Table 52.1 Classification of vWD | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
Table 52.2 Selected laboratory features of vWD subtypes | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||
stress, and where possible, tests should not be performed in proximity to hemorrhagic events, pregnancy, acute infection, or strenuous exercise. Optimal testing strategies have not yet been validated experimentally, and the approach described here is an attempt to synthesize various expert opinions.
Table 52.3 Frequency (%) of bleeding symptoms in vWD | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Table 52.4 Influence of ABO blood groups on vWF:Ag in volunteer blood donors | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
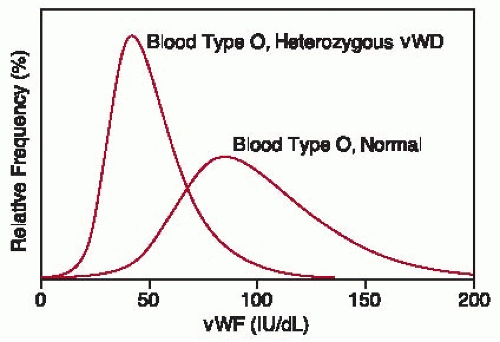 FIGURE 52.1 Distribution of vWF:Ag levels in normal blood donors of blood type O and hypothetical distribution of vWF:Ag levels in individuals of blood type O who are heterozygous for vWF null alleles. The reference normal value of 100 IU/dL is based on the World Health Organization standard pooled plasma. (Adapted from tabulated data of Gill JC, Endres-Brooks J, Bauer PJ, et al. The effect of ABO blood group on the diagnosis of von Willebrand disease. Blood 1987;69:1691-1695.) |
time,55,56,57 a prolonged PFA-100 closure time was not found to predict increased surgical bleeding.61
connective tissue. Mutations in vWD type 2 can disrupt any of these properties, and vWD type 2 accounts for between 7% and 30% of all vWD in various reports. Such qualitative abnormalities of vWF often are associated with discrepancies between the level of vWF:Ag and functional assays such as vWF:RCo, vWF:CB, or vWF:FVIIIB. Four subtypes are currently recognized (Tables 52.1 and 52.2). The prevalence of the subtypes appears to be increasing as awareness of them increases, although the distribution of subtypes differs considerably among treatment centers. A survey in France found that patients with vWD type 2 were divided as 30% type 2A, 28% type 2B, 8% type 2M (or unclassified), and 34% type 2N.93 A similar study in Germany found 74% type 2A, 10% type 2B, 13% type 2M, and 3.5% type 2N.94
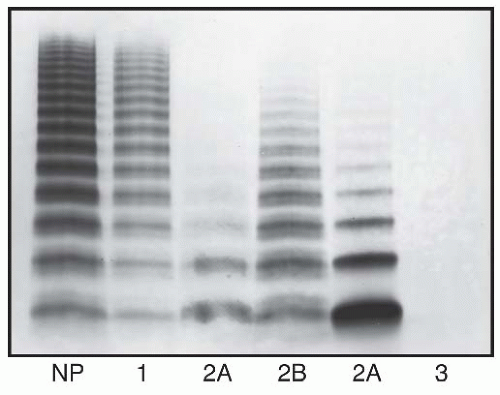 FIGURE 52.2 Multimer patterns in vWD. Samples of plasma from a normal individual (N) and from patients with vWD type 1, type 2A (two unrelated patients), type 2B, and type 3 were electrophoresed through a 1.3% agarose/sodium dodecyl sulfate (SDS) gel and von Willebrand factor (vWF) was detected by Western blotting. (Reprinted from Sadler JE. von Willebrand disease. In: Bloom AL, Forbes CD, Thomas DP, et al., eds. Haemostasis and thrombosis, 3rd ed. Edinburgh, UK: Churchill-Livingstone, 1994:843-857, with permission.) |
platelet vWF is relatively protected from proteolysis. The plasma metalloprotease, a disintegrin and metalloprotease with thrombospondin type 1 motifs member 13 (ADAMTS13), cleaves the Tyr1605-Met1606 bond within domain A2 of vWF,107,108 and this cleavage accounts for the major proteolytic fragments of the vWF subunit that are found in plasma vWF.109 Mutations that increase the susceptibility of vWF to ADAMTS13 cause the loss of large, hemostatically effective multimers from plasma and result in bleeding. Conversely, deficiency of ADAMTS13 causes thrombotic thrombocytopenic purpura associated with unusually large vWF multimers, as discussed in Chapter 99.
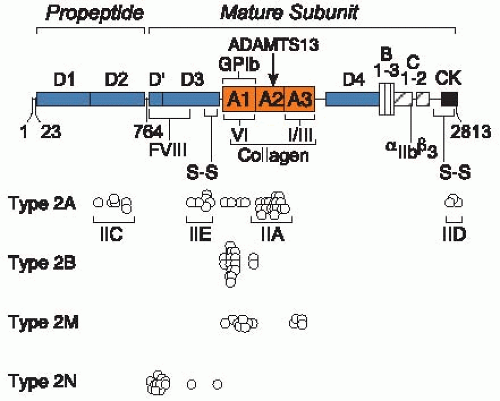 FIGURE 52.3 Mutations in vWD type 2. Amino acid residues are numbered by codon number 1 to 2,813 for prepro-vWF. The signal peptide contains residues 1 to 22, the propeptide contains residues 23 to 763, and the mature subunit contains residues 764 to 2,813. The locations of conserved structural domains (A, B, C, D, and CK) are indicated. Intersubunit disulfide bonds (not shown) connect CK to CK domains and D3 to D3 domains. The locations of binding sites are indicated for FVIII, platelet glycoprotein (GP)Ib, collagen, and platelet αIIbβ3. The metalloprotease ADAMTS13 cleaves a Tyr-Met bond in domain A2 (arrow). Circles mark the positions of mutations that cause the vWD phenotypes noted at the left. For vWD type 2A, the labeled brackets show the location of mutations that correspond to variants with characteristic multimer patterns discussed in the text. These include variants with increased sensitivity to proteolysis (IIA) or defective multimer assembly due to mutations in the propeptide (IIC), the D3 domain (IIE), or the CK domain (IID). References to the individual mutations can be found in the database of vWD mutations20 maintained by the International Society on Thrombosis and Haemostasis at the University of Sheffield and accessible at http://www.shef.ac.uk/vwfvwf.group.shel.ac.uk/index.html. (Adapted from White GC II, Sadler JE. Von Willebrand disease: clinical aspects and therapy. In: Hoffman R, Benz EJ, Shattil SJ, eds. Hematology: basic principles and practice, 4th ed. Philadelphia, PA: Elsevier Churchill Livingstone, 2005:2121-2136, with permission.)
Related posts: The Field of Hemostasis and Thrombosis: Selected Translational Achievements The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Integrín αIIbβ3 and Platelet Aggregation Integrín αIIbβ3 and Platelet Aggregation
 Acquired Nonimmune Thrombocytopenia Acquired Nonimmune Thrombocytopenia
 Pathogenesis and Treatment of Biomaterial-Associated Thrombosis Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
 Blood Management in the Cardiovascular Surgical Patient Blood Management in the Cardiovascular Surgical Patient
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|