Wilms Tumor
Wilms tumor (WT, nephroblastoma) is a highly curable childhood neoplasm. The prognosis of children with WT has improved considerably from a very high mortality rate at the beginning of the 20th century to the current cure rate of >90%.1 The management of WT is a paradigm for successful interdisciplinary treatment of solid tumors of childhood to maximize cure rates and minimize treatment-related complications.
 EPIDEMIOLOGY
EPIDEMIOLOGY
WT is the most common malignant renal tumor of childhood. It occurs with an annual incidence of 7 cases per million children <15 years of age. Approximately 500 new cases are diagnosed each year in North America. The peak incidence is between 3 and 4 years of age. WT may arise as sporadic or hereditary tumors or in the setting of specific genetic disorders.2 Most WTs are solitary lesions, multifocal within a single kidney in 12% and bilateral in 7%.3 The clinical syndromes associated with WT include WAGR syndrome (WT, Aniridia, Genitourinary malformations, mental Retardation), Denys-Drash syndrome (pseudohermaphroditism, mesangial sclerosis, renal failure, and WT), and overgrowth syndromes like Beckwith-Wiedemann syndrome (somatic gigantism, omphalocele, macroglossia, genitourinary abnormalities, ear creases, hypoglycemia, hemihypertrophy, and a predisposition to WT and other malignancies) and Simpson-Golabi-Behmel syndrome.4,5
 BIOLOGY
BIOLOGY
Among the various genetic changes implicated in the development of WT, the most widely studied involves WT1, which is a tumor suppressor gene at chromosome 11p13 that was isolated from a child with WAGR syndrome.6 WT1 is likely to play a specific role in glomerular and gonadal development.7 WT1 can also act as a dominant negative oncogene resulting in abnormal cell growth such as in Denys-Drash syndrome.8 Germline WT1 mutations are observed in approximately 82% of WT patients who have genitourinary anomalies or renal failure. The frequency of WT1 mutations in sporadic and familial WT is much lower at ~20% and ~4%, respectively.9 Beckwith-Wiedemann syndrome maps to chromosome 11p15.5; this locus is also referred to as WT2.10
Patients with loss of heterozygosity (LOH) at 16q and 1p have higher relapse and mortality rates.11 The National Wilms Tumor Study-5 (NWTS-5) prospectively evaluated the prognostic significance of LOH on 16q and 1p. Analysis of these data revealed that the relative risks (RR) for relapse for patients with stages I to IV favorable histology (FH) tumors with LOH stratified by stage were 1.8 for LOH 1p (P <.01) and 1.4 for LOH 16q (P = .05). When the effects of LOH for both 1p and 16q were considered jointly, the RR for relapse in stages I and II FH disease was 2.9 (P = .001) and for stages III and IV FH disease was 2.4 (P = .01). The RR for death for patients with stages I and II FH disease with LOH for both regions was 4.3 (P = .01) and for stages III and IV was 2.7 (P = .04). Based on these results, it was proposed that in future WT trials, the therapy for children with LOH at both 1p and 16q be augmented by the addition of doxorubicin to regimen EE4A (discussed below) for early-stage (stages I and II) tumors and cyclophosphamide/etoposide to regimen DD4A (discussed below) for advanced-stage tumors (stages III and IV).12
A novel Wilms tumor suppressor gene on the X chromosome, WTX, was recently discovered. This gene is inactivated in approximately one-third of sporadic WT cases.13 Anaplastic tumors have shown changes on 17p consistent with TP53 deletion and specific genomic loss or underexpression on 4q and 14q and focal gain of MYCN.14 Rhabdoid tumors are characterized by the genetic loss of the SMARCB1/hSNF5/INI-1 gene located at chromosome 22q11. Global gene expression studies have shown that loss of SMARCB1 results in repression of neural crest development and loss of cyclin-dependent kinase inhibition.15 In children with very low-risk WT treated with just surgery alone, the presence of WT1 mutation and 11p15 loss have been prospectively validated to be an important predictor of relapse. These biomarkers may be used to stratify patients to receive reduced chemotherapy in the future.16
 PATHOLOGIC CLASSIFICATION
PATHOLOGIC CLASSIFICATION
Although histopathologists had attempted to relate appearance to prognosis, no generally acceptable classification was available until the report of Beckwith and Palmer17 from the National Wilms Tumor Study-1 (NWTS-1). The NWTS classifies all tumors as having either FH or unfavorable histology (UH). The UH tumors include anaplastic tumors, clear cell sarcoma, and rhabdoid tumor of kidney. Of 1,465 patients randomly assigned on NWTS-3, 163 (11.1%) had UH.18 WTs are usually sharply demarcated, spherical masses with a “pushing” border and a surrounding distinct intrarenal pseudocapsule. Histologically, WT reflects the development of the normal kidney, consisting of three components: blastemal, epithelial (tubules), and stromal elements, in varying proportions.17 The proportion of the different components has prognostic significance.19 Nephrogenic rests consist of embryonal nephroblastic tissue and are found in 35% of kidneys with unilateral WT and in nearly 100% of kidneys with bilateral WT.20 Nephrogenic rests may be intralobar or perilobar based on their location within the kidney.21 Most nephrogenic rests undergo spontaneous regression and only a small proportion (1% to 5%) transform into WT.22 The histologic feature of greatest clinical significance in WT is anaplasia.23 Anaplasia may be focal (FA) or diffuse (DA). The definitions of FA and DA have been revised to reflect the distribution of anaplastic cells in the tumor rather than their quantitative density. These revised definitions are of prognostic significance. The 4-year survival rates for patients with stages II, III, and IV FA were 90%, 100%, and 100%, compared with 55%, 45%, and 4%, respectively, for patients with similar stage DA WT.24
Clear cell sarcoma of kidney (CCSK) and malignant rhabdoid tumor of kidney (RTK) are no longer considered true WT, but they have been included in NWTS protocols.17 CCSK has a propensity to metastasize to bone, and a skeletal survey and bone scan should be performed. RTK is the most lethal renal neoplasm in children. Primitive neuroepithelial tumors of the cerebellum or pineal region may be seen in 10% to 15% of patients with RTK.25
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
The classic presentation for WT is that of a healthy child in whom abdominal swelling is discovered by the child’s mother or by a physician during a routine physical examination. A smooth, firm, nontender mass on one side of the abdomen is felt. Gross hematuria occurs in as many as 25% of these cases.26 The child may be hypertensive or have nonspecific symptoms such as malaise or fever.27 Only rarely does a patient present with symptomatic metastases.
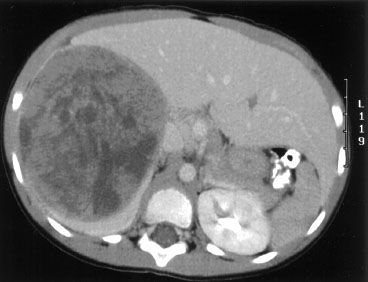
FIGURE 85.1. Computed tomography scan of a 4-year-old girl with a large right-sided Wilms’ tumor measuring 10.5 é 8.4 é 13.5 cm. The left kidney did not show any lesions. At laparotomy, the tumor was found to invade the diaphragm. She underwent a right radical nephrectomy. Surgical margins of resection were positive and she had metastases in the para-aortic lymph nodes. The tumor was classified as stage III favorable histology and received 10.8 Gy to the right flank and chemotherapy with vincristine, dactinomycin, and doxorubicin.
