1 and Dkk4 antagonize Wnt signaling, while Dkk2 can function as antagonist or agonist based on the cellular context . Indeed, it appears that in the absence of Krm, Dkk2 binding to LRP5/6 activates canonical Wnt signaling. In contrast, in the presence of Krm, Dkk2 binding to LRP5/6 blocks the β-catenin signaling. This is probably due to the fact that while in Dkk1 the N-terminal CRD inhibits the C-terminal CRD, in Dkk2 the CRDs do not antagonize each other . Importantly, since Dkk1 binds to the extracellular domain of LRP5/6 but not to Fzd receptors, it blocks only the canonical β-catenin pathway, leaving the cells able to respond to alternative Wnt signaling cascades.
Another crucial Wnt inhibitor is sclerostin, the product of the Sost gene. While Sost is expressed in several tissues during embryonic development, sclerostin is expressed postnatally in mineralized tissues such as bone, mineralized cartilage and teeth. In the skeleton, sclerostin is mainly produced by osteocytes, but also by hypertrophic chondrocytes . Based on its amino acid sequence, sclerostin belongs to the Dan family of glycoproteins, many members of which have been reported to antagonize bone morphogenetic protein (BMP) activity . Sclerostin was originally believed to act as BMP antagonist in bone, but it was then shown to only weakly bind BMPs and inhibit BMP signaling. On the other hand, in vitro studies have demonstrated that sclerostin binds LRP5 and LRP6 preventing their interaction with Wnts .
Wise, another Wnt antagonist, belongs to the Dan/Cerberus family of proteins, like sclerostin. In addition to neutralizing BMP activity, Wise inhibits Wnt signaling by binding to LRP4, LRP5, and LRP6. Notably, Wise has been shown not only to inhibit the activities of some Wnt proteins but also to enhance the activity of others, dependent on the cell context .
Antagonists that bind to ligands
Wnt inhibitors that bind to Wnt ligands include Wif-1 and sFRPs. These secreted antagonists bind directly to Wnts, interfering with their ability to interact with the receptors ( Fig. 12.1 ). sFRPs were identified in Xenopus due to their homology with the Fzd receptors and subsequently associated with Wnt signaling because of their ability to bind Wnt proteins. sFRPs contain two domains: a N-terminus with a signal peptide followed by a CRD similar to that of Fzd and Ror, and a C-terminal domain, important for heparin-binding, followed by six cysteine residues that form three disulfide bridges. The sFRP family consists of five members in humans, sFRP1 to 5, with sFRP1, sFRP2, and sFRP5 sharing high sequence homology. sFRPs can bind to Fzd receptors, antagonize one another’s activity, and interact with other molecules and receptors and BMP signaling through inhibition of Tolloid action on Chordin .
The Wnt-interacting domain of Wif-1 is similar to that of the Ryk receptors, while the sFRPs contain a CRD similar to the extracellular Wnt-binding domain of the Fzd receptors. Because the CRDs of Fzd receptors can bind Wnts and form homodimers or heterodimers, sFRPs can modulate Wnt signaling by sequestering Wnts through the CRD or by forming inactive complexes with the Fzd receptors. Thus, differently from Dkk1, sclerostin and wise, these antagonists affect all Wnt-activated pathways, canonical and noncanonical.
12.2.5
Intracellular effectors and regulators
The core of the canonical Wnt signaling pathway is the β-catenin destruction complex, which has a pivotal role in maintaining proper β-catenin levels in the cytoplasm and in the nucleus by promoting its ubiquitination and degradation. As proper levels of β-catenin are required for many biological events, the components of the destruction complex, GSK3–Axin–Ck1–APC, are important intracellular regulators of canonical Wnt signaling. Importantly, some of these intracellular regulators are also implicated in the regulation of noncanonical Wnt signaling and other signaling pathways, including BMP and FGF signaling.
GSK3, a serine/threonine kinase, is a negative regulator of several signaling pathways, including both canonical and noncanonical Wnt signaling. GSK3 phosphorylates β-catenin, thereby promoting its degradation through recruitment of beta-transducin repeat containing protein, ubiquitination and proteosomal degradation. Indeed, inhibition of GSK3-mediated β-catenin phosphorylation is the major mechanism that Wnt signals use to prevent β-catenin degradation . However, GSK3 also has a positive role in activating Wnt signaling, once it is recruited to the plasma membrane. Upon binding of Wnt ligands to Fzd/LRP5/6 receptor complexes, GSK3 and Axin move to the membrane where GSK3 phosphorylates LRP5/6 cytoplasmic tail, which in turn sequesters GSK3 thereby allowing β-catenin translocation to the nucleus and activation of gene transcription .
Axin1 and/or Axin2 act as a scaffold for all the components necessary for β-catenin degradation. In the presence of Wnt ligands, Axin together with other components of the destruction complex are recruited to LRP5/6 thereby activating downstream β-catenin signaling. In addition to bringing together the β-catenin destruction complex, Axin2 expression is a direct target of β-catenin, thus this scaffold protein functions as a negative feedback inhibitor of the canonical Wnt pathway .
APC is a tumor suppressor that is mutated in more than 80% of colon cancers. It has been established that the major function of APC is to antagonize canonical Wnt signaling. In addition to associating with β-catenin in the cytoplasm, APC also interacts with β-catenin in the nucleus where it prevents β-catenin from associating with Lef/Tcf transcription factors .
The CKI family of serine/threonine protein kinases is involved in many diverse and important cellular functions. Mammals have seven family members: α, β, γ1, γ2, γ3, δ, and ε. This family of kinases has been shown to regulate Wnt signaling both positively and negatively by interacting with and phosphorylating multiple components of this pathway. Targets for positive regulation of CKI include Dvl, Tcf3, and the coreceptor LRP6, whereas negative regulation occurs via APC and β-catenin where phosphorylation leads to complete or partial proteolysis of β-catenin and thereby inactivation of the pathway in the absence of ligand .
