Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Kanagasabai Vadivel
Amy E. Schmidt
Victor J. Marder
Sriram Krishnaswamy
S. Paul Bajaj
Physiologic coagulation is initiated when, upon vascular damage, blood comes into contact with tissue factor (TF) expressed by cells in the extravascular compartment.1,2,3,4,5,6,7 TF exposed at the injury site complexes with factor VIIa (FVIIa, ˜100 pM) as well as with factor VII (FVII, ˜10 nM).8,9 The FVIIa/TF complex activates both factors X (FX) and IX (FIX), leading to thrombin (IIa) generation and fibrin formation (FIGURE 14.1). In positive feedback loops, factor Xa (FXa), factor IXa (FIXa), and FVIIa/TF generate additional FVIIa/TF from FVII/TF.10,11,12,13,14,15 Similarly, factor XIa (FXIa), formed by the activation of FXI by thrombin, produces additional FIXa for sustained coagulation.16,17,18,19,20 FXa initially formed by FVIIa/TF generates small amounts of thrombin, which activates platelets as well as factor V (FV) and factor VIII (FVIII).21 FIXa and FVIIIa assembled on the surface of the activated platelets convert additional FX to FXa, which complexes with FVa to further augment thrombin generation.21 In addition to the four procoagulant proteins (FVII, FIX, FX, and FII), three other vitamin K-dependent (VKD) proteins, protein C (PC), protein Z (PZ), and protein S (PS), control the process by acting as anticoagulants. Activated PC (APC) generated by the action of thrombin/thrombomodulin complex inactivates FVa and FVIIIa,22,23 PZ serves as a cofactor for inhibition of FXa by the protein Z-dependent inhibitor (PZI),24 and PS serves as a cofactor for inactivation of FVa and FVIIIa by APC.25,26,27 Other VKD proteins have been described, including osteocalcin (bone metabolism),28 periostin (matrix homeostasis), and Gas6 (endothelial homeostasis),29,30 plus others of unknown function.31 This chapter focuses on the structural and functional aspects of the VKD proteins involved in hemostasis and thrombosis.
MOLECULAR ORGANIZATION OF THE VKD PROTEINS
Domain Organization
Zymogen FVII, FIX, FII, PZ, and PS circulate as single-chain molecules, whereas FX and PC circulate as two-chain molecules.32,33,34,35,36,37,38,39 FVII, FIX, FX, and PC have similar structural architecture, in which an N-terminal γ-carboxyglutamic acid (Gla)-rich domain is followed by two epidermal growth factor (EGF)-like domains and a C-terminal serine protease domain (FIGURE 14.2).40,41,42,43,44,45 PZ has similar domain organization, but it contains a pseudoprotease domain without catalytic activity at the C-terminus.37,38 PS consists of six domains, an N-terminal Gla domain, a thrombin-sensitive region (TSR), four EGF-like domains, and a sex hormone globulin binding domain.46,47,48 Prothrombin (FII) has a distinct domain architecture, in which the N-terminal Gla domain and the C-terminal protease domain are linked by the two kringle domains.49
Zymogen Activation
The VKD zymogens are synthesized in the liver and secreted into the blood; the molecular mass and plasma concentrations are shown in Table 14.1 (see also Chapter 9).
FVII consists of an N-terminus Gla domain (residues 1 to 38), a short hydrophobic segment (residues 39 to 45) followed by two EGF-like domains (residues 47 to 84 and 31 to 85), and a C-terminus latent protease domain (residues 153 to 406)12,34,50 (FIGURE 14.2A). FXa, FIXa, and the FVIIa/TF complex activate FVII by cleaving a single peptide bond between Arg152 and Ile153 located in the connecting region between the EGF2 and the protease domain,15,51,52,53 leading to a two-chain molecule (FIGURE 14.3A), a light chain of 152 residues, and a heavy chain of 254 residues linked by a single disulfide bond between Cys-135 and Cys-262.
FIX consists of N-terminus Gla domain (residues 1 to 40), a short hydrophobic segment (residues 41 to 46), two EGF-like domains (residues 47 to 84 [EGF1] and 85 to 127 [EGF2]), an activation peptide region (residues 146 to 180), and a latent serine protease module (residues 181 to 415) (FIGURE 14.2A).33,54,55 Activation of FIX requires two peptide bond cleavages, one at Arg145-Ala146 and Arg180 and one at Val-181, releasing a 35-residue activation peptide (FIGURE 14.3A).32,56 The resulting FIXa consists of a light chain containing Gla, EGF1, and EGF2 domains (residues 1 to 145) and a heavy chain containing the protease domain (181 to 415) held together by a single disulphide bond.