 DIAGNOSTIC WORK-UP
DIAGNOSTIC WORK-UP
The differential diagnosis of WT includes other malignant childhood lesions of the kidney, neuroblastoma, and benign conditions such as hydronephrosis, polycystic disease, and splenomegaly in left-sided tumors. Plain films of the abdomen may demonstrate calcifications, which occur in 60% to 70% of neuroblastomas but in only 5% to 10% of WT. Excretory urography (intravenous pyelography) was once the mainstay of imaging in WT and now has largely been replaced by ultrasonography and computed tomography (CT) scanning. Ultrasonography is very useful because it is readily available and is cost-effective.28 A specific advantage of ultrasonography is its ability to assess vessels for flow and tumor thrombus with duplex and color Doppler.29 Routine use of Doppler sonography after abdominal CT scans was not found to be useful in detecting cavoatrial thrombus in a Children’s Oncology Group (COG) study.30 Abdominal CT scans can demonstrate gross extrarenal spread, lymph node involvement, liver metastases, and the status of the opposite kidney (Fig. 85.1).31 Magnetic resonance imaging (MRI) has several advantages over CT scans, especially in identifying renal origin and vascular extension of the tumor.32 CT and MRI are useful in the detection and follow-up of patients with nephrogenic rests.33 Clinical and imaging impressions do not, however, obviate the need for inspection at laparotomy.34 Plain chest radiography and chest CT are also essential because asymptomatic pulmonary metastases are common.35 A complete blood cell count and urinalysis should be performed. Patients with WT can be anemic from hematuria. Serum blood urea nitrogen and creatinine levels and liver function tests are routine. If neuroblastoma is not ruled out, a test for urinary catecholamines should be performed. Table 85.1 outlines the pretreatment investigations recommended by the COG.
 NATURAL HISTORY
NATURAL HISTORY
The disease is often localized at diagnosis, as evidenced by the fact that surgery and radiation therapy is curative in almost 50% of cases.36 The first signs of local tumor spread beyond the pseudocapsule are invasion into the renal sinus or the intrarenal blood and lymphatic vessels. Spread throughout the peritoneal cavity may also occur, especially if there has been preoperative rupture or the disease has been spilled at surgery.37,38 The most common sites of metastases of WT are in the lungs, lymph nodes, and liver. Among patients with stage IV disease, lungs were the only metastatic site in approximately 80% and 15% have liver metastases.39 The NWTS-2 study demonstrated the prognostic importance of lymph node involvement. The 2-year relapse-free survival (RFS) with and without lymph node involvement was 54% and 82%, respectively.37
TABLE 85.1 PRETREATMENT WORK-UP
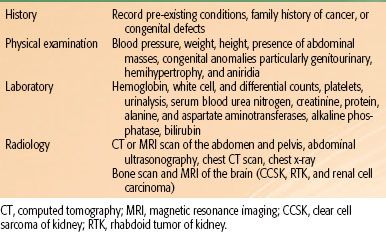
TABLE 85.2 CHILDREN’S ONCOLOGY GROUP STAGING OF WILMS’ TUMOR, RHABDOID TUMOR, AND CLEAR CELL SARCOMA OF THE KIDNEY
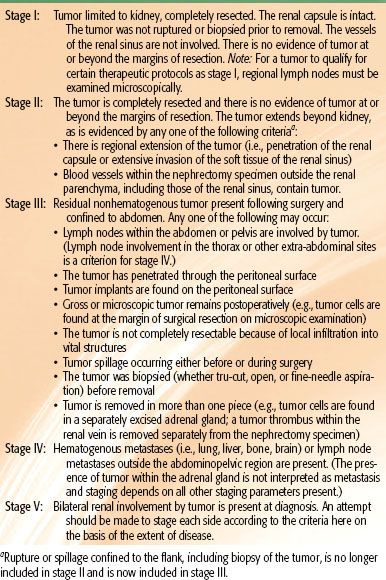
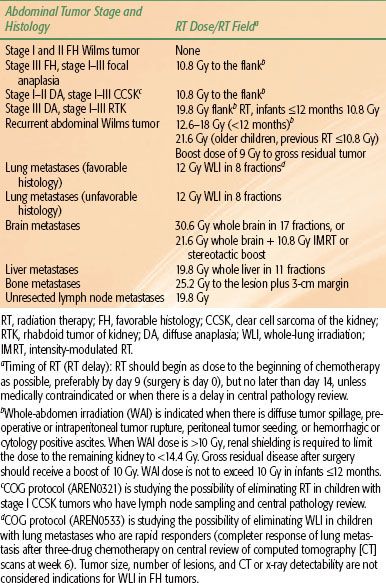
TABLE 85.3 CHILDREN’S ONCOLOGY GROUP RISK GROUP CLASSIFICATION FOR FAVORABLE HISTOLOGY WILMS’ TUMORS
 STAGING
STAGING
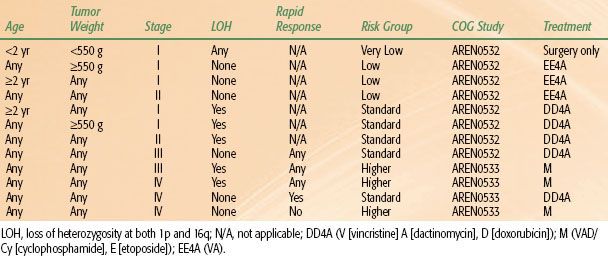
Tumor staging is performed after examining the radiologic, operative, and histopathologic findings.38,39 In NWTS-1 and NWTS-2, a tumor grouping system was used for staging and treatment stratification. After analyzing the prognostic significance of several clinicopathologic factors in NWTS-1 and NWTS-2, a new staging system was adopted in NWTS-3. The presence of lymph node involvement was upstaged to stage III instead of group II, and local tumor spill was downstaged from group III to stage II.38 In NWTS-5, the most significant change was the distinction between stages I and II. The criteria for stage I was revised to accommodate an important subset of WT that is being managed by nephrectomy alone. Before NWTS-5, the distinction between stages I and II in the renal sinus was established by the hilar plane, which was an imaginary plane connecting the most medial aspects of the upper and lower poles of the kidney. This criterion was difficult to apply because of tumor distortion, and thus the hilar plane criterion has been replaced with renal sinus vascular or lymphatic invasion. This definition includes not only the involvement of vessels within the hilar soft tissue, but also the vessels located in the radial extensions of the renal sinus into the renal parenchyma.40,41 The COG staging guidelines for WT are shown in Table 85.2. The major change from NWTS-5 is that children with tumor spillage are upstaged from stage II to stage III because of the higher risk for relapse with two-drug chemotherapy alone.42 The COG risk group classification for treatment assignment in the new generation of WT protocols is shown in Table 85.3. In addition to tumor stage, this classification will also consider the patient’s age, tumor weight, presence or absence of LOH at 1p and 16q, and response to chemotherapy in children with FH tumors and lung metastases.
 GENERAL MANAGEMENT
GENERAL MANAGEMENT
The diagnosis of WT is usually made before surgery and confirmed at surgery. A transverse transabdominal, transperitoneal incision is recommended for adequate exposure and thorough abdominal exploration.43 The surgeon must excise all tumors without spillage, if possible. Lymph node sampling from the para-aortic, celiac, and iliac areas must be performed. The use of titanium clips to identify residual tumor and margins of resection is also recommended. Routine exploration of the contralateral kidney was mandated in the past, but it is no longer recommended due to better imaging of the contralateral kidney with CT and MRI scans.
The chemotherapy and radiation therapy (RT) regimens for WT in the COG protocols are outlined in Tables 85.4 and 85.5.
TABLE 85.4 OUTLINE OF CHILDREN’S ONCOLOGY GROUP RENAL TUMOR STUDY
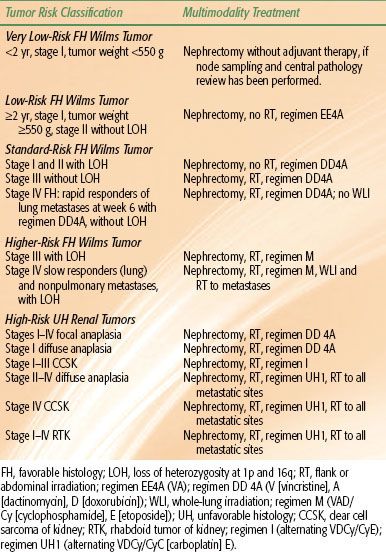
TABLE 85.5 CHILDREN’S ONCOLOGY GROUP RENAL TUMOR PROTOCOL RADIATION THERAPY GUIDELINES
 RADIATION THERAPY TECHNIQUES
RADIATION THERAPY TECHNIQUES
RT guidelines used for primary and recurrent WT in the COG protocols are shown in Table 85.5.