Another important intracellular regulator is Dvl, which has a central role in transmitting Wnt signaling from receptors to downstream effectors in both the canonical and noncanonical pathways . Three Dvl homologues (Dvl1, 2, and 3) have been identified in humans and mice. They contain three conserved domains: an N-terminal DIX (Dvl, Axin) domain, a central PDZ (postsynaptic density-95, discs large, zonula occludens-1) domain, and a C-terminal DEP (Dvl, Egl-10, Pleckstrin) domain. Studies in mouse models have demonstrated that Dvl is required for proper cardiac development, neural tube formation, and cochlear polarity . Dvl proteins can interact with a wide array of proteins and are thought to mediate protein–protein interactions and protein phosphorylation. Upon activation of the canonical Wnt pathway, Dvl is recruited by the receptor Fzd and prevents the destruction of cytosolic β-catenin by the proteosome. In the noncanonical Wnt pathways, Dvl is involved in the activation of several alternative pathways, including the Wnt/PCP signaling, calcium-dependent pathways, Wnt-Ror signaling, and Wnt/G protein signaling ( Fig. 12.1 ).
12.2.6
Ligand posttranslational modifiers
Wnts are known to be acylated. This posttranslational modification is essential for Wnt activity and is carried out by membrane-bound O-acyl transferases (MBOATs). Porcupine (Porcn), a Wnt–MBOAT, attaches palmitoleate and shorter cis unsaturated fatty acids onto Wnts . Palmiteoylation is required for (1) binding to Wntless, a protein that mediates Wnt intracellular transport; (2) secretion of Wnt ligands; and (3) binding to Frizzled receptors. Small-molecule inhibitors that block Porcn-mediated Wnt modification and Wnt signaling have been reported and have recently been shown to exhibit therapeutic efficacy in treating Wnt-driven cancers. However, it has been recently shown that in mice these porcupine inhibitors lead to decreased trabecular and cortical bone mass and strength due to both decreased bone formation and increased bone resorption On the other hand, a lipase that deacetylases Wnt proteins, Notum, has also been recently identified to negatively affect Wnt signaling . In particular, it has been shown that Notum enzymatically inhibits Wnt signaling activity by removing the palmitoleate moiety of Wnts, which contributes directly to Frizzled receptor binding . Deletion of the Notum gene in mice has been shown to increase cortical bone mass .
12.3
WNT signaling in skeletal development, homeostasis, and diseases
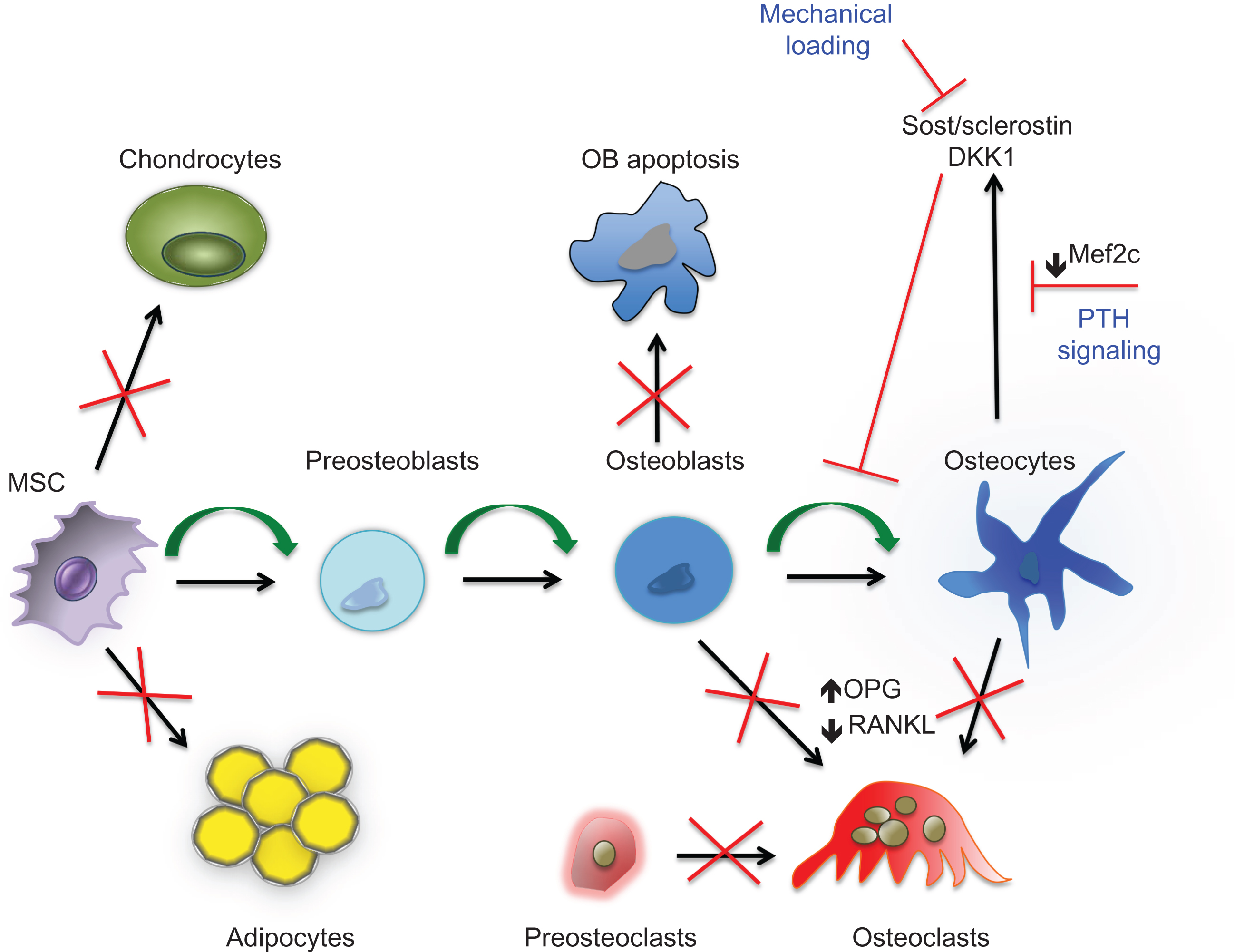
The discovery that mutations in LRP5 and sclerostin in humans alter bone mass constituted a major breakthrough in bone research, opening new avenues for the treatment of bone diseases. Since then, a number of KO mouse studies have very firmly established that the Wnt pathway is required for proper bone development and for postnatal bone homeostasis. With more advanced techniques in mouse genetic engineering, components of the Wnt pathway have been deleted or activated at specific stages of osteoblast differentiation, from early limb mesenchymal precursors to osteocytes. Overall these studies have demonstrated a direct role for Wnt signaling pathway in osteoblast commitment, proliferation, survival, and differentiation as well as a role in osteoclastogenesis both indirectly, via effects on cells of the osteoblast lineage, and directly ( Figs. 12.2 and 12.3 ).
