Waldenström Macroglobulinemia
Rafael Fonseca
Suzanne R. Hayman
Stephen M. Ansell
INTRODUCTION
Waldenström macroglobulinemia (WM) is a malignancy of mature B cells characterized by a monoclonal IgM in the serum and the presence of lymphoplasmacytic lymphoma (LPL) in the bone marrow.1, 2, 3 The disease is now defined at the molecular level by the near-universal presence of mutations of the myeloid differentiation primary response gene MYD88, believed to be the key step in disease pathogenesis. The classical clinical findings in this disease include anemia, organomegaly, lymphadenopathy, and hyperviscosity. The vast majority of patients typically present with a monoclonal IgM in the serum and anemia.1, 2, 3 In this chapter, we review the biology and management of WM, including discussions of its preceding state IgM monoclonal gammopathy of undetermined significance (IgM MGUS).
EPIDEMIOLOGY
General Principles
WM is rare in the United States, with only 1,500 new cases diagnosed per year,4, 5 an incidence of only one sixth that of multiple myeloma (MM). Although it is now possible to conclusively identify patients by the presence of the MYD88 mutation, this aberration is not exclusively seen in WM6 and can be present in other B-cell neoplasms. In future classification schemes for the disease, it is likely that molecular confirmation will be needed. Patients with WM need to be distinguished from patients with a monoclonal IgM in the serum, but no evidence of LPL and no disease-related symptoms or signs (Table 100.1). These patients are felt to have an IgM MGUS.7 The disease is slightly more common in males than females, and more common with advancing age (median age at diagnosis 63 years).4 WM is rare in patients of Mexican-mestizo or African descent, and more common among Caucasians.4, 5, 8
Kristinsson identified an association between WM and autoimmunity using a population-based registry from Sweden, finding that WM patients were more likely to have a history of autoimmunity.9 This risk was observed both in patients with a personal history of WM, but also in those with a familial predisposition. Among the various autoimmune disorders, Sjögren syndrome and autoimmune hemolytic anemia were notable risk factors for subsequent development of WM. Recent observations by Koshiol and colleagues using a large database from the US Veterans Administration showed that there is a heightened relative risk (RR) for WM among patients who have a history of autoimmunity (RR, 2.23), autoimmune antibodies (RR, 2.3 to 2.5), hepatitis (RR, 3.39), human immunodeficiency virus infection (RR, 12.05), and rickettsiosis (RR, 3.35).10 Because of the rarity of the disease, these increased relative risks do not pose major consequences for individuals with these conditions. However, these observations present an hypothesis that chronic stimulation may either drive excessive traffic through the germinal center, with an increased risk of clonal establishment or that a cytokine-enriched milieu allows for further expansion of WM precursor clones normally dormant.
TABLE 100.1 RELATION AMONG IGM-MONOCLONAL GAMMOPATHY OF UNDETERMINED SIGNIFICANCE, SMOLDERING WALDENSTRÖM MACROGLOBULINEMIA, AND WALDENSTRÖM MACROGLOBULINEMIA | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||
Familial Disease and Genetic Predisposition
In some WM, there appears to be a familial component with more than one member of the family affected.11, 12, 13, 14, 15, 16, 17 This implies that a single gene defect may be capable of creating heightened susceptibility. The relationship between this heightened susceptibility and MYD88 mutations has yet to be elucidated. McMaster has shown that the prevalence of IgM monoclonal gammopathies is as high as 10% among relatives of individuals diagnosed with WM.18 They also showed that the risk of monoclonal gammopathy decreases with genetic distance, further indicating genetic predisposition. Linkage analyses that include IgM MGUS have shown positive associations.19 McMaster studied 11 high-risk WM families with WM informative for linkage and collected DNA samples from 122 individuals. Genotypic analysis using microsatellite mapping showed linkage at chromosomes 1q and 4q. In addition, chromosomes 3 and 6 were also suggested as linked.19
In another study, Kristinsson and colleagues looked at genetic predisposition for WM by studying 2,144 LPL/WM patients diagnosed in Sweden (1,539 cases with WM (72%) and 605 cases of LPL (28%).20 They compared them with 8,279 population-based matched controls, and linkable first-degree relatives of patients
(n = 6,177) and controls (n = 24,609). They reported that first-degree relatives of LPL/WM patients have a 20-fold increased risk for LPL/WM.
(n = 6,177) and controls (n = 24,609). They reported that first-degree relatives of LPL/WM patients have a 20-fold increased risk for LPL/WM.