Timing of Radiation Therapy
The NWTS has shown that although RT does not need to be given immediately after surgery,36 a delay of ≥10 days after surgery was associated with a significantly higher abdominal relapse rate, particularly among patients with UH tumors.44–47 Because the pathologist cannot always rule out UH quickly, all patients with WT should be scheduled to start RT no later than day 9, the day of surgery being day 0. Although most patients may not be irradiated, it is easier to cancel than to make arrangements to start RT for a small child on short notice. The influence of RT delay on abdominal tumor recurrence in patients with FH tumors treated on NWTS-3 and NWTS-4 has been reported. The mean RT delay was 10.9 days. Although univariate and multivariate analysis did not reveal RT delay of ≥10 days to adversely influence flank and abdominal recurrence, it is important to note that in 59% of children the RT delay ranged from 8 to 12 days.48 For the COG protocols, it is recommended that RT be given preferably by day 9 but no later than day 14 after surgery.
Radiation Therapy Dose
In NWTS-1 and NWTS-2 RT dosages to the operative bed were given according to the age of the patient, however, no significant dose–response association was detected.45,47 In NWTS-3, there was a randomization for patients with FH tumors that resulted in elimination of RT for stage II FH, and a reduction of dose to 10 Gy for stage III patients.46 NWTS-3 and NWTS-4 data showed no RT dose response for CCSK and anaplastic tumors.49 Therefore, it was decided to treat all abdominal disease with 10 Gy. In the COG protocols, the dose is 10 Gy for most indications except for stage III DA and stages I to III RTK, where a higher dose of 19.8 Gy is recommended (Table 85.5).50,51
Radiation Therapy Volume
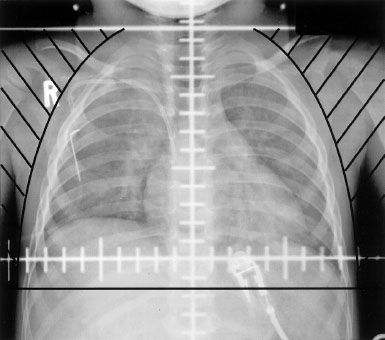
Parallel-opposed fields using 4 or 6 MV photons are preferred. The flank RT field is determined by the CT or MRI scan performed at diagnosis before any chemotherapy is administered. The planning target volume is the tumor bed (outline of the kidney and associated tumor on the initial CT or MRI) with a 1-cm margin. The medial border must cross the midline to include the entire width of the vertebrae so as to minimize growth disturbances. An example of a flank RT portal is shown in Figure 85.2.44 When whole-abdomen RT is administered, the femoral heads and acetabulum must be shielded (Fig. 85.3). Whole-lung Irradiation (WLI) portals are shown in Figure 85.4. If the lungs and either the flank or whole abdomen have to be treated simultaneously, it is preferable to include them in one treatment portal.
FIGURE 85.2. Anteroposterior flank irradiation portal in a 2-year-old child with a left-sided stage III favorable histology Wilms tumor (WT), showing inclusion of the entire width of the vertebral body in the irradiated volume. The outline of the right kidney (RK) and the WT from the preoperative computed tomography scan is shown.
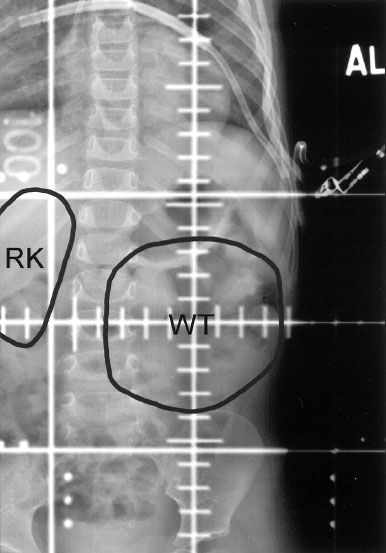
FIGURE 85.3. Anteroposterior whole-abdomen irradiation portal in a patient with stage III Wilms tumor and diffuse peritoneal tumor spillage. The upper margin of the abdominal field must include the diaphragm. The acetabulum and femoral head should be excluded from the irradiated volume to decrease the probability of slipped femoral capital epiphysis. The pants zipper can be seen low on the hips. In general, it is advisable to remove the trousers completely to ensure a reproducible setup.
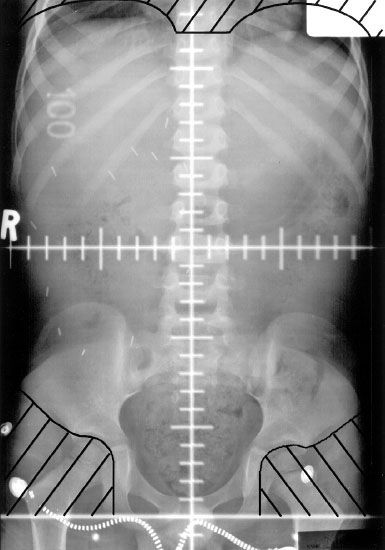
FIGURE 85.4. Anteroposterior whole-lung irradiation portal in a patient with stage IV favorable histology Wilms tumor. The axial, coronal, and sagittal chest computed tomography simulation scans should be carefully reviewed to ascertain inclusion of the anterior and posterior costophrenic angles at the inferior edge of the treatment volume with a 1-cm margin.
 SUMMARY OF CLINICAL TRIALS
SUMMARY OF CLINICAL TRIALS
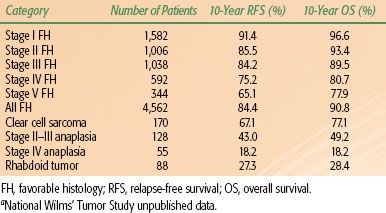
No tumor has been studied by clinical trials as thoroughly and effectively as WT. The NWTS has been active in North America since 1969. There have also been successful studies run by the International Society for Pediatric Oncology (SIOP). The long-term results of NWTS-3 and NWTS-4 are shown in Table 85.6.
First National Wilms Tumor Study (1969–1974)
NWTS-1 showed that postoperative RT was not necessary for children younger than 2 years of age with group I tumors, and that combined dactinomycin and vincristine for irradiated patients with group II and III tumors was better than therapy with either agent alone. The RFS with and without RT among patients with group I tumors younger than 2 years of age was 90% and 88%, respectively.44
TABLE 85.6 LONG-TERM RESULTS OF NATIONAL WILMS TUMOR STUDIES 3 AND 4A
Second National Wilms Tumor Study (1974–1979)
NWTS-2 showed that in patients with group I tumors there was no survival difference between 6 months or 15 months of dactinomycin plus vincristine. Patients with groups II to IV tumors had a superior 2-year RFS of 77% with doxorubicin, dactinomycin, and vincristine compared with 63% with dactinomycin and vincristine alone.37
Third National Wilms Tumor Study (1979–1985)
The overall objective of NWTS-3 was to reduce therapy for low-risk patients (stages I to III FH) and to intensify treatment by adding a fourth drug, cyclophosphamide, for stage IV tumors with FH and all UH tumors. The results of this study demonstrated that RT and doxorubicin could be eliminated in children with stage II FH tumors. Patients with stage III FH tumors who received doxorubicin or 20 Gy had fewer abdominal relapses than those receiving 10 Gy without doxorubicin.46 The addition of cyclophosphamide in high-risk patients did not improve outcomes.49
Fourth National Wilms Tumor Study (1986–1994)
By the conclusion of NWTS-3, it was clear that the treatment of WT had been refined for the majority of patients; 62% of patients with WT have stage I or II FH disease and therefore require neither flank RT nor the potentially cardiotoxic doxorubicin. NWTS-4 was designed with cost containment in mind. The results proved that the survival was similar among patients who received standard-course (5 days) or single-dose, pulse-intensive dactinomycin chemotherapy. Further, pulse-intensive therapy was associated with less hematologic toxicity and marked reduction of treatment costs.52,53
Fifth National Wilms Tumor Study (1995–2001)
One of the major goals of the NWTS-5 trial was to prospectively analyze the prognostic significance of LOH at chromosomes 1p and 16q. These results have been discussed elsewhere.12 Patients with stage I FH and anaplastic histology and stage II FH tumors were treated with 18 weeks of dactinomycin and vincristine without RT. Stage I FH tumors in children younger than 24 months of age and with tumor weight less than 550 g were treated with surgery alone. Stages III and IV FH and stages II to IV focal anaplastic tumors were treated with 24 weeks of dactinomycin, vincristine, doxorubicin, and irradiation. Stages II to IV diffuse anaplastic tumors and stages I to IV CCSK were treated with cyclophosphamide, vincristine, doxorubicin, and etoposide along with irradiation. Stages I to IV RTK were treated with carboplatin, etoposide, cyclophosphamide, and irradiation. The RT guidelines in NWTS-5 were similar to those used in NWTS-4 except for anaplastic tumors, where a dose of 10.8 Gy to the flank and abdomen was recommended compared to an age-adjusted schedule used in NWTS-1 to NWTS-4.