12.3.1
LRPs plays a key role in bone
The first study to show a link between Wnt signaling and bone mass reported that inactivating mutations in LRP5 in human cause osteoporosis pseudoglioma syndrome (OPPG, OMIM259770), an autosomal recessive disorder characterized by severe early-onset osteoporosis . Shortly after these observations, two groups independently reported a specific point mutation (G171V) in LRP5 resulting in high bone mass (HBM) . Most interestingly, this mutation decreased the LRP5 binding affinity of two endogenous Wnt inhibitors, Dkk1 and sclerostin, resulting in increased canonical Wnt signaling. Further support for a key role of LRP5 in skeletal homeostasis was provided by several GWAS, which showed association between LRP5 and bone mass and fracture risk in diverse human populations .
Studies in animal models have confirmed that loss- and gain-of-function of LRP5 recapitulate the OPPG and the HBM phenotype. Thus while mice with germline deletion of LRP5 develop low bone mass due to impaired osteoblast proliferation and bone formation rate, transgenic mice expressing the G171V mutation in immature osteoblasts develop an HBM phenotype because of increased bone formation and decreased osteoblast and osteocyte apoptosis . However, no changes in osteoclastogenesis and bone resorption were observed in these mice. Thus overall these studies suggested that LRP5, expressed in osteoblasts, regulates bone formation via proliferation and apoptosis of cells of the osteoblast lineage.
These findings were challenged when Yadav et al. reported that neither conditional deletion of LRP5 in osteoblast progenitors or mature osteoblasts nor conditional knock-in of the G171V mutation into mature osteoblasts resulted in changes in bone formation . These authors suggested that it is LRP5 expressed in the duodenum that, via serotonin secretion, regulates bone formation. In support of this hypothesis were the findings that OPPG patients have high levels of circulating serotonin, whereas patients with HBM have lower serotonin levels . These findings were very surprising in view of the phenotype of transgenic mice expressing the G171V mutation in LRP5 targeted to osteoblasts . Independent of whether gut-derived serotonin affects bone formation or not, a very thorough recent study strongly supports the earlier findings that LRP5 functions locally in bone to regulate bone mass . In these studies, conditional deletion or knock-in of the HBM mutation of LRP5 in osteocytes led to a marked decrease or increase in bone mass, respectively, while LRP5 deletion in the intestinal stem cells did not affect bone mass. In addition, knock-in of the HBM mutation in LRP5 in limb mesenchymal progenitors resulted in increased bone mass only in the appendicular skeleton, firmly establishing the local nature of these events, and excluding a significant role of gut-derived factors. Although the reason(s) for this inconsistency is still unknown, the results from Cui et al. unequivocally establish that the HBM mutation exerts its effects in cells of the osteoblast lineage, and most probably osteocytes.
Other members of the LDLRs family have also been shown to be involved in the regulation of bone mass. Mutations in LRP6 have been linked to changes in bone mass and haploinsufficiency of LRP6 in LRP5 null mice leads to more severe osteopenia, indicating that these receptors are partially redundant . In addition, it has recently been shown that LRP4 acts as a coreceptor for Dkk1 and sclerostin in vitro and that LRP4-deficient mice develop shortened femurs, reduced cortical femoral perimeter, reduced total femur bone mineral content, and BMD .
12.3.2
β-Catenin and bone
Since LRP5/6 are coreceptors for β-catenin-dependent Wnt signaling, the role of these receptors in the regulation of bone mass requires consideration of how β-catenin affects bone mass. Gain-and-loss-of-function studies in mice have shown an essential role for β-catenin in osteoblast differentiation and in the regulation of bone mass and emphasized that the ultimate response to activation of the canonical Wnt signaling is cell stage-dependent.