FX consists of a light chain (amino acids 1 to 139) and a heavy chain (amino acids 143 to 448) held together by a single disulfide bond between Cys-132 and Cys-c302 (FIGURE 14.2A).35 The release of a 52-residue activation peptide after cleavage at Arg194-Ile195 (by FVIIa/TF, FIXa/FVIIIa, or Russell’s viper venom protease) in the heavy chain leads to the generation of FXa (FIGURE 14.3A).57 The light chain of FXa contains N-terminus Gla domain (residues 1 to 39), a hydrophobic stack of residues (residues 40 to 45), EGF1 domain (residues 46 to 84), and EGF2 domain (residues 85 to 128), and the heavy chain contains the protease domain (residues 195 to 448).
Prothrombin (FII) is the most abundant VKD protein in plasma and circulates as a single-chain molecule consisting of an
N-terminal Gla domain, two kringle domains, and a latent serine protease domain (FIGURE 14.2A).49,58,59 In vitro, thrombin cleavage of prothrombin at Arg155-Ser156 generates prothrombin fragment-1 (PF1) and prethrombin-1 (PT1) (FIGURE 14.3B).60,61 PF1 consists of Gla domain (residues 1 to 40) and kringle-1 domain (residues 65 to 155), whereas PT1 consists of prothrombin fragment-2 (PF2, residues 156 to 271), and prethrombin-2 (PT2), which contains the A chain (residues 272 to 320) and B chain (residues 321 to 579) of the C-terminus latent protease domain (FIGURE 14.3C). Cleavage of PT1 by FXa at Arg271-Thr272 releases PF2 from PT2. Proteolysis at Arg320-Ile321 by FXa then results in the two-chain active thrombin, which cleaves itself at R284-T285 to generate stable α-thrombin.32,62,63 Physiologic activation of prothrombin by the prothrombinase complex (FXa, Ca2+/Mg2+, PL, FVa) occurs by sequential proteolysis at Arg271-Thr272 and Arg320-Ile321.32,63,64 Initial cleavage at R320-I321 between the A and B chains is the preferred pathway under physiologic conditions and generates the active intermediate meizothrombin that retains all four domains of prothrombin. The alternative initial cleavage at Arg271-Thr272 generates inactive precursor PT2 (residues 272 to 579) and a combined PF1 and PF2 (residues 1 to 271). Cleavage of PT2 by FXa at Arg320-Ile321 generates a two-chain active thrombin composed of a 49 residue A chain and 259 residue B chain. This two-chain molecule, further processed by thrombin which cleaves chain A at Arg284, generates a stable α–thrombin with 36 residues A chain (FIGURE 14.3B).
N-terminal Gla domain, two kringle domains, and a latent serine protease domain (FIGURE 14.2A).49,58,59 In vitro, thrombin cleavage of prothrombin at Arg155-Ser156 generates prothrombin fragment-1 (PF1) and prethrombin-1 (PT1) (FIGURE 14.3B).60,61 PF1 consists of Gla domain (residues 1 to 40) and kringle-1 domain (residues 65 to 155), whereas PT1 consists of prothrombin fragment-2 (PF2, residues 156 to 271), and prethrombin-2 (PT2), which contains the A chain (residues 272 to 320) and B chain (residues 321 to 579) of the C-terminus latent protease domain (FIGURE 14.3C). Cleavage of PT1 by FXa at Arg271-Thr272 releases PF2 from PT2. Proteolysis at Arg320-Ile321 by FXa then results in the two-chain active thrombin, which cleaves itself at R284-T285 to generate stable α-thrombin.32,62,63 Physiologic activation of prothrombin by the prothrombinase complex (FXa, Ca2+/Mg2+, PL, FVa) occurs by sequential proteolysis at Arg271-Thr272 and Arg320-Ile321.32,63,64 Initial cleavage at R320-I321 between the A and B chains is the preferred pathway under physiologic conditions and generates the active intermediate meizothrombin that retains all four domains of prothrombin. The alternative initial cleavage at Arg271-Thr272 generates inactive precursor PT2 (residues 272 to 579) and a combined PF1 and PF2 (residues 1 to 271). Cleavage of PT2 by FXa at Arg320-Ile321 generates a two-chain active thrombin composed of a 49 residue A chain and 259 residue B chain. This two-chain molecule, further processed by thrombin which cleaves chain A at Arg284, generates a stable α–thrombin with 36 residues A chain (FIGURE 14.3B).