To further investigate familial predisposition and environmental factors, Royer and colleagues used a questionnaire-based survey in the hope of identifying environmental influences that could modulate this predisposition.21 They analyzed data on 103 WM patients and 272 unaffected relatives from 35 multiple-case WM and 46 mixed WM/B Cell disorders (BCD) kindred and 28 nonfamilial (sporadic) WM patients. Once more, autoimmunity was notable (overall response [OR], 2.27), as well as infections (OR, 2.13). Other factors identified included farming (OR, 2.70), pesticide exposure (OR), exposure to wood dust (OR, 2.86), and exposure to organic solvents (OR, 4.21). Although the net effect of these risks is small, they do point to possible environmental modification of germline genetic susceptibility.
DIAGNOSIS AND LABORATORY TESTING
The diagnosis in WM is confirmed by the presence of a monoclonal IgM protein in the serum in association with bone marrow infiltration by monoclonal lymphoplasmacytic cells. These diagnostic criteria are the consequence of various consensus meetings to establish standardized diagnostic criteria for WM (see below).22 As mentioned above, the utilization of molecular techniques will likely become essential in the final confirmation of the diagnosis.
The use of the serum monoclonal protein is a cornerstone of disease diagnosis and monitoring, similar to its use in MM. There are multiple laboratory tests that exist for monitoring the serum concentration of the protein. The treating physician needs to be consistent in using the tests that are used for monitored. The serum protein electrophoresis (SPEP) is the preferred method of detecting the monoclonal protein, but quantitative detection of the IgM is also appropriate because the baseline concentration of the normal IgM (polyclonal) is low enough that it does not substantially alter the monoclonal protein concentration. Initially, immunofixation is needed to characterize a new monoclonal protein (in cases where no IgM has been measured) and also to confirm a complete response (CR). In our practice, we measure both the quantitative IgM and the SPEP because discrepancies are sometimes associated with technical aspects of the measurement. The β2-microglobulin should be determined, in addition to standard laboratory testing, as it is a prognostic factor for the disease (see below).23, 24
At the time of diagnosis, serum viscosity should be determined and repeated as needed. For any given patient, it is usually possible to determine a level at which an IgM protein will result in hyperviscosity.25 Determining viscosity, therefore, at each visit is not really needed (see section “Hyperviscosity, Neurologic and Retinal Complications”). Urine collections can be performed to measure the monoclonal light chain excreted, but the clinical value of this is not clear. Many series have shown that a significant fraction of patients who have WM can have Bence-Jones proteinuria (i.e., light chains).7 For unknown reasons, WM patients rarely develop intrinsic renal failure despite elevated serum-free light chains (SFLC) in some. Perhaps the large size of the monoclonal IgM prevents most of the intact molecule from reaching kidney tubules, unlike myeloma where the intact immunoglobulin molecule can also be detected in the urine, and possibly contributes to cast formation when large amounts of free light chains are excreted. Studies are not available that address the clinical significance of serum-free light-chain detection; however, it is clear that this assay can be used as a surrogate tumor marker as well.
The use of the serum-free light chain (SFLC) has also been explored in WM.26 Leleu studied 72 patients (15 new diagnosis and 57 previously treated) and found a correlation between the serum concentration of sFLC and negative prognostic indicators.27 Itzykson reported on 42 cases of WM (all at diagnosis) and observed similar associations, with elevated levels predicting shorter times to treatment initiation.28 Larger studies are needed to better quantify these associations and associateds clinical implications.
It is useful to obtain a computed tomographic (CT) scan of the chest, abdomen, and pelvis at baseline to assess the spleen and liver size and the presence or absence of lymphadenopathy. Obtaining a metastatic bone survey for patients with WM has little value because, in most cases of WM, bone disease is not present. Obtaining a bone survey is recommended in cases with bone symptoms or in those where the bone marrow pathology is purely plasmacytic.29, 30, 31, 32, 33 The use of positron emission tomographic scan is not considered routine, but can be useful in determining the extent of disease bulk in selected patients.34 In some cases, discordant results have been observed, however, a global recommendation for its use has not been made. In patients with proven bone lesions, a diagnosis of IgM myeloma rather than WM needs to be considered. Until recently, the entity of IgM myeloma had not been defined at the genetic level, making the distinction between WM and IgM myeloma extremely difficult (see below).