Deletion of β-catenin in early skeletal progenitor cells leads to impaired osteoblast differentiation and ectopic chondrogenesis in the mouse embryo demonstrating a role for β-catenin in osteoblast proliferation and differentiation and in the balance between osteoblasts and chondrocytes ( Fig. 12.3 ). On the other hand, mice expressing constitutively active β-catenin in early mesenchymal cells display an absence of appendicular and skull bones, suggesting that increased levels of β-catenin during embryonic development negatively affect skeletal development . Moreover, activation of the canonical Wnt pathway regulates the expression of OPG, the decoy RANK receptor that inhibits osteoclast differentiation by interacting with RANKL. Disruption of β-catenin in mature osteoblasts and in osteocytes in mice affects bone resorption rather than bone formation because of elevated expression of RANKL and decreased expression of OPG . Confirming these findings, constitutive activation of β-catenin in osteoblasts and osteocytes results in decreased osteoclast numbers . Thus canonical Wnt signaling controls osteoblasts at different levels: commitment, proliferation/apoptosis, and function. In addition, β-catenin can directly regulate osteoclastogenesis . Using different Cre-mice to delete or constitutively express β-catenin in osteoclast progenitors, macrophage precursors, and mature osteoclasts, these studies have suggested that constitutive activation of β-catenin blocks osteoclast differentiation and β-catenin deletion impairs osteoclast proliferation, both causing osteopetrosis. On the other hand, 50% deletion of β-catenin leads to osteoporosis due to increased osteoclast differentiation. Thus deregulation of β-catenin has a biphasic effect on osteoclasts, inhibiting osteoclastogenesis when activation is either low or high.
Because the phenotypes of the β-catenin and LRP5 null mice are different, it is important to mention that, on the one hand, β-catenin activation may not be the only signaling event downstream of Wnt receptor complexes and, on the other hand, β-catenin may also function independently of the Wnt signaling pathway to regulate osteoblast proliferation and differentiation and bone formation. For instance, β-catenin interacts with N-cadherin, which has been shown to regulate osteoblast proliferation and survival, and mechanical strain in osteoblasts and osteocytes can activate β-catenin directly to promote bone formation .
12.3.3
Wnts and bone
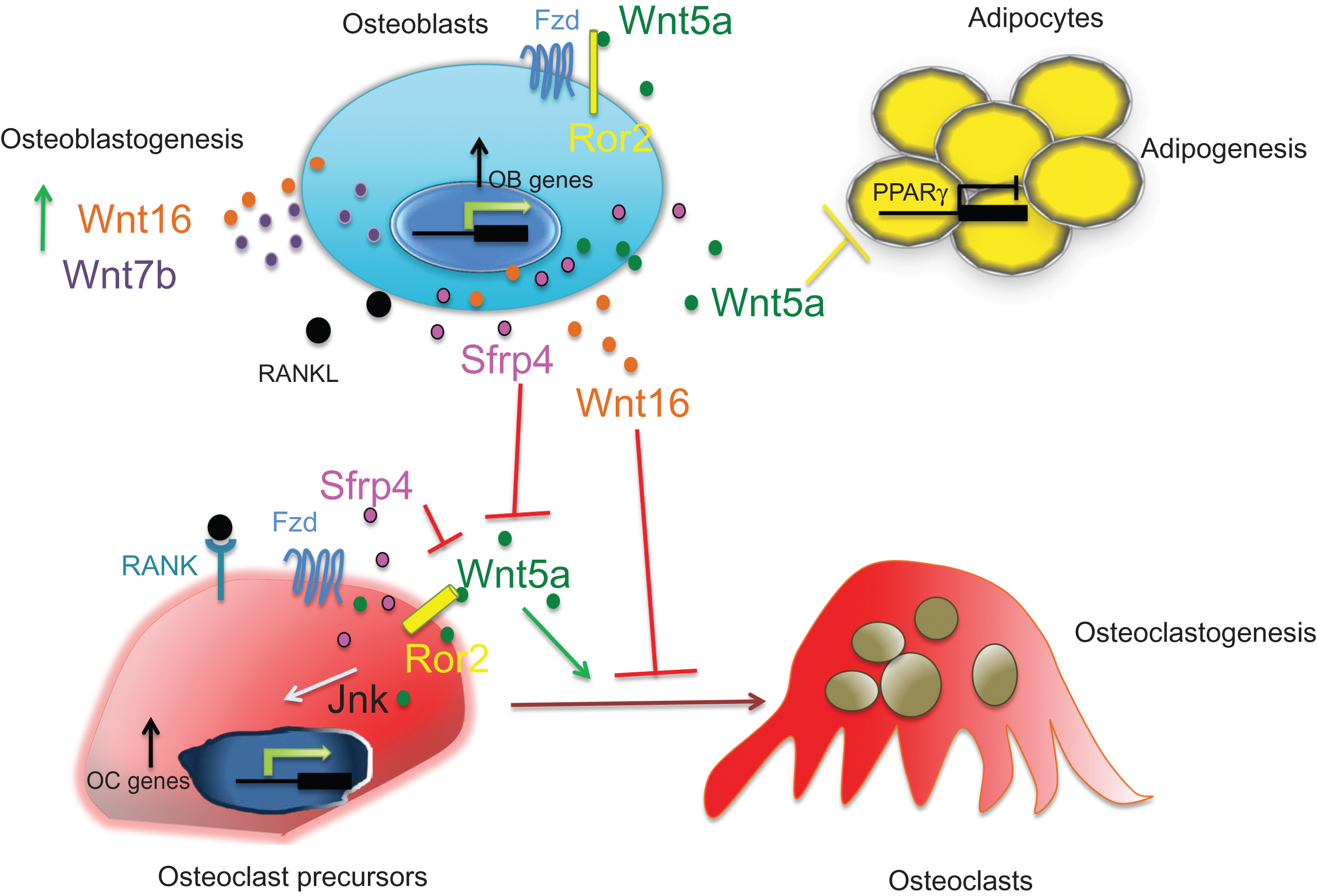
Many Wnts have been shown to impact bone biology. Early experiments in chickens and in mice have demonstrated that ligands such as Wnt2b, Wnt3a, Wnt4, Wnt5a, Wnt8c, and Wnt7a are expressed early during limb development and are crucial for proper limb outgrowth and patterning along the proximal–distal and dorsoventral axes . Studies in mice have shown that several Wnt proteins are required for chondrocyte and osteoblast differentiation during limb development as well as for bone homeostasis. Importantly, several GWAS showed association of some of the Wnt proteins, including, Wnt4, Wnt5b, and Wnt16 with BMD and/or fracture risk . Thus loss- and gain-of-function studies have demonstrated that ligands such as Wnt1, Wnt3a, Wnt4, Wnt5a, Wnt7b, Wnt10b, and Wnt16 affect osteoblastogenesis, osteoclastogenesis, and adipogenesis thereby contributing to bone mass via β-catenin-dependent or β-catenin-independent pathways . In particular, the first functional characterization of Wnt16 has confirmed its critical role in the regulation of cortical bone mass and bone strength in mice, confirming Wnt16’s strong association with cortical bone fragility in GWAS . These studies also supported the emerging hypothesis that the homeostasis of the trabecular and cortical bone compartments is, at least in part, differentially regulated. Most interestingly, Wnt16 appears to be a strong antiresorptive soluble factor acting on both osteoblasts and osteoclast precursors.