Children’s Oncology Group Studies (2002–)
The COG renal tumors committee is the successor of the NWTS. The COG staging and risk-group classification for treatment assignment in the new generation of WT protocols are shown in Tables 85.2 and 85.3. This classification will, in addition to tumor stage, also consider the patient’s age, tumor weight, presence or absence of LOH at 1p and 16q, and response to chemotherapy in children with FH tumors and lung metastases. The main objectives of the first generation of COG protocols are listed below. The COG chemotherapy regimens and RT guidelines are outlined in Tables 85.4 and 85.5, respectively.
AREN0321
This is a study for children with high-risk renal tumors. This study will determine whether a regimen of cyclophosphamide/carboplatin/etoposide alternating with vincristine/doxorubicin/cyclophosphamide improves the survival of patients with DA and RTK. This study will also determine whether the excellent event-free survival in stage I CCSK can be maintained without the use of abdominal RT.
AREN0532
This is a study for children with very low and standard risk FH WT. The main objectives are: (a) to demonstrate that very low-risk patients treated by nephrectomy and observation alone will have a 4-year RFS of ≥85% and overall survival (OS) of ≥95%; (b) to document continued excellent outcome for patients with stage III WT without LOH of 1p and 16q treated with vincristine, dactinomycin, doxorubicin, and RT (regimen DD4A); (c) to improve the current 4-year RFS for patients with stages I and II WT with LOH of 1p and 16q by adding doxorubicin but not RT to the standard dactinomycin and vincristine regimen.
AREN0533
This is a study for higher risk FH WT. The main objectives are: (a) to demonstrate that patients with stage IV tumors with pulmonary metastases only, who have complete resolution of the pulmonary lesions after 6 weeks of regimen DD4A chemotherapy (rapid complete responders) will have a 4-year RFS of 85% with additional chemotherapy (regimen DD4A) and without WLI; (b) to demonstrate that stage IV patients who do not have resolution of pulmonary metastases by week 6 (slow incomplete responders) will have a 4-year RFS of 85% with the addition of cyclophosphamide and etoposide to a modified regimen DD4A (regimen M) and WLI; (c) to improve the 4-year RFS to 75% for patients with stage III or IV FH WT with LOH of chromosomes 1p and 16q.
AREN0534
This is a study for patients with bilateral, multicentric, or bilaterally predisposed unilateral WT. The main objectives are: (a) to improve 4-year RFS to 73% for patients with bilateral Wilms tumor (BWT); (b) to prevent complete removal of at least one kidney in 50% of patients with BWT by using prenephrectomy three-drug chemotherapy induction with vincristine, dactinomycin, and doxorubicin; (c) to have 75% of children with BWT undergo definitive surgical treatment by 12 weeks after initiation of chemotherapy.
Outcomes of Children with Wilms Tumor
Lung Metastases
Patients with stage IV FH with lung metastases had a 4-year survival of 80% on NWTS-3, whereas survival for those with stage IV UH was 55%.18,39,49 In a United Kingdom Children’s Cancer Study Group (UKCCSG) trial, patients with stage IV FH were spared WLI if they had complete resolution of pulmonary metastases after chemotherapy. The 6-year RFS and OS were 50% and 65%, respectively.54 These results appear to be somewhat worse than the 4-year survival rate of 82% on NWTS-3 and 75% in the second UKCCSG Wilms tumor study due to the greater use of WLI.18,55 In children with FH tumors enrolled in NWTS-3 and NWTS-4 who had negative chest radiographs and CT scans positive for pulmonary metastases, the 4-year RFS with and without WLI was similar at 89% and 80%, respectively.56 In a report from NWTS-4 and NWTS-5, among children with lung metastases detected only by CT scans, the 5-year RFS after three drugs with or without WLI was significantly higher than those receiving two drugs (80% vs. 56%). There was no difference seen in 5-year OS between the three-drug and two-drug subsets (87% vs. 86%). There were no significant differences in RFS (82% vs. 72%) or OS (91% vs. 83%) based on whether these patients did or did not receive WLI. This report concluded that in patients with CT-only lung lesions, the addition of doxorubicin may improve RFS but not OS, and there was no added benefit from WLI.57 In COG protocol AREN0533, chemotherapy response at week 6 will be used to determine whether WLI is delivered or not. Patients with FH tumors and lung metastasis who achieve a complete radiologic response to three-drug chemotherapy at week 6 will not receive WLI, all others will receive WLI. All patients with UH WT and lung metastases will receive WLI, regardless of response to chemotherapy (Tables 85.4 and 85.5).
Liver Metastases
Patients with liver metastasis undergo hepatic RT if the metastatic lesions are not completely resected at the time of initial diagnosis before any chemotherapy is delivered. Whole-liver RT is given for diffuse disease, with supplementary boosts to gross disease as indicated. When possible, however, more limited RT fields are used if the disease is more localized in the liver. In a report from NWTS-4 and NWTS-5, the RFS for patients with FH WT and liver metastases was 76%, and this was similar to the RFS in patients with lung metastases (76%), liver and lung metastases (70%), and metastases to other sites (64%).58
Bilateral Wilms Tumor
The goals of treatment in BWT are to maximize cure rates and to preserve functional renal parenchyma; thus, the role of radical nephrectomy and RT has been restricted. Initial surgical resections should be performed only if more than two-thirds of each kidney can be preserved.59 The initial surgery should confirm histologic diagnosis, assess extent of disease in the kidney, and perform a lymph node biopsy. Systemic chemotherapy is then delivered, after which second-look surgery is done in order to perform tumor resection with maximal preservation of renal function. Unpublished 10-year data from the NWTS-3 and NWTS-4 showed that BWT patients, compared with those who have stages I to IV unilateral FH tumors, had lower RFS (65% and 86%) and OS (78% and 92%). In NWTS-4, the 8-year RFS and OS were 74% and 89%, respectively, for FH and 40% and 45%, respectively, for UH BWT. The incidence of end-stage renal disease was 12% among stage V patients.60 In NWTS-5, BWT patients had a 4-year RFS and OS of only 61% and 80%, respectively. The factors that might have contributed to these poor outcomes were understaging or undertreatment, delay in definitive surgical resection, and increased incidence of anaplasia.61,62 The current COG protocol (AREN0534) recommends earlier biopsies or resection of nonresponsive tumors so that ineffective therapies for patients with DA could be avoided. This study will intensify chemotherapy upfront (three drugs), require second-look surgery at 6 weeks and definitive surgery at 12 weeks, and recommends chemotherapy based on histologic response after definitive surgery. Radiation therapy is indicated for stage III FH tumors, stages I to III UH tumors, or when chemotherapy and several surgeries do not result in complete tumor resection with negative margins. Unlike in unilateral WT, the performance of a tumor biopsy or the use of chemotherapy before definitive surgery is not an indication for flank RT in BWT.
Effect of Tumor Spillage on Outcome in Patients with Stages II and III Disease
Operative tumor spillage was identified in 24% of patients in NWTS-3 and NWTS-4, and 22% of the spills were classified as diffuse.42,63,64 An analysis was undertaken to determine the influence of RT and chemotherapy on abdominal tumor recurrence caused by spilled cells in abdominal stages II and III FH WT. The odds ratio for the risk of recurrence relative to no RT was 0.35 for 10 Gy (P = .01) and 0.08 for 20 Gy (P = .01). Thus, RT (10 or 20 Gy) significantly reduced abdominal tumor recurrence rates following tumor spillage. After adjusting for RT, the effect of doxorubicin on tumor recurrence was not significant. For stage II patients (NWTS-4), the 8-year RFS and OS with and without spillage were 74% and 85% (P = .02) and 90% and 94% (P = .4), respectively. The higher relapse rate among stage II children with spillage was the reason for upstaging patients with tumor spillage to stage III in the new COG staging system (Table 85.2). These patients will now receive three drugs and flank RT.42
Nephrectomy Only for Patients with Stage I Favorable Histology Wilms Tumor
In NWTS-5, a single-arm study was conducted to evaluate the efficacy of nephrectomy alone in children younger than 24 months of age with small (<550 g) stage I FH WT. A total of 75 children were enrolled and the 2-year RFS and OS were 87% and 100%, respectively. This study was ended because of stringent stopping rules.65 In a recent update, the 5-year RFS and OS for these children treated with just surgery alone were 85% and 98%, respectively. These outcomes were similar after treatment with surgery and two-drug chemotherapy (regimen EE4A).66
The COG will again examine the possibility of avoiding any chemotherapy or irradiation in these children. However, only those children (<24 months, tumors <550 g) who have stage I FH tumors after central pathology review, lymph nodes sampling, and CT scan staging will be eligible for the surgery-only therapy.