IgM MONOCLONAL GAMMOPATHY OF UNDETERMINED SIGNIFICANCE AND SMOLDERING WALDENSTRÖM MACROGLOBULINEMIA
Diagnosis
Having a minimal concentration of a monoclonal protein (e.g., 1.5 or 3.0 g/dl) has classically been required to establish the diagnosis of WM; however, a consensus panel eliminated this requirement.22 The rationale was that having a required minimal concentration could underdiagnose patients early in the course of their disease who may already be symptomatic. It is still unclear how to differentiate IgM MGUS from WM.7 Inasmuch as most patients with a monoclonal IgM do not necessarily have WM, better diagnostic tools are needed, perhaps including molecular detection of MYD88 mutations. Asymptomatic patients with a small monoclonal protein and minimal involvement of the bone marrow by lymphoplasmacytic cells or clonal lymphocytes (e.g., less than 10%), still need to be considered as having an IgM MGUS. Patients like these, according to many epidemiology studies, may have no disease progression for decades.35 One study found that patients with IgM MGUS, as well as patients with asymptomatic WM, have a better survival than patients with symptomatic WM, emphasizing the critical importance of symptoms over laboratory values.36 The importance of segregating IgM MGUS from symptomatic WM is highlighted by this last study.
Evolution to Waldenström Macroglobulinemia from Asymptomatic Stages
The evolution of the monoclonal gammopathy is different for patients with non-IgM disease versus those with IgM MGUS.35 IgM MGUS, in contrast to non-IgM disease, tends to evolve to WM and not to myeloma35 (Table 100.1). Therefore, the greatest risk factor for the development of WM is having an IgM MGUS.35 These patients have a 46-fold greater risk of developing WM than the general population.35 Identification of risk factors for the progression of IgM MGUS is therefore needed. Identification of such risk factors will not only shed new light on the mechanisms of progression, but will also provide a mechanism by which to monitor patients at risk for progression.
In another series, Kyle reported on the outcome of patients with IgM MGUS.37 In contrast to those with non-IgM MGUS, these
patients have a heightened risk of progression to WM (and not myeloma per se). He identified a total of 213 patients with IgM MGUS seen at the Mayo Clinic—Rochester from 1960 to 1994.37 These patients were followed for a median of 6.3 years (cumulative 1,567 person-years). Interestingly, a lymphoma not otherwise specified developed in 17 patients (RR 14.8), and WM developed in 6 (RR, 262). Risk factors predictive of progression included the concentration of the serum monoclonal protein and albumin at diagnosis (for progression to lymphoma or a related disorder).
patients have a heightened risk of progression to WM (and not myeloma per se). He identified a total of 213 patients with IgM MGUS seen at the Mayo Clinic—Rochester from 1960 to 1994.37 These patients were followed for a median of 6.3 years (cumulative 1,567 person-years). Interestingly, a lymphoma not otherwise specified developed in 17 patients (RR 14.8), and WM developed in 6 (RR, 262). Risk factors predictive of progression included the concentration of the serum monoclonal protein and albumin at diagnosis (for progression to lymphoma or a related disorder).
Morra reported on risk factors for progression from IgM MGUS to WM.38 She studied 384 asymptomatic IgMMGUS and a total of 74 other IgM-related disorders.38 At 5 and 10 years, the risk of progression was 8% and 29%, respectively for IgM MGUS. A total of 41 patients evolved to WM after a median follow-up of 45 months. Factors predictive of progression included the extent of bone marrow infiltration, sedimentation rate, hemoglobin, serum concentration of the IgM, and lymphocytosis. However, in the multivariate analysis, only the serum IgM concentration and lymphocytosis remained predictive. Among patients with smoldering Waldenström macroglobulinemia (SWM), the risk of progression seems to be greater in patients who have a monoclonal spike greater than 3 g/dl, and/or a lymphoplasmacytic infiltrate of the bone marrow, greater than 30%, or diffuse infiltration of the bone marrow, or anemia.39 Likewise, Alexanian reported that a hemoglobin less than 11.5, M-spikes greater than 3 g/dl, and an elevated β2-microglobulin correlate with a heightened risk of progression.40
Gobbi and colleagues reported on a large series of patients with various IgM monoclonal gammopathies.36 A large cohort of 698 patients with IgM gammopathy was subdivided into 4 unique clinical entities; IgMMGUS, SWM, WM, and other IgM-related disorders (excluded from further analysis). Not surprisingly, IgM MGUS and SWM had median survivals similar to that of the general population. The standardized mortality rate for patients with symptomatic WM was 5.4.
Baldini and colleagues evaluated 217 patients with IgM MGUS and another 201 patients with SWM.41 The median time to progression was not reached for IgM MGUS, and was 142 months for those with SWM (median duration of follow-up of 56 and 60 months). Progression was more likely in patients with elevated serum monoclonal proteins, anemia, and male sex.