12.3.4
Secreted Wnt antagonists and bone: sFRPs, Dkks, and Sclerostin
As mentioned earlier, gain-of-function mutations in LRP5 decrease the ability of antagonists such as Dkk1 and sclerostin to bind to LRP5, thereby leading to HBM. Sclerostin is the protein encoded by the Sost gene located on the human chromosome 17q12-q21. Mutations in the Sost gene cause sclerosteosis (MIM269500) and van Buchem disease (MIM239100) and polymorphisms of the Sost promoter have been linked to osteoporosis . Genetically engineered mouse models designed to mimic the mutations seen in sclerosteosis and van Buchem’s patients reproduce the HBM phenotypes seen in these patients, while transgenic expression of sclerostin in mice leads to osteopenia . Sclerostin is secreted by osteocytes and it is believed to reach the bone surface through the network of osteocyte canaliculi and to regulate osteoblast and osteoclast differentiation and function . However, recent studies in which Sost was deleted in mesenchymal progenitor cells, mature osteoblasts or osteocytes have shown that, although Sost deletion in each cell types leads to a significant increase in trabecular bone mass, only Sost deletion in mesenchymal progenitor cells (Prx1-Cre) fully recapitulated the high trabecular and cortical bone mass phenotype seen in the appendicular skeleton of the global KO as well as the B-cell phenotype. Importantly, despite normal expression of Sost in the axial skeleton of Prx1-Cre deleted mice, these mice developed HBM in the vertebrae, while the sclerostin released in circulation by the axial skeleton did not affect bone mass in the appendicular skeleton. Thus these findings add new insights into sclerostin and the cells secreting it and suggest that the only Sost expression affecting bone mass is derived from descendants of Prx1 -positive limb bud progenitors . As discussed later, sclerostin expression is regulated by mechanical loading and parathyroid hormone (PTH) and it is thought that the anabolic effects of both mechanical stimulation and PTH, are due, at least in part, to suppression of sclerostin expression and secondary enhancement of canonical Wnt signaling. In addition to expressing sclerostin, osteocytes also express high levels of RANKL , suggesting that sclerostin might function in osteocytes to support osteoclastogenesis by repressing the Wnt-induced increase in OPG .
Dkk1 also binds to LRP5/6 and suppresses canonical Wnt signaling, inhibiting bone formation. Dkk1 null mice die shortly after birth with severe developmental abnormalities . However, Dkk1 haploinsufficient mice are viable and develop increased bone formation and bone mass without changes in bone resorption . Recent studies have shown that Dkk1 deletion in cells of the osteoblastic lineage results in HBM with increased bone formation, despite increased levels of sclerostin and that osteoprogenitors but not mature osteoblasts or osteocytes are the main source of systemic Dkk1 . Consistent with these findings and confirming a negative role for Dkk1 in the regulation of bone mass, transgenic overexpression of Dkk1 in osteoblasts significantly decreases osteoblast number and bone formation rate and tends to increase bone resorption . Although Dkk1 mutations affecting bone mass in humans have not been discovered, high levels of Dkk1 have been implicated in the development of osteolytic lesions in metastatic bone disease and multiple myeloma . Similar to Dkk1, Dkk2 blocks canonical Wnt signaling and inhibits osteoblast differentiation pathways in osteoarthritic osteoblasts . However, it has been reported that Dkk2 activates β-catenin in Xenopus and that mice lacking Dkk2 develop osteopenia , suggesting that in contrast to the inhibitory functions of Dkk1 on bone mass, Dkk2 can stimulate bone formation, possibly by favoring the last stages of osteoblast maturation.