Wilms Tumor with Peritoneal Implants
The outcome of 57 patients with FH WT and peritoneal implants at the time of nephrectomy in NWTS-4 and NWTS-5 were analyzed. All children received multimodality therapy with three-drug chemotherapy and RT. Forty-seven patients (82%) received whole-abdominal RT to a dose of 10.5 Gy. The overall abdominal and systemic tumor control rates were 97% and 93%, respectively. The detection of peritoneal implants was not associated with inferior survival. The 5-year RFS with and without peritoneal implants was 90% and 83%, respectively.67
Clear Cell Sarcoma
In the NWTS-1 through NWTS-4 experience for 351 patients with CCSK, the OS rate was 69%. Multivariate analysis revealed four independent prognostic factors for survival: treatment with doxorubicin, tumor stage, age at diagnosis, and tumor necrosis.68 In NWTS-4, there was no significant difference among those patients initially randomized to pulse-intensive (PI) or standard chemotherapy with vincristine, dactinomycin, and doxorubicin. The 8-year RFS and OS were 72% and 87% for PI and 70% and 84% for standard chemotherapy, respectively. The second randomization to short-duration and long-duration chemotherapy also did not show any significant difference in survival between the two arms. The survival in NWTS-4 was significantly superior to that of NWTS-3 (83% vs. 67%). The PI chemotherapy administration of dactinomycin and doxorubicin in NWTS-4 was presumed to be one of the reasons for the improvement in outcomes.69
Rhabdoid Tumor of Kidney
A total of 142 children with RTK were enrolled in the NWTS-1 through NWTS-5 trials. The OS at 4 years was 23%. The survival rate for children with stages I or II tumors (42%) was significantly higher than for those with stages II or III tumors (16%; P = .014). The survival rate in infants <6 months of age was 9% compared with 41% in children >2 years of age (P <.001). Children who received a higher dose of RT (>25 Gy) had a significantly better outcome. However, the dose of RT was not an independent predictor of survival.51
Anaplastic Wilms Tumor
In NWTS-5, among 2,596 patients who were enrolled, 281 (11%) had anaplastic WT. The 4-year RFS and OS for patients with stage I anaplasia treated with vincristine and dactinomycin without RT were 70% and 83%, respectively. The 4-year RFS for anaplastic tumor patients who underwent immediate nephrectomy and regimen-I chemotherapy was 83%, 65%, and 33% for stages II, III and IV tumors, respectively. The 4-year RFS and OS for stage V tumors were 44% and 55%, respectively. Based on these results, the therapy for stages I, III, IV, and V tumors will be augmented in the new COG protocols (Tables 85.4 and 85.5).50
Recurrent Wilms Tumor
Children with relapsed FH WT have a variable prognosis depending on the site of relapse, the time from initial diagnosis to relapse, and their previous therapy. The favorable prognostic factors include no previous treatment with doxorubicin; relapse more than 12 months after diagnosis; and intra-abdominal relapse in a patient not previously treated with abdominal RT.70 In NWTS-5 relapse protocol, patients who relapsed after initial treatment with vincristine and dactinomycin only without RT were treated on stratum “B” with regimen “I” chemotherapy, surgery, and RT. The 4-year RFS and OS were 71% and 81% for all patients, 68% and 81% for those who relapsed in the lung only, and 78% and 83% for those who relapsed in the operative bed with or without lung metastasis.71 Patients who relapsed after treatment with vincristine, dactinomycin, doxorubicin, and RT were treated on stratum “C” of NWTS-5 protocol, with alternating courses of drug pairs; cyclophosphamide/etoposide and carboplatin/etoposide, surgery, and RT. The 4-year RFS and OS were 42% and 48% for all patients, and 49% and 53% for those who relapsed in the lung only.72 The COG protocol (Table 85.5) recommends postoperative RT for all children with abdominal relapse because these tumors are generally large and infiltrative, and surgical resection with negative margins is unlikely.
Wilms Tumor in Older Patients
WT is rarely seen in patients ≥16 years of age. Their survival is similar to that of children and they should be treated similarly.73,74
International Society of Pediatric Oncology Trials
The SIOP studies have primarily used preoperative therapy. The goals of administering preoperative therapy are to facilitate surgical removal of the tumor without rupture, to allow for early treatment of micrometastases, and to stratify patients for postoperative therapy based on pathologic tumor response at the time of surgery. The first SIOP trial found that preoperative RT reduced the incidence of tumor spillage but did not increase survival.75 SIOP-5, reported in 1983, showed that preoperative chemotherapy with vincristine and dactinomycin was as effective as preoperative RT plus dactinomycin in preventing tumor rupture.76 In SIOP-6, patients with stage I disease were randomly assigned to either 17 or 38 weeks of vincristine and dactinomycin and showed no difference in survival. Among patients with stage II disease and negative lymph nodes (SIOP staging is not identical to NWTS staging) who were randomly assigned to not receive RT, there was a higher recurrence rate.77 In SIOP-9, there was a randomization of the duration of prenephrectomy therapy with vincristine and dactinomycin (4 weeks vs. 8 weeks). No advantage was noted for 8 weeks of therapy. Among patients with stage II disease with negative lymph nodes, the rate of abdominal relapse was reduced to 7% by the addition of epirubicin.78 SIOP-93–01 further stratified treatment according to the pathologic response to preoperative chemotherapy. The recommended dose of RT in SIOP-9 and SIOP-93-01 was 15 Gy in patients with low- and intermediate-risk stage III disease and 30 Gy in high-risk patients.79 SIOP-93-01 showed that the amount of postoperative chemotherapy of stage I patients with either intermediate-risk histology or anaplasia could be reduced to four doses of vincristine and one course of dactinomycin with 5-year RFS and OS rates of 87% and 95%, respectively.80
United Kingdom Children’s Cancer Study Group
The first UKCCSG Wilms tumor study (UKW1) showed that vincristine could be used alone in patients with stage I FH disease. Among patients with lung metastases, the 6-year survival rate of 65% was significantly worse than the 4-year survival rate of 82% on NWTS-3, probably because of the inclusion of routine WLI in the NWTS.54 In the second UKCCSG WT study (UKW2), stage I FH patients had similar survival rates as NWTS stage I patients after 10 weekly doses of vincristine. The 4-year survival rate in patients with stage IV disease was higher than in UKW1, at 75%, probably due to the greater use of WLI. The flank RT doses used for stage III FH and UH tumors are 20 Gy and 30 Gy, respectively.55 The UKW3 trial conducted a randomized comparison of a primary nephrectomy followed by adjuvant therapy based on surgical stage (NWTS approach) and a preoperative chemotherapy followed by nephrectomy and adjuvant therapy (SIOP approach). The 4-year RFS and OS were equivalent in the primary nephrectomy arm (80% and 85%) and in the preoperative chemotherapy arm (79% and 95%), respectively. The UKCCSG has now joined the current SIOP clinical study.81
 LATE EFFECTS OF TREATMENT
LATE EFFECTS OF TREATMENT
The study of late effects is of paramount importance to prevent survivors of childhood cancer from becoming chronically sick adults.
Scoliosis
A series from Washington University showed a high incidence of scoliosis in 54% of patients who were treated with a median dose of 30 Gy. However, there was minimal functional disability.82 In another report, the incidence of scoliosis after 10 to 12 Gy, 12.1 to 23.9 Gy, and 24 to 40 Gy was 8%, 46%, and 63%, respectively.83 Thus, at present, with the use of megavoltage x-rays, lower doses and coverage of the entire width of the vertebra, the incidence of scoliosis should be low.