Likewise, Greco et al. evaluated risk factors for progression from IgM MGUS and SWM to active disease.42 Their series included a total of 287 patients: 201 with IgM MGUS, and 86 with SWM. After a median of 50 months, 32 cases evolved (11.1%): 26 to WM and 6 to other non-Hodgkin lymphoma. In the univariate analysis, risk factors for progression included the degree of bone marrow infiltration, elevated sedimentation rate, the concentration of the serum monoclonal protein, and IgA levels. The highest risk of progression was identified for patients with SWM, serum monoclonal protein ≥3 g/dl and/or those with ≥10% BM infiltration.
Kyle presented a recent update on the risk of transformation from SWM to WM.43 SWM was defined as those patients having an elevated monoclonal IgM to greater than 3 g/dl and/or greater than 10% involvement of the bone marrow by the clonal process. By definition, these patients must have no evidence of end organ damage (anemia, constitutional symptoms, hyperviscosity, lymphadenopathy, or hepatosplenomegaly). A total of 48 patients were identified who fulfilled these criteria. After a median follow-up of 15.4 years, a total of 34 patients progressed to WM. In two other patients, other complications were observed (amyloidosis [AL] in one and lymphoma in the other). Based on these data, the cumulative risk of progression was estimated at 6% at 1 year, 39% at 3 years, 59% at 5 years, and 68% at 10 years. Risk factors predictive of progression included the degree of bone marrow involvement (hazard ratio [HR] 1.31), concentration of the serum M-spike (HR 2.1), reduction of the serum IgA (HR 2.4), and hemoglobin (HR 0.7). As in the Greco et al. study, it seems that progression is a stochastic event and, therefore, tumor bulk (i.e., cells at risk) predicts likelihood of progression.
PATHOLOGY
The WHO classification and the Revised European-American Lymphoma classify WM as LPL because of its immunophenotypic and morphologic characteristics.44 Some cases of LPL have no associated monoclonal protein,45 a more aggressive clinical course, and a different set of genetic abnormalities.23 It is now believed that there are several subsets of LPL, of which WM is only one subtype.
Estimation of clonal involvement in the bone marrow is routinely performed at diagnosis with a unilateral trephine aspirate and biopsy. The disease is typically a pleomorphic infiltrate of lymphoplasmacytic cells,46, 47, 48 and variability in cell morphology is common (Fig. 100.1). The infiltrate can range from purely lymphocytic to one that is predominantly plasmacytic.46, 47, 48 The co-existence of mast cells in association with the lymphomatous infiltrate is a unique feature of WM.46, 47, 48, 49, 50 The role of these mast cells as part of the disease pathogenesis has not been fully elucidated. A complex network of interactions between the WM cells and the bone marrow microenvironment has been suggested in preliminary observations.50 In addition, infiltration by malignant B cells can be seen in lymphoid structures and multiple other organs (giving rise to organomegaly and lymphadenopathy).44 The detection of clonal cells in other organs is not known to have particular prognostic implications; however, clinically evident organomegaly is usually a negative prognostic factor for the disease51, 52 and usually indicates a large tumor burden.
The nature of the clonal B-cell populations is remarkable.53, 54 Sahota has identified the normal counterpart of WM cells as mature B cells that have undergone somatic hypermutation, but that have not yet completed isotype class switching.55 In that particular study, they did not find evidence of intraclonal heterogeneity. It is notable that in many cases of WM, one can observe co-existent populations of monoclonal B cells and plasma cells. Recently, Zehentner and colleagues demonstrated that only in 40% of cases are the clonal B-cell and plasma cell populations related.56 Conversely, in 60% of cases, they represent two separate and independent clones. By performing sequencing of the immunoglobulin genes, and in some cases using fluorescence
in situ hybridization (FISH) analysis, they conclusively show the separate nature of these clones in 60% of cases.
in situ hybridization (FISH) analysis, they conclusively show the separate nature of these clones in 60% of cases.