As mentioned previously, in contrast to sclerostin and Dkk1, sFRPs and Wif-1 bind to Wnt ligands preventing their functional association with both LRP5/6 and Fzd receptors. Although, sFRPs are all expressed in bone , they play quite distinct roles in skeletal development and homeostasis. Mice with germline deletion of sFRP1 develop modest increases in trabecular bone mass, mineral apposition rate, and total volume, but not in cortical BMD in adult mice , whereas transgenic sFRP1 overexpression decreased bone density . Mechanistically, sFRP1 deletion reduces osteoblast and osteocyte apoptosis in vivo, and cell proliferation and differentiation in vitro . It has also been suggested that sFRP1 binds RANKL thereby impairing osteoblast-induced osteoclastogenesis . In addition to sFRP1, other members of the family are involved in skeletal development and/or bone homeostasis. sFRP2 inactivation results in brachydactyly, without other obvious skeletal changes , while sFRP3 has been associated with the development of osteolysis and heterotopic ossification . sFRP4 is expressed in cells of the osteoblast lineage and it inhibits osteoblast proliferation by suppressing Wnt3a activity in vitro . Studies in mice have shown that sFRP4 expression is markedly increased in osteopenic accelerated aging SAMP6 mice and that overexpression/deletion of sFRP4 results in specific skeletal phenotypes . Directly relevant to osteoporosis in humans, several independent GWAS have linked sFRP4 to BMD, including cortical sites . Recent studies have established that SFRP4 loss-of-function mutations in humans are linked to Pyle’s disease , a rare genetic skeletal disease first described in 1931 by Edwin Pyle as a “case of unusual bone development” . The clinical hallmarks of Pyle’s disease are genu valgum and long bones characterized by wide and expanded metaphyses with increased trabecular bone, while cortical bone is thinner than normal. As a result, the bones are fragile and fracture easily. Affected individuals also present with mild cranial sclerosis, thin calvarium with expanded diploe and impaired formation of cranial sinuses. In addition, some patients present with a smaller head circumference (>2 SD smaller) or with an abnormal shape, characterized by prominence of the frontal bones and prognathism. Dental problems are also common in Pyle’s disease, including delayed eruption of permanent teeth, cavities, and malocclusion . Loss-of-function studies in mice have demonstrated that sFRP4 deletion results in a skeletal phenotype similar to that observed in Pyle’s disease and uncovered that, while activation of canonical Wnt signaling leads to increased trabecular bone mass, activation of the noncanonical Wnt/Jnk signaling cascade, together with increased BMP signaling and sclerostin levels, leads to decreased cortical thickness and homeostasis . sFRP4 has also been described as a phosphate regulator both in vivo and in vitro . However, ablation of sFRP4 in mice does not result in changes in serum or urine phosphate levels, thus dampening the potential role of sFRP4 in phosphate homeostasis .
12.3.5
Noncanonical Wnt signaling and bone
Unlike canonical Wnt signaling, the role of noncanonical Wnt signaling in bone remains to be fully elucidated, albeit a number of recent papers have indicated that it also plays a role in skeletal development and homeostasis. The Wnt7b–Gα q/11 –PKCδ noncanonical cascade has been shown to be required for proper osteoblast differentiation and bone formation through the regulation of Runx2 expression . Oishi et al. reported that Wnt5a binds to Ror2 and activates JNK phosphorylation, suggesting that during embryonic development Ror2 functions as a receptor for Wnt5a to activate noncanonical signaling. Indeed, Wnt5a or Ror2 null mice display a similar phenotype characterized by dwarfism, facial abnormalities, and shortened limbs . In humans, Ror2 mutations cause brachydactyly type B, a dominant skeletal disorder characterized by hypoplasia/aplasia of distal phalanges , and Robinow syndrome, a recessive condition characterized by short stature and limb bone shortening . Confirming these findings, Wnt5a is required not only for osteoblastogenesis but also for proper osteoclastogenesis via a Ror2–JNK signaling, suggesting a role for noncanonical Wnt signaling in the regulation of postnatal bone homeostasis . In addition, recent work has shown that Sfrp4-dependent suppression of the noncanonical Wnt5a/Ror2/Jnk cascade in osteoclasts, but not that of β-catenin signaling, regulates osteoclastogenesis and protects cortical bone from excessive endocortical resorption . Furthermore, Takada et al. have shown that Wnt5a heterozygous mice display an osteopenic phenotype, with decreased trabecular bone mass and increased bone marrow adipogenesis. Both of these effects were shown to be due to CAMKII–TAK1–TAB2–NLK-dependent activation of PPARγ transcriptional activity .
Thus collectively these studies suggest that, similar to canonical Wnt signaling, activation of noncanonical Wnt signaling can affect osteoblastogenesis, bone marrow adipogenesis, and osteoclastogenesis ultimately regulating bone mass ( Fig. 12.3 ).
12.4
WNT signaling interacts with other pathways in bone mass regulation
The skeleton is continuously modeled during development and growth and remodeled in the adult. This complex regulation of bone geometry and homeostasis involves multiple signaling pathways, which implies significant cross talk. Not surprisingly, Wnt signaling interacts with other key pathways involved in bone biology in particular BMP and PTH signaling.
12.4.1
Wnt and bone morphogenetic protein signaling
BMP and Wnt signaling are essential signals implicated in multiple events during skeletal development and bone formation. BMPs share high homology with the decapentaplegic (dpp) protein in Drosophila , and with the Xenopus Vg-1, which are proteins involved in early embryo pattern formation . BMPs were originally identified as molecules derived from bone, which were capable of inducing new bone formation ectopically when subcutaneously injected in animal models . In vitro, BMP-2 has been reported to induce, the expression of many osteoblast-specific genes in different cell types and analysis of mutations of mammalian BMP signaling have confirmed the critical role for members of this family both in skeletal development and postnatal bone . In addition, human disorders such as chondrodysplasia and fibrodysplasia ossificans progressiva confirm that BMP signaling plays an important role in regulating bone formation.
Although, the role of BMP signaling in promoting bone formation is clear, preclinical studies in animals have shown that inhibition of BMP signaling has an anabolic effect on bone and that activation of BMP signaling by deleting Noggin results in osteopenia . This can be explained by the findings that BMP signaling represses Wnt canonical signaling through induction of sclerostin and subsequently bone formation. Indeed, accumulated evidences suggest that the BMP and Wnt signaling pathways regulate one another both synergistically and antagonistically in a context and age-dependent fashion . In vitro studies have demonstrated that canonical Wnt signaling induces BMP expression and that BMPs induce Wnt expression and in vivo studies have confirmed the interaction between these two signaling pathways. It has been shown that BMP signaling inhibits Wnt signaling by increasing Sost expression and decreases bone mass by supporting osteoclastogenesis through the RANKL–OPG pathway .