Congestive Heart Failure
The cumulative frequency of congestive heart failure among patients on NWTS-1 through NWTS-4 was 4.4% at 20 years among patients treated initially with doxorubicin and 17.4% among patients treated with doxorubicin for their first or subsequent relapse. The factors that were significantly associated with the incidence of heart failure were female sex, cumulative doxorubicin dose, WLI, and left abdominal RT.84
Pregnancy Outcome in Wilms Tumor Survivors
The NWTS Long-Term Follow-Up Study analyzed pregnancy outcomes among WT survivors. Malposition of the fetus and premature labor were significantly more frequent among previously irradiated women. The offspring of female patients who received flank RT were more likely to be of low birth weight (<2,500 g), premature (<36 weeks of gestation), and to have congenital malformations. A flank RT dose response was identified with higher complication rates at doses >25 Gy. A number of radiation-induced side effects involving the spine, uterus, and ovaries may all have been responsible.85–87 The pregnancy outcomes in survivors who received abdominal RT on NWTS protocols were also analyzed. Fertility could be preserved in children with upper abdominal RT that did not include the pelvis. In rare instances, fertility could be preserved after whole-abdominal RT to 10.5 Gy. However, higher doses to the abdomen and pelvis resulted in miscarriages and fetal deaths.88
 END-STAGE RENAL DISEASE
END-STAGE RENAL DISEASE
The 20-year cumulative incidence of end-stage renal disease among WT survivors after unilateral nephrectomy on NWTS protocols was 74% for children with Denys-Drash syndrome, 36% for children with WAGR syndrome, 7% for children with genitourinary anomalies, and 0.6% for patients with none of these conditions. The importance of long-term screening for high-risk children to facilitate early detection and treatment of impaired renal function was emphasized.89
Second Malignant Neoplasm
Among NWTS patients, the 15-year cumulative risk of second malignant neoplasm (SMN) was 1.6%. The risk of developing a lymphoma or leukemia was 0.4% at 8 years, after which no cases occurred. However, the risk of developing a solid tumor continued to rise sharply with time. Approximately 73% of solid tumors arose within a previous RT field. Higher doses of abdominal RT, doxorubicin use, and treatment for relapse were the significant factors correlated with the development of second tumors.90 In another NWTS report, the standardized mortality ratio was 24.3 within 5 years of diagnosis, 12.6 for the next 5 years, and >3.0 thereafter. The main cause of mortality within the first 5 years was the original disease (91%). However, beyond 5 years the two important causes of mortality were the original disease (40%) and late effects of treatment (39%). The three common treatment-related late effects that contributed to mortality were SMNs, congestive heart failure, and end-stage renal disease. The risk of death, particularly from treatment-related late effects, remained elevated even 20 years after diagnosis.91 Similar results have been shown recently by the Childhood Cancer Survivor Study after a follow-up of 25 years.92 In the British Cancer Survivor Study, the cumulative incidence of a second primary neoplasm at 30, 40, and 50 years of age was 2%, 7%, and 12%, respectively.93
 FUTURE DIRECTIONS
FUTURE DIRECTIONS
The first generation of WT protocols conducted by the COG is expected to close in 2013. Study proposals are presently being considered for the second generation of COG protocols. All of these proposals are aimed at further refining the treatment of low-risk patients to decrease treatment-related toxicity and intensify treatment of high-risk tumors to improve outcomes. The following are some of the proposals under consideration: to include tumor molecular signatures as part of the COG risk stratification system to further refine the definition of very low-risk and low-risk WT that will be treated either by surgery alone or surgery followed by two-drug chemotherapy; to use cardiac-sparing intensity-modulated radiation therapy (IMRT) in children receiving WLI to reduce cardiac toxicity94; to use IMRT to reduce renal toxicity in children receiving whole-liver irradiation95; to re-evaluate the necessity of irradiating all children who receive chemotherapy before nephrectomy; to re-evaluate the current recommendation for using whole-abdomen RT in children with localized preoperative tumor rupture limited to the flank without any ascites or peritoneal implants; to add new biologic agents to the currently used chemotherapy regimens in children with diffuse anaplastic WT and rhabdoid tumors; and to intensify therapy for children with FH WT and lymph node metastases who have a higher risk of tumor relapse. The biologic samples banks of COG will continue to be a valuable source of tissue for studies aimed at identifying new biologic markers that may be of prognostic significance.
 REFERENCES
REFERENCES
1. Coppes MJ, Ritchey ML, D’Angio GJ. Preface: the path to progress in medical science: a Wilms’ tumor conspectus. Hematol Oncol Clin North Am 1995;9:xiii–xviii.
2. Birch JM, Breslow N. Epidemiologic features of Wilms’ tumor. Hematol Oncol Clin North Am 1995;9:1157–1178.
3. Breslow N, Beckwith JB, Ciol M, et al. Age distribution of Wilms’ tumor: report from the National Wilms’ Tumor Study. Cancer Res 1988;48:1653–1657.
4. Breslow NE, Olshan A, Beckwith JB, et al. Epidemiology of Wilms’ tumor. Med Pediatr Oncol 1983;21:172–181.
5. Coppes MJ, Egeler RM. Genetics of Wilms’ tumor. Semin Urol Oncol 1999;17:2–10.
6. Call KM, Glaser T, Ito CY, et al. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumor locus. Cell 1990;60:509–520.
7. Pritchard-Jones K, Fleming S, Davidson D, et al. The candidate Wilms’ tumor gene is involved in genitourinary development. Nature 1990;346:194–197.
8. Pelletier J, Bruening W, Kashtan CE, et al. Germline mutations in the Wilms’ tumor suppressor gene are associated with abnormal urogenital development in Denys-Drash syndrome. Cell 1991;67:437–447.
9. Huff V. Wilms’ tumor genetics. Am J Hum Genet 1998;79:260–267.
10. Koufos A, Grundy P, Morgan K, et al. Familial Wiedemann-Beckwith syndrome and a second Wilms’ tumor locus both map to 11p15.5. Am J Hum Genet 1989;44:711–719.
11. Grundy PE, Telzerow PE, Breslow N, et al. Loss of heterozygosity for chromosomes 16q and 1p in Wilms’ tumors predicts an adverse outcome. Cancer Res 1994;54:2331–2333.
12. Grundy PE, Breslow NE, Li S, et al. Loss of heterozygosity for chromosomes 1p and 16q is an adverse prognostic factor in favorable histology Wilms tumor: a report from the National Wilms Tumor Study Group. J Clin Oncol 2005;23:7312–7321.
13. Rivera MN, Kim WJ, Wells J, et al. An X chromosome gene, WTX, is commonly inactivated in Wilms tumor. Science 2007;315:642–645.
14. Williams RD, Al-Saadi R, Natrajan R, et al. Molecular profiling reveals frequent gain of MYCN and anaplasia-specific loss of 4q and 14q in Wilms’ tumor. Genes Chromosomes Cancer 2011;50:982–995.
15. Gadd S, Sredni ST, Huang CC, et al. Rhabdoid tumor: gene expression clues to pathogenesis and potential therapeutic targets. Lab Invest 2010;90:724–738.
16. Perlman EJ, Grundy PE, Anderson JR, et al. WT1 mutation and 11p15 loss predict relapse in very low-risk Wilms’ tumor treated with surgery alone: a Children’s Oncology Group Study. J Clin Oncol 2010;26:698–703.
17. Beckwith JB, Palmer NJ. Histopathology and prognosis of Wilms’ tumor: results from the first National Wilms’ Tumor Study. Cancer 1978;41:1937–1948.
18. D’Angio GJ, Breslow N, Beckwith JB, et al. The treatment of Wilms’ tumor: results of the Third National Wilms’ Tumor Study. Cancer 1989;64:349–360.
19. Beckwith JB, Zuppan CE, Browning NG, et al. Histological analysis of aggressiveness and responsiveness in Wilms’ tumor. Med Pediatr Oncol 1996;27:422–428.
20. Beckwith JB. Precursor lesions of Wilms’ tumor: clinical and biological implications. Med Pediatr Oncol 1993;21:158–168.