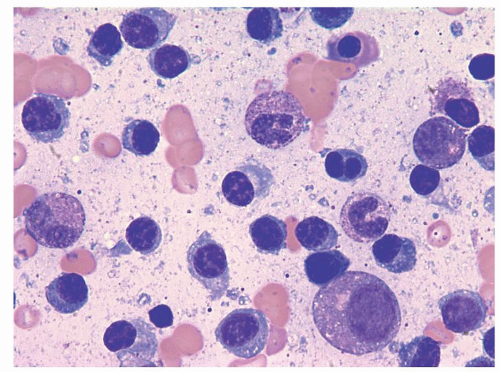 FIGURE 100.1. Lymphoplasmacytic morphology of the clonal cells of Waldenström macroglobulinemia. As shown in the graph, the cells have variable morphology but most show transition between mature lymphocytes and plasma cell morphology. The cells have been called in the past “plymphs.” The morphology can be variable with some cases showing more extreme plasma cell morphology, whereas others have more lymphocytic predominance. |
Flow cytometry is usually indicated as part of the pathologic work-up to make the diagnosis of WM. Flow cytometry is helpful in differentiating WM from other morphologically similar neoplasms, such as mantle cell lymphoma, marginal zone lymphomas, and B-cell chronic lymphocytic leukemia (CLL).49, 57, 58, 59 WM cells are characterized by the surface expression of CD19, CD20, CD22,49, 57, 58, 59, 60, 61 are light chain restricted, and commonly express CD79a. They are also typically CD10 and CD23 negative, in contrast to follicular lymphoma and B-cell CLL, respectively. The associated plasma cells are monoclonal and express CD138.49, 57, 58, 59, 60 CD5 is not expressed on the malignant cells (in contrast to mantle cell lymphoma and B-cell CLL) in the majority of cases; however, some cases can be positive.49, 57, 58, 59, 60
Cell proliferation markers are usually indicative of the indolent nature of the disease. The fraction of cells incorporating 5-bromo-2′-deoxyuridine (BrdU),62 indicating cell replication, is usually minimal, and frequently no such fraction is detected at all. The evolution of WM to a more aggressive lymphoproliferative disorder has been previously reported,63, 64 and the presence of a highly proliferative fraction may suggest transformed disease. The role of karyotype analysis in the clinical management of patients is not felt to be necessary and, in most patients, the metaphases will be normal.65, 66, 67 Therefore, this analysis should be reserved for patients suspected of having treatment-associated myelodysplasia.68
WM variants have been described where patients have a similar pathology to WM, but present with IgG monoclonal proteins.69 These patients appear to be clinically different from WM and tend to have a lymphocytosis present in the peripheral blood. It is not clear if these diseases have a similar pathogenesis, as only one series of patients with this variant tumor has been reported. Also, some patients have been described as having WM with associated bone lesions.29, 30, 33 This makes distinguishing WM from IgM myeloma very difficult. Given the availability of molecular testing that can identify MYD88 mutations and also chromosome abnormalities seen in IgM MM, this distinction should now be possible. If some cases of true WM indeed have bone lesions, it is likely that these will be due to the same mechanisms operative for myeloma.70 In a recent study of patients with IgM myeloma, it was found that the t(11;14)(q13;q32) was nearly universally present.71 Another recent study did not find it as universal, but still common in IgM myeloma patients (38%).72 Also, the t(14;16) (q32;q23) has been reported in one case of IgM myeloma.73 In addition, we have found that translocations of the IGH locus seen in IgM myeloma are not typically observed in patients with WM, and when present, they are secondary genetic events, seen only in cell subsets.67 This, along with the new-found mutation of MYD88, clearly separates WM and IgM myeloma as separate disease entities.
BIOLOGY AND GENETICS
Somatic Mutations
There is compelling evidence for a postgerminal center origin of the clonal B cells in WM.31, 32, 74, 75, 76 To ascertain the lack of class switch rearrangements involving the mu switch region, we studied cases using a Southern blot assay, with no rearrangements identified (concordant 5′ and 3′ switch µ bands after digestion).67 The aggregate of these studies thus shows that cell arrest just prior to class switching is the likely stage of neoplastic transformation, and genetic defects of the class switch machinery may be involved in the disease pathogenesis. It is important to note that lymphoplasmacytic cells of WM can mature to clonal plasma cells in vitro77 suggesting that additional mechanisms may play a role.