12.4.2
Wnt and parathyroid hormone signaling
It is well established that PTH controls the rate of bone remodeling in both humans and animal models. PTH, which exerts its action via the PTH/PTH-related peptide type 1 receptor (PTH1R), functions both as an anabolic and catabolic effector in bone depending upon whether its action is continuous, as in hyperparathyroidism, or intermittent when used in therapeutic intervention for osteoporosis. Intermittent administration of PTH in rodents induces osteoblast differentiation, prolongs the life span of mature osteoblasts, and favors bone formation. In contrast, continuous administration of PTH, which also increases osteoblast number and bone formation, increases RANKL production by these cells thereby increasing bone resorption as well .
In the last few years, several studies have demonstrated that PTH signaling interacts with and regulates canonical Wnt signaling through multiple mechanisms, supporting the hypothesis that the anabolic effect of PTH in bone is mediated, at least in part, via activation of the canonical Wnt signaling pathway in osteocytes . Most importantly, PTH represses the expression of Sost in vitro and in vivo by inhibiting Mef2c (myocyte enhancer factor 2), a transcription factor required for activation of Sost expression . PTH also inhibits the expression of other Wnt signaling agonists, such as Dkk1 and Wif-1 . In addition, similar to the findings in Sost null mice, sFRP1 affects, in a gene dosage–dependent fashion, the PTH-induced increase in trabecular bone, indicating that activation of Wnt signaling contributes to the anabolic action of PTH . Mice lacking PTH1R in osteocytes develop osteopenia with increased Sost expression and decreased canonical Wnt signaling , whereas osteocyte-specific constitutive activation of the PTHR1 leads to increased canonical Wnt signaling, increased cortical bone area, and a higher rate of periosteal and endocortical bone formation . Deletion of the Wnt receptor LRP5 in these mice reduces the increase in bone mass and PTH-induced bone gain is blunted in Sost-deficient or overexpressing mice, although with different effects on bone formation and resorption . Thus these findings demonstrate that PTH signaling regulates bone homeostasis in large part through osteocytes and emphasize the role of Wnt signaling in PTH responses.
12.4.3
Wnt and Hippo signaling
An emerging signaling cascade involved in cell fate, tissue development and homeostasis, the Hippo/YAP–TAZ signaling, has been recently implicated in bone biology . The Hippo pathway is a critical regulator of cell proliferation and organ size, and uncontrolled activity of this signaling cascade leads to tissue overgrowth, mainly due to increased cell proliferation . In mammals the Hippo pathway is a kinase cascade comprising the Mst1/2 and Lats1/2 kinases that end up phosphorylating and inhibiting YAP and TAZ in the cytoplasm. In this cascade, YAP and TAZ shuffle between the cytoplasm and the nucleus where they mediate the transcription of genes involved in cell proliferation, apoptosis, and differentiation. As such, their nuclear accumulation is the key determinant of their function. It has been shown that TAZ is a coactivator of Runx2 activity and several studies have now reported that deletion of TAZ and/or YAP in mature osteoblasts leads to impaired osteoblast differentiation and decreased bone mass in mice. Confirming these findings, Zebrafish embryos with decreased TAZ levels have defective bone formation, and mice overexpressing TAZ in osteoblasts display increased bone mass. The regulation of the Hippo cascade by upstream cues remains captivating in that no specific ligands have yet been identified. Multiple layers of interaction between WNT signaling and TAZ and YAP have been described . However, the mechanisms of the WNT–Hippo interaction or of how TAZ and YAP function independent of Hippo are not clear, especially in skeletal homeostasis. It has been proposed that cytoplasmic interactions between β-catenin and TAZ/YAP as well as TAZ and DVL inhibit canonical WNT signaling . Azzolin et al. have proposed a WNT/YAP/TAZ axis in which both TAZ and YAP are part of the destruction complex and WNT signaling activation leads to their translocation into the nucleus with subsequent activation of YAP/TAZ biological responses . Although these studies clearly show that YAP/TAZ intersects with canonical WNT signaling, these interactions have mainly been examined in the light of their ability to influence cell fate decisions, while their effects in the context of skeletal homeostasis remain unclear.
12.5
WNT signaling and mechanical loading
Establishing a potential link between bone remodeling, hormonal regulation, and skeletal homeostasis, one of the most important observations about the role of Wnt signaling in bone biology is its tight association with mechanosensing . Mechanical loading is of pivotal importance in the regulation of bone modeling and the maintenance of bone quality through bone remodeling. Osteocytes possess primary cilia residing in their lacunae and mechanical loading increases fluid shear stress along the canalicular network, activating signaling downstream of the cilium . Osteocytes are the main source of sclerostin and the expression of this canonical Wnt inhibitor is strongly repressed by mechanical stimuli . As mentioned earlier, mutations in Sost have dramatic effects on the skeleton and mice lacking sclerostin are protected from the bone loss induced by mechanical unloading .
Interestingly, since sclerostin expression is also repressed by PTH, this establishes a link between loading, PTH, and Wnt signaling. Moreover, LRP5 has also been implicated in the response of bone to mechanical loading. Indeed, LRP5 mutant mice exhibit altered sensitivity to mechanical loading with decreased osteogenic response in LRP5 null mice and increased response in the gain-of-function transgenic mouse . Furthermore, deletion of β-catenin in osteocytes reduces new bone formation in response to mechanical loading . Thus there is compelling evidence for a central role for canonical Wnt signaling in osteocytes in the response to loading, a link that might be of particular importance when considering the possibilities of targeting Wnt signaling for the treatment of bone diseases.
12.6
Targeting the WNT pathway for therapeutic intervention
The observations made in humans with mutations in components of the canonical Wnt pathway and several aspects of the Wnt signaling machinery have made this pathway a highly desirable target for therapeutic intervention in clinical situations where bone mass is low and bone formation needs to be enhanced, such as osteoporosis, osteogenesis imperfecta, and fracture healing.