21. Beckwith JB. Nephrogenic rests and the pathogenesis of Wilms’ tumor: developmental and clinical considerations. Am J Med Genet 1998;79:268–273.
22. Coppes MJ, Arnold M, Beckwith JB, et al. Factors affecting the risk of contralateral Wilms’ tumor development: a report from the National Wilms’ Tumor Study Group. Cancer 1999;85:1616–1625.
23. Bonadio JF, Storer B, Norkool P, et al. Anaplastic Wilms’ tumor: clinical and pathologic studies. J Clin Oncol 1985;3:513–520.
24. Faria P, Beckwith JB, Mishra K, et al. Focal versus diffuse anaplasia in Wilms’ tumor: new definitions with prognostic significance. A report from the National Wilms’ Tumor Study Group. Am J Surg Pathol 1996;20:909–920.
25. Schmidt D, Beckwith JB. Histopathology of childhood renal tumors. Hematol Oncol Clin North Am 1995;9:1179–1200.
26. Ledlie EM, Mynors LS, Draper GJ, et al. Natural history and treatment of Wilms’ tumor: an analysis of 335 cases occurring in England and Wales 1962–1966. BMJ 1970;4:195–200.
27. Sukarochana K, Tolentino W, Kiesewetter WB. Wilms’ tumor and hypertension. J Pediatr Surg 1972;7:573–576.
28. Hartman DS, Sanders RC. Wilms’ tumor versus neuroblastoma: usefulness of ultrasound in differentiation. J Ultrasound Med 1982;1:117–122.
29. Ramos IM, Taylor KJW, Kier R, et al. Tumor vascular signals in renal masses: detection with Doppler US. Radiology 1988;168:633.
30. Khanna G, Rosen N, Anderson JR, et al. Evaluation of diagnostic performance of CT for detection of tumor thrombus in children with Wilms tumor: a report from the Children’s Oncology Group. Pediatr Blood Cancer 2012;58(4):551–555.
31. Reiman TAH, Siegel MJ, Shackelford GD. Wilms’ tumor in children: abdominal CT and US evaluation. Radiology 1986;160:501–505.
32. Belt TG, Cohen MD, Smith JA, et al. MRI of Wilms’ tumor: promise as the primary imaging modality. AJR Am J Roentgenol 1986;146:955–961.
33. Gylys-Morin V, Hoffer FA, Kozakewich H, et al. Wilms’ tumor and nephroblastomatosis: imaging characteristics at gadolinium-enhanced MR imaging. Radiology 1993;188:517–521.
34. Ritchey ML, Green DM, Breslow NB, et al. Accuracy of current imaging modalities in the diagnosis of synchronous bilateral Wilms’ tumor: a report from the National Wilms’ Tumor Study Group. Cancer 1995;75:600–604.
35. Cohen MD. Current controversy: is computed tomography scan of the chest needed in patients with Wilms’ tumor? Am J Pediatr Hematol Oncol 1994;16:191–193.
36. Gross RE, Neuhauser EBD. Treatment of mixed tumors of the kidney in childhood. Pediatrics 1950;6:843.
37. D’Angio GJ, Evans AE, Breslow NE, et al. The treatment of Wilms’ tumor: results of the Second National Wilms’ Tumor Study. Cancer 1981;47:2302–2311.
38. Farewell VT, D’Angio GJ, Breslow N, et al. Retrospective validation of a new staging system for Wilms’ tumor. Cancer Clin Trials 1981;4:167–171.
39. Breslow N, Churchill G, Nesmith B, et al. Clinicopathologic features and prognosis for Wilms’ tumor patients with metastases at diagnosis. Cancer 1986;58:2501–2511.
40. Beckwith JB. National Wilms’ Tumor Study: an update for pathologists. Pediatr Dev Pathol 1998;1:79–84.
41. Weeks DA, Beckwith JB, Luckey DW. Relapse-associated variables in stage I favorable histology Wilms’ tumor: a report of the National Wilms’ Tumor Study. Cancer 1987;60:1204–1212.
42. Kalapurakal JA, Li SM, Breslow NE, et al. Intraoperative spillage of favorable histology Wilms tumor cells: influence of irradiation and chemotherapy regimens on abdominal recurrence: a report from the National Wilms Tumor Study. Int J Radiat Oncol Biol Phys 2010;76:201–206.
43. Leape LL, Breslow NE, Bishop HC. The surgical treatment of Wilms’ tumor: results of the National Wilms’ Tumor Study. Ann Surg 1978;187:351–356.
44. D’Angio GJ, Evans AE, Breslow NE, et al. The treatment of Wilms’ tumor: results of the National Wilms’ Tumor Study. Cancer 1976;38:633–646.
45. D’Angio GJ, Tefft M, Breslow NE, et al. Radiation therapy of Wilms’ tumor: results according to dose, field, postoperative timing and histology. Int J Radiat Oncol Biol Phys 1978;4:769–780.
46. Thomas PRM, Tefft M, Compaan PJ, et al. Results of two radiotherapy randomizations in the third National Wilms’ Tumor Study (NWTS-3). Cancer 1991;68:1703–1707.
47. Thomas PRM, Tefft M, Farewell VT, et al. Abdominal relapses in the Second National Wilms’ Tumor Study patients. J Clin Oncol 1984;2:1098–1101.
48. Kalapurakal JA, Li SM, Breslow NE, et al. Influence of radiation therapy delay on abdominal tumor recurrence in patients with favorable histology Wilms tumor treated on NWTS-3 and -4: a report from the National Wilms Tumor Study Group. Int J Radiat Oncol Biol Phys 2003;57:495–499.
49. Green DM, Beckwith JB, Breslow NE, et al. The treatment of children with stages II–IV anaplastic Wilms’ tumor: a report from the National Wilms’ Tumor Study Group. J Clin Oncol 1994;12:2126–2131.
50. Dome JS, Cotton CA, Perlman EJ, et al. Treatment of anaplastic histology Wilms tumor: results from the fifth National Wilms Tumor Study. J Clin Oncol 2006;24:2352–2358.
51. Tomlinson GE, Breslow NE, Dome J, et al. Rhabdoid tumor of the kidney in the National Wilms Tumor Study: age at diagnosis as a prognostic factor. J Clin Oncol 2005;23:7641–7645.
52. Green DM, Breslow NE, Beckwith JB, et al. Comparison between single-dose and divided-dose administration of dactinomycin and doxorubicin for patients with Wilms’ tumor: a report from the National Wilms’ Tumor Study Group. J Clin Oncol 1998;16:237–245.
53. Green DM, Breslow NE, Evans I, et al. The effect of chemotherapy dose intensity on the hematological toxicity of the treatment for Wilms’ tumor: a report from the National Wilms’ Tumor Study. Am J Pediatr Hematol Oncol 1994;16:207–212.
54. Pritchard J, Imeson J, Barnes J, et al. Results of the United Kingdom Children’s Cancer Study Group First Wilms’ Tumor Study. J Clin Oncol 1995;13:124–133.
55. Mitchell C, Jones PM, Kelsey A, et al. The treatment of Wilms’ tumor: results of the United Kingdom Children’s Cancer Study Group (UKCCSG) second Wilms’ tumor study. Br J Cancer 2000;83:602–608.
56. Meisel JA, Guthrie KA, Breslow NE, et al. Significance and management of computed tomography detected pulmonary nodules: a report from the National Wilms’ Tumor Study Group. Int J Radiat Oncol Biol Phys 1999;44:579–585.
57. Grundy P, Li SM, Green DM, et al. Event free but not overall survival is improved for Wilms tumour patients with pulmonary lesions detectable only by computed tomography by the addition of doxorubicin but not from pulmonary irradiation: results of National Wilms Tumour Studies 4 and 5. Pediatr Blood Cancer 2012;59(4):631–635.
58. Ehrlich PF, Ferrerar F, Ritchey M, et al. Hepatic metastasis at diagnosis in favorable histology Wilms tumor is not an independent adverse prognostic factor. A report from the National Wilms Tumor Study Group. Ann Surg 2009;250:642–648.