MYD88 L265P
Just recently, a mutation of the gene MYD88 has been described by Treon et al. in almost all cases of WM.78 The mutation is associated with intracellular signaling and is believed to be key in the pathogenesis of the disease.78 The prevalence of this mutation in IgM MGUS cases is still being investigated. In the initial reports it was felt to be uncommon in IgM MGUS cases, but the actual prevalence is still being investigated, with some reports showing up to 50% of IgM cases harboring the mutation. The MYD88 L265P activating mutation has been reported in 90% of WM cases.78 MYD88 encodes for an adapter protein that affects the interleukin (IL)-1 and Toll-like receptor pathway, and the L265P leads to the dysregulation of the nuclear factor kappa B (NF-κB) and JAK signaling pathways.6
Other Mutations
Two other genes have been found to be inactivated in WM: TNFAIP3 and TRAF3. TNFAIP3 is a tumor suppressor gene whose inactivation results in the constitutive activation of NF-κB.79 We reported biallelic inactivation/deletion of TNFAIP3 in 5% of WM patients.80 In addition, 38% of patients were found to have monoallelic deletions of TNFAIP3 with associated diminished transcription of the gene, with proposed associated haploinsufficiency. Inactivating mutations of TRAF3 (located at 14q32.32) have been described and also result in the constitutive activation of NF-κB in about (˜5%) of patients.79 TRAF3 is a negative regulator of the noncanonical NF-κB signaling pathway, with a consequent increase in serine/threonine protein kinase NIK.79, 81, 82 Similar observations have been made using novel high-throughput genomic tools.83
6q Deletions and Other Genetic Changes
The most common cytogenetic abnormality in WM is deletion of 6q. We know through multiple studies that in at least one half of cases, large deletions of chromosome 6 are observed in WM by a combination of FISH and aCGH (array comparative genomic hybridization).66, 67, 84, 85 Usually, these deletions involve chromosome bands 6q21-q23, with the most commonly deleted being the q23 region. These deletions are clonally selected in the majority of cases.67 The clinical relevance of the presence of a 6q deletion has not been well defined, but a recent study suggested that these patients tend to have more aggressive disease and a shorter survival.86 These observations, however, need to be confirmed in additional studies.67 Although the data are limited, chromosome 6q deletions seem to be rare in cases of IgM MGUS, suggesting that deletions in 6q are involved in the progression to WM87 (Fig. 100.2). Although there is no direct link yet made between 6q deletions and MYD88 mutations, and given the ubiquitous nature of the latter,
and the high prevalence of the former, it is only logical to conclude that both are important in disease pathogenesis. The second most common observation by aCGH was the gain of 6p (16.6%), always occurring as a secondary event following the loss of 6q.79
and the high prevalence of the former, it is only logical to conclude that both are important in disease pathogenesis. The second most common observation by aCGH was the gain of 6p (16.6%), always occurring as a secondary event following the loss of 6q.79
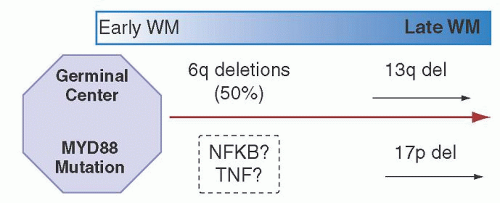 FIGURE 100.2. Biology of progression for Waldenström macroglobulinemia (WM). Mutations of MYD88 are likely founder lesions for the disease. In 50% of cases, deletion of the long arm of chromosome 6 may be the culprit in the initiation of the clonal process of Waldenström macroglobulinemia. In some cases, deletions of chromosomes 13 and 17 may represent progression events. Emerging data suggest that abnormalities of tumor necrosis factor (TNF) and nuclear factor kappa B (NF-κB) signaling may also be contributing factors in the pathogenesis of the disease. |
Originally, due to the presence of LPL in the bone marrow, it was believed that WM must contain the t(9;14)(p13;q32) (described originally in at least 50% of LPL cases, none of which had detectable IgM monoclonal protein).88 The t(9;14)(p13;q32) results in upregulation of PAX-5,89–91 implicating this gene as a putative oncogene in the pathogenesis of LPL cases without monoclonal IgM.45, 92 PAX-5 prevents expression of high levels of immunoglobulin and diminishes production of the J-peptide needed for the pentameric IgM assembly; therefore, the presence of PAX-5 up-regulation by this translocation is inconsistent with the phenotype of WM. In contrast to many other B-cell malignancies, it has been shown that WM never harbors the t(9:14), or any other 14q32 translocations.67 In a significant number of cases, review of the karyotype also failed to identify IGH translocations on chromosome 14.65, 66, 67 Although some rare cases have clones with IGH translocations,93 these events are now believed to be secondary genetic events associated with disease progression. In most WM cases, the karyotype results are usually normal.65, 66, 67, 94, 95, 96
In sporadic cases of WM, the t(11;18)(q21;q21), usually associated with a subset of marginal zone lymphomas, has been reported.97 Using an interphase FISH-based strategy, others have been unable to detect this translocation.67 Again, molecular confirmation of MYD88 mutations will be important in redefining whether these entities should be considered under the WM diagnosis. Other chromosomal abnormalities, such as deletions of 17p13.1 and 13q14, are not common at the time of diagnosis, but at the time of disease progression, may be observed in 15% of patients.98 The clonal cells of WM are usually diploid,67 and this has been confirmed by DNA content analysis in a subset of patients.99
Additional data have suggested that WM may have features in common with other disease entities, as well as with MM. At the gene expression level, WM resembles B-cell CLL more than it does MM.100 In addition, studies of patients with IgM MGUS show clustering together with WM.100 Furthermore, the clonal cells of WM express high levels of IL-6, which likely would explain the clinical observation of high C-reactive protein levels in the serum of WM patients.100, 101 In a manner similar to the anemia of chronic disease, the high levels of IL-6 likely contribute to the anemia, of WM.102 Experiments of gene expression profiling in WM were performed, separating the CD19+ cells from the CD138+ cells, and have found remarkably similar patterns of gene expression.103