Of particular importance are the observations made in heterozygote sclerosteosis or Van Buchem patients carrying mutations in Sost in only one allele, and thereby expressing about 50% of normal sclerostin levels . In these patients, bone mass is increased at both trabecular and cortical sites but none of the deleterious effects of full deletion of Sost, such as increased intracranial pressure and closure of foramens, are observed. Furthermore, biochemical markers of bone formation indicate that if bone formation is high at younger ages it appears to decrease after 30–35 years of age to be within the upper limits of normal. Similarly, HBM patients, in which mutations in LRP5 or 6 affect sclerostin affinity, exhibit HBM but no deleterious effects from the increased bone formation . These findings therefore suggest that a moderate increase in canonical Wnt signaling, possibly affecting mostly bone because of the restricted pattern of expression of sclerostin, increases bone mass but remains relatively safe. Moreover, the fact that sclerostin is regulated by mechanical loading suggests the possibility that bone formation is “directed” to the right places in the skeleton, preventing bone being formed in unwanted areas.
Another property of the Wnt signaling pathway that makes it a very favorable target for therapeutic intervention is the fact that it can be modulated by intervention in the extracellular milieu, where several endogenous inhibitors reside. Moreover, as mentioned, one of these inhibitors, sclerostin, appears to be quite restricted to the bone microenvironment.
Antibodies to sclerostin have been generated and one has been recently approved by FDA to treat postmenopausal women with osteoporosis and high risk of fractures. Over the last decade, preclinical and clinical studies have shown that sclerostin antibodies increase bone formation and bone mass dramatically. One of the most striking aspects of all these studies was the fact that bone formation is increased not only in the context of bone remodeling but also, and predominantly, in the context of de novo bone formation along quiescent surfaces, that is, bone modeling. Furthermore, this increase in bone formation occurs along trabecular, endocortical, and periosteal surfaces, increasing cortical thickness markedly and thereby potentially affecting nonvertebral fracture risk, if translated in the human . Indeed, Phase-I and Phase-II clinical trials in healthy human subjects confirmed that decreasing sclerostin levels with a single dose of a neutralizing antibody to sclerostin results in a marked increase in serum procollagen type 1 amino-terminal propeptide (P1NP), a bone formation marker, and a significant reduction in CTX-1, a serum resorption marker . Consistent with these findings, Phase III clinical trials: the FRAME study, the ARCH study, and the Bridge study have confirmed the role of blocking sclerostin on bone mass . Importantly, while the FRAME trial did not show any safety issue , the ARCH trial showed cardiovascular adverse events in the Romosozumab-treated group compared with the Alendronate-treated group, suggesting that the expression of sclerostin in blood vessels and in particular in vascular pathophysiology associated with vascular calcification might have a role in these pathologies . Thus although these safety issues need to be taken in consideration, it is clear that activation of the canonical Wnt signaling pathway by the endogenous inhibitor sclerostin increases both trabecular and cortical bone formation and bone strength to reduce fracture risk in humans. Given the very striking effects of inhibiting sclerostin on bone formation, antibodies to sclerostin have also been tested in animal models of fracture healing where they have been shown to markedly enhance callus formation as well to treat osteogenesis imperfecta . In addition, sclerostin antibody therapy prevents bone loss in mice undergoing partial weight-bearing, which might be of clinical importance for the prevention of bone loss in bed-rest patients as well as in astronauts subjected to spaceflight-induced bone loss . Although less restricted to the bone environment and to osteocytes, Dkk1 is also an extracellular soluble antagonist to Wnt signaling and therefore a potential target for therapeutic intervention. Animal studies have shown that Dkk1 haploinsufficiency results in increased bone formation and bone mass in mice and preclinical studies suggest that Dkk1 is implicated in glucocorticoid- and estrogen deficiency–mediated osteoporosis . In addition, there is evidence that Dkk1 plays a role in inflammatory arthritis . Dkk1 protein expression is enhanced in synovial fibroblasts and is increased in the serum of patients with rheumatoid arthritis (RA). Although Dkk1 is probably also a negative factor in the pathogenesis of osteoporosis, it is mostly in multiple myeloma that it has been shown to be a key component of the pathological process, inhibiting bone formation locally and enhancing bone resorption . Consequently, neutralizing Dkk1 antibodies hold potential for inhibition of osteolytic lesions in multiple myeloma . Consistent with the genetic mice data, preclinical animal studies suggest that inhibition of DKK1 induces bone gain in preclinical animal models and may decrease erosive bone destruction observed in RA and could thereby serve as an adjunct treatment in this disease.
12.7
Conclusion
The last decade has provided a large number of experimental and clinical data demonstrating that Wnt signaling is a major regulator of postnatal bone homeostasis. An enormous amount of work and studies have been compiled, clearly demonstrating that different players in the pathway are involved in the regulation of bone mass. In mammals, there are 19 Wnt proteins that, by engaging various receptor complexes, activate different signaling cascades to orchestrate the activity of mesenchymal progenitors, osteoblasts, osteocytes, and osteoclasts and favor bone formation while suppressing bone resorption. Collectively, mouse genetic and clinical studies have opened novel avenues for therapeutic intervention in bone diseases and show great promise for the treatment of osteoporosis, fracture healing, and other skeletal diseases associated with bone fragility.
References
Related posts:
 Osteoclast biology
Osteoclast biology
 Impact of physical characteristics and lifestyle factors on bone density and fractures
Impact of physical characteristics and lifestyle factors on bone density and fractures
 Clinical and epidemiological studies: skeletal changes across menopause
Diabetes, diabetic medications, and risk of fracture
Clinical and epidemiological studies: skeletal changes across menopause
Diabetes, diabetic medications, and risk of fracture
 Relationship between periodontal disease, tooth loss, and osteoporosis
Relationship between periodontal disease, tooth loss, and osteoporosis
 On the evolution and contemporary roles of bone remodeling
On the evolution and contemporary roles of bone remodeling
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