59. Blute ML, Kelalis PP, Offord KP, et al. Bilateral Wilms’ tumor. J Urol 1987;138:968–973.
60. Hamilton TE, Ritchey ML, Haase GL, et al. The management of synchronous bilateral Wilms tumor: a report from the National Wilms’ Tumor Study Group. Ann Surg 2011;253:1004–1010.
61. Shamberger RC, Haase GC, Argani P, et al. Bilateral Wilms tumors with progressive or nonresponsive disease. J Pediatr Surg 2006;41:652–657.
62. Hamilton TE, Green DM, Perlman EJ, et al. Bilateral Wilms tumor with anaplasia: lessons from the National Wilms Tumor Study. J Pediatr Surg 2006;41:1641–1644.
63. Breslow NE, Beckwith JB, Haase GM, et al. Radiation therapy for favorable histology Wilms’ tumor: prevention of flank recurrence did not improve survival on National Wilms Tumor Studies 3 and 4. Int J Radiat Oncol Biol Phys 2006;65:203–209.
64. Shamberger RC, Guthrie KA, Ritchey ML, et al. Surgery-related factors and local recurrence of Wilms’ tumor in National Wilms’ Tumor Study 4. Ann Surg 1999;229:292–297.
65. Green DM, Breslow NE, Beckwith JB, et al. Treatment with nephrectomy only for small, stage I/favorable histology Wilms’ tumor: a report from the National Wilms’ Tumor Study Group. J Clin Oncol 2001;19:3719–3724.
66. Shamberger RC, Anderson JR, Breslow NE, et al. Long term outcomes of infants with very low risk Wilms tumor treated with surgery alone on National Wilms Tumor Study-5. Ann Surg 2010;251:555–558.
67. Kalapurakal JA, Green DM, Haase G, et al. Outcomes of children with favorable histology Wilms ‘tumor and peritoneal implants treated on National Wilms’ Tumor Studies-4 and -5. Int J Radiat Oncol Biol Phys 2010;77:554–558.
68. Argani P, Perlman EJ, Breslow NE, et al. Clear cell sarcoma of the kidney: a review of 351 cases from the National Wilms’ Tumor Study Group Pathology Center. Am J Surg Pathol 2000;24:4–18.
69. Seibel N, Li S, Breslow NE, et al. Effect of duration of treatment on treatment outcome for patients with clear-cell sarcoma of the kidney: a report from the National Wilms’ Tumor Study Group. J Clin Oncol 2004;22:468–473.
70. Grundy P, Breslow NE, Green DM, et al. Prognostic factors of children with recurrent Wilms’ tumor: results from the second and third National Wilms’ Tumor Study. J Clin Oncol 1989;7:638–647.
71. Green DM, Cotton CA, Malogolowkin M, et al. Treatment of Wilms tumor relapsing after initial therapy with vincristine and actinomycin D. A report from the National Wilms Tumor Study Group. Pediatr Blood Cancer 2007;48:493–499.
72. Malogolowkin M, Cotton CA, Green DM, et al. Treatment of Wilms tumor relapsing after initial treatment with vincristine, actinomycin D and doxorubicin. A report from the National Wilms tumor Study Group. Pediatr Blood Cancer 2008;50:236–241.
73. Kalapurakal JA, Nan B, Norkool P, et al. Treatment outcomes in adults with favorable histologic type Wilms tumor: an update from the National Wilms Tumor Study Group. Int J Radiat Oncol Biol Phys 2004;60:1379–1384.
74. Segers H, van den Heuvel-Eibrink MM, Pritchard-Jones K, et al Management of adults with Wilms’ tumor: recommendations based on international consensus. Expert Rev Anticancer Ther 2011;11:1105–1113.
75. Lemerle J, Vote PA, Tournade MF, et al. Preoperative versus postoperative radiotherapy, single versus multiple courses of actinomycin D in the treatment of Wilms’ tumor. Cancer 1976;38:647–654.
76. Lemerle J, Vote PA, Tournade MF, et al. Effectiveness of preoperative chemotherapy in Wilms’ tumor: results of an International Society of Pediatric Oncology (SIOP) trial. J Clin Oncol 1983;1:604–609.
77. Jereb B, Burgers MV, Tournade M-F, et al. Radiotherapy in the SIOP (International Society of Paediatric Oncology) nephroblastoma studies: a review. Med Pediatr Oncol 1994;22:221–227.
78. DeKraker J, Weitzman S, Vote PA. Preoperative strategies in the management of Wilms’ tumor. Hematol Oncol Clin North Am 1995;9:1275–1285.
79. Graf N, Tournade MF, de Kraker J. The role of preoperative chemotherapy in the management of Wilms’ tumor. The SIOP studies. Urol Clin North Am 2000;27:443–454.
80. De Kraker J, Graf N, van Tinteren H, et al. Reduction of postoperative chemotherapy in children with stage I intermediate-risk and anaplastic Wilms tumor (SIOP-93–01): a randomized trial. Lancet 2004;364:1229–1235.
81. Mitchell C, Shannon R, Vujanic GM, et al. The treatment of Wilms tumor: results of the United Kingdom Children’s Cancer Study group Third Wilms Tumor Study. Med Pediatr Oncol 2003;41:289.
82. Thomas PRM, Griffith KD, Fineberg BB, et al. Late effects of treatment for Wilms’ tumor. Int J Radiat Oncol Biol Phys 1983;9:651–657.
83. Paulino AC, Wen BC, Brown CK, et al. Late effects in children treated with radiation therapy for Wilms’ tumor. Int J Radiat Oncol Biol Phys 2000;46:1239–1246.
84. Green DM, Grigoriev YA, Nan B, et al. Congestive heart failure after treatment for Wilms’ tumor: a report from the National Wilms’ Tumor Study Group. J Clin Oncol 2001;19:1926–1934.
85. Critchley HOD. Factors of importance for implantation and problems after treatment for childhood cancer. Med Pediatr Oncol 1999;33:9–14.
86. Green DM, Peabody EM, Nan B, et al. Pregnancy outcome after treatment for Wilms’ tumor. J Clin Oncol 2002;20:2506–2513.
87. Green DM, Lange JM, Peabody EM, et al Pregnancy outcome after treatment for Wilms tumor: a report from the national Wilms tumor long-term follow-up study. J Clin Oncol 2010;28:2824–2830.
88. Kalapurakal JA, Peterson S, Peabody EM, et al. Pregnancy outcomes after abdominal irradiation that included or excluded the pelvis in childhood Wilms tumor survivors: a report of the National Wilms Tumor Study. Int J Radiat Oncol Biol Phys 2004;58:1364–1368.
89. Breslow NE, Collins AJ, Ritchey ML, et al. End stage renal disease in patients with Wilms tumor: results from the National Wilms Tumor Study Group and the United States renal data system. J Urol 2005;174:1972–1975.
90. Breslow NE, Takashima JR, Whitton JA, et al. Second malignant neoplasms following treatment for Wilms’ tumor: a report from the National Wilms’ Tumor Study Group. J Clin Oncol 1995;13:1851–1859.
91. Cotton CA, Peterson S, Norkool PA, et al. Early and late mortality after diagnosis of Wilms tumor. J Clin Oncol 2009;27:1304–1309.
92. Termhuhlen AM, Tersak JM, Liu Q, et al. Twenty-five year follow up of childhood Wilms tumor. A report from the Childhood Cancer Survivor Study. Pediatr Blood Cancer 2011;57:1210–1216.
93. Taylor AJ, Winter DL, Pritchard-Jones K, et al. Second primary neoplasms in survivors of Wilms tumor: a population-based cohort study from the British Cancer Survivor Study. Int J Cancer 2008;122:2085–2093.
94. Kalapurakal JA, Gopalakrishnan M, Zhang Y, et al. Advantages of cardiac-sparing whole lung IMRT in children with lung metastases from Wilms’ tumor, rhabdomyosarcoma or Ewing Sarcoma: a dosimetry study based on 4-D gated chest CT scans. Int J Radiat Oncol Biol Phys 2011;18(2):S662.
95. Kalapurakal JA, Zhang Y, Sathiaseelan V, et al. Advantages of whole liver IMRT compared to standard AP-PA technique in children with liver metastasis from right or left sided unilateral Wilms’ tumor: a 3–4D CT dosimetry study Int J Radiat Oncol Biol Phys 2011;18(2):S663.
< div class='tao-gold-member'>