Epigenetic Aberrations; Micro-ribonucleic Acid and Histone Acetylation
Roccaro et al. reported on a unique miRNA profile in WM cells, something that was different from the normal B-cell counterparts.104 Their studies showed that miRNA-363*, -206, -494, -155, -184, -542-3p were up-regulated and miRNA-9* was underexpressed.104 Many of the predicted targets for these miRNAs include genes associated with the cell cycle, apoptosis, transcription factors and oncogenes. Because of its previous descriptions in other B-cell malignancies, miRNA-155 has been more extensively studied in WM.104 For a more detailed analysis, the reader is referred to a recent review article.105, 106 Likewise, alteration of the balance between histone acetyl transferases and histone deacetylases has been postulated as important in the pathogenesis of WM. This is important as two miRNAs are associated with histone acetylation: miRNA-206 and -9*.107
Cytokines and B-cell Signaling
A tumor necrosis factor family member, B-lymphocyte stimulator (BLyS), is critical for maintenance of normal B-cell development and homeostasis. Elsawa and colleagues found that WM cells variably express the BLyS receptors BAFF-R, TACI, and BCMA and bind soluble BLyS.108 Also, they documented elevated serum BLyS levels in patients with WM and expression of BLyS in bone marrow samples by immunohistochemistry.108 BLyS, in vitro, increased the viability and proliferation of WM cells and stimulated secretion of the monoclonal IgM. She also found that chemokine (C-C motif) ligand 5 (CCL5), granulocyte colony-stimulating factor, and soluble IL-2 receptor are overexpressed in WM, with CCL5 correlating quite closely with the serum levels of IL-6. As mentioned earlier, up to 50% of WM have up-regulation of IL-6, a cytokine critical in B-cell development and implicated in various B-cell malignancies. Furthermore, WM cells also express the IL-6 receptor, and CCL5 results in increased secretion of IL-6 by bone marrow stromal cells in WM, further enhancing IL-6 stimulation, occurring both in an autocrine and paracrine fashion. This stimulation resulted in increased secretion of IgM in a process mediated via JAK/STAT signaling.
CLINICAL FEATURES
It is best to differentiate the clinical symptoms of this disease based on their causes: those signs and symptoms secondary to the effects of the monoclonal protein and other paraneoplastic phenomena, and those related to the clonal proliferation in the bone marrow and other lymphoid organs.3 It should be noted that most patients with WM have a limited symptom complex at the time of diagnosis, consisting of a monoclonal IgM and various degrees of anemia.24, 110 This anemia is often secondary to the clonal expansion of cells in the bone marrow, but may also be due to hyperviscosity (by reducing erythropoietin production) or the increased plasma volume.111 In many cases, anemia can be a consequence of treatment. Recent data suggest that some patients may have an anemia secondary to elevated levels of IL-6 emanating from the clonal cells, and will present with a picture reminiscent of the anemia of inflammation.100 Given the high level of expression of IL-6 and IL-6 receptors in at least half of WM, it is not surprising that elevated levels of hepcidin have been postulated as key mediators in the anemia of WM.102 In cases with significant involvement of the bone marrow by the WM clonal cells, thrombocytopenia may also be observed.24, 110
In 20% of cases, the clonal cells of WM can infiltrate other organs and result in hepatomegaly and splenomegaly in 15% of patients.23, 24, 51, 52, 110, 112 Splenomegaly can result in hypersplenism, with worsening pancytopenia. Although patients may have liver involvement, this is usually asymptomatic. Lymphadenopathy can be seen in 15% to 20% of patients,23, 24, 51, 110, 112 and involvement of other organs, such as the lung, can be seen in WM, although this is present in only a minority of cases.113, 114
Hyperviscosity, Neurologic and Retinal Complications
Hyperviscosity is one of the most characteristic features of WM; however, it is only seen in 15% of cases.25, 115, 116 The details of the physical properties of the pentameric IgM molecule leading to hyperviscosity have been previously described.25, 116 The large dimensions of the IgM molecule make the serum more viscous, and results in slower transit in capillaries.25 The increased viscosity is not just the result of the large size of the IgM molecule, as
other large particles such as albumin do not have the same effect on the serum viscosity.25 Although the overt clinical syndrome of hyperviscosity is obvious, subtler symptomatology can also be seen. Typical symptoms include neurocognitive effects (sometimes with overt hemorrhage and neurologic abnormalities), retinal bleeding, and mucosal bleeding in gums and nose. In different patients, different serum concentrations of the monoclonal IgM will give rise to hyperviscosity, and there is no absolute value at which hyperviscosity will be observed.25, 115, 116 However, for a given patient, a given IgM level will usually again result in symptoms when the serum IgM reaches that level again.25, 115, 116 Despite having a high serum viscosity, some patients will have no symptoms, whereas others will have prominent symptoms at lower viscosity levels. To see symptoms of hyperviscosity at values less than 4 cps is uncommon. In contrast, nearly all patients with a viscosity greater than 8 will be symptomatic, and most patients with a viscosity of 5 to 8 cps will have symptoms.25, 115, 116 Also, to have hyperviscosity with an IgM less than 4,000 mg/dl is very rare.25 The patient’s hydration and the red cell mass are other factors that contribute to blood viscosity. Red cell transfusions should therefore be used with caution, and sometimes in conjunction with pre-transfusion plasmapheresis, in patients with high IgM concentrations.
other large particles such as albumin do not have the same effect on the serum viscosity.25 Although the overt clinical syndrome of hyperviscosity is obvious, subtler symptomatology can also be seen. Typical symptoms include neurocognitive effects (sometimes with overt hemorrhage and neurologic abnormalities), retinal bleeding, and mucosal bleeding in gums and nose. In different patients, different serum concentrations of the monoclonal IgM will give rise to hyperviscosity, and there is no absolute value at which hyperviscosity will be observed.25, 115, 116 However, for a given patient, a given IgM level will usually again result in symptoms when the serum IgM reaches that level again.25, 115, 116 Despite having a high serum viscosity, some patients will have no symptoms, whereas others will have prominent symptoms at lower viscosity levels. To see symptoms of hyperviscosity at values less than 4 cps is uncommon. In contrast, nearly all patients with a viscosity greater than 8 will be symptomatic, and most patients with a viscosity of 5 to 8 cps will have symptoms.25, 115, 116 Also, to have hyperviscosity with an IgM less than 4,000 mg/dl is very rare.25 The patient’s hydration and the red cell mass are other factors that contribute to blood viscosity. Red cell transfusions should therefore be used with caution, and sometimes in conjunction with pre-transfusion plasmapheresis, in patients with high IgM concentrations.
Since the original reports by Waldenström,117 neurologic abnormalities in patients with WM have been described (Bing-Neel syndrome). Plasma exchange should be entertained in any patient presenting with a focal or global neurologic deficits that potentially could be attributed to hyperviscosity, even if a clear causal association is not immediately established. The symptoms may be nonspecific and the spectrum can include headache, fatigue, impaired cognition, confusion, stroke, or frank dementia.118, 119
Peripheral neuropathies associated with the presence of monoclonal proteins include, demyelinating polyneuropathy (both associated and not with antimyelin-associated glycoprotein antibodies),120, 121 other demyelinating neuropathies, cryoglobulinemia-associated symptoms, and neuropathies associated with light-chain AL. The monoclonal protein, in most cases, has affinity for the myelin sheath and results in demyelinization,122 which can be observed in up to one half of WM patients with peripheral neuropathy.123 Patients can also have abnormalities of proprioception resulting in ataxia, in addition to the sensory deficits.123, 124 Some of the new therapeutic agents, namely bortezomib, can also be associated with treatment-induced peripheral neuropathy. As a direct consequence of the increased viscosity, patients can have retinal complications of WM, and these present on physical examination as venous engorgement, hemorrhage, and cotton-wool exudates.125, 126 In some severe cases, WM can precipitate retinal vein occlusion.125, 126
Related posts:
 The Diagnostic and Therapeutic Approach to Hematologic Problems
Mast Cells and Basophils: Ontogeny, Characteristics, and Functional Diversity
Platelet Structure and Function in Hemostasis and Thrombosis
Hemochromatosis
Sickle Cell Anemia and Other Sickling Syndromes
Anemias Secondary to Chronic Disease and Systemic Disorders
The Diagnostic and Therapeutic Approach to Hematologic Problems
Mast Cells and Basophils: Ontogeny, Characteristics, and Functional Diversity
Platelet Structure and Function in Hemostasis and Thrombosis
Hemochromatosis
Sickle Cell Anemia and Other Sickling Syndromes
Anemias Secondary to Chronic Disease and Systemic Disorders
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