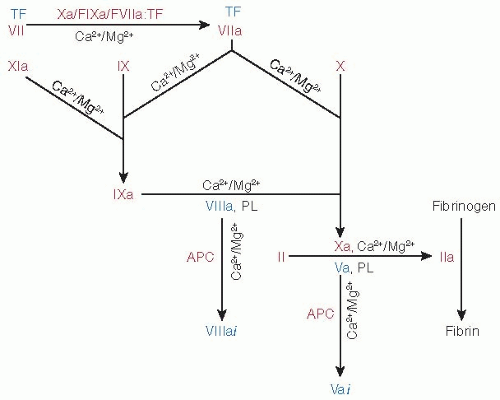 FIGURE 14.1 The role of VKD coagulant and anticoagulant proteins in hemostasis. The FVIIa/TF (extrinsic tenase complex) converts zymogens FX and FIX into their respective active FXa and FIXa forms. In positive feedback loops (top left), FXa, FIXa, and FVIIa/TF activate FVII/TF. FIXa bound to FVIIIa (intrinsic tenase complex) activates FX to FXa on the phospholipid (PL) surfaces, FXa then binding to FVa (prothrombinase complex) to convert prothrombin (II) to thrombin (IIa). APC inactivates FVa (to FVai) and FVIIIa (to FVIIIai). Activation of PC to APC by the IIa/-thrombomodulin complex and that of FXI to FXIa by IIa are not shown. Under physiologic conditions, all activation and inactivation processes involving VKD enzymes are Ca2+/Mg2+ dependent. Although Ca2+ alone can function in these reactions, free Mg2+ can substitute for Ca2+. Enzymes and inactive precursors are shown in red, cofactors in blue. |
APC circulates as a disulfide-linked two-chain zymogen PC.65,66,67 The N-terminal light chain contains the Gla domain and two EGF-like domains (EGF1 and EGF2), and the heavy chain contains the latent serine protease domain (FIGURE 14.2B).22,68,69 Although α–thrombin can activate PC to APC, the rate is extremely slow for a physiologically significant reaction. The physiologic activator of PC is the thrombin/thrombomodulin complex at the endothelial surface.23,70,71 During conversion of PC to APC, a short 12-peptide fragment is removed from the heavy chain N-terminus to generate the active protease (FIGURE 14.3A).60
Cofactor Participation
The catalytic activity of each VKD proteolytic protein is augmented by binding to their respective cofactors (Table 14.2). TF functions as a cofactor for FVIIa in the activation of FX and FIX. TF is an integral membrane protein and consists of an extracellular region, a transmembrane region, and a cytoplasmic region, which is structurally homologous to the class 2 cytokine receptor family.72 The extracellular region is made up of two fibronectin type-III domains that interact with all FVIIa domains.
FIXa requires FVIIIa as a cofactor for effective activation of FX. FVIII circulates in plasma in an inactive form consisting of a heavy chain (Mr varies between 90,000 and 200,000) that contains A1, A2, and B domains and a light chain (Mr ˜ 80,000) that contains A3, C1, and C2 domains.73,74 The activated form of FVIII is generated by the removal of B domain, either by FXa or by thrombin.75 Similarly, FVa acts as a cofactor for FXa to augment prothrombin activation. FV (Mr ˜ 330,000) circulates in plasma as a single-chain molecule having a similar domain organization as FVIII.76,77 The release of domain B by the action of thrombin generates FVa,78 a two-chain molecule consisting of a heavy chain (A1-A2 domains) and a light chain (A3-C1-C2 domains) (see Chapter 12).
PZ circulates in complex with PZI, as a single-chain molecule,79 consisting of an N-terminus Gla domain (residues 1 to 41), two EGF-like domains (EGF1, residues 42 to 82; EGF2, residues 83 to 139), and a pseudocatalytic domain (residues 149 to 360) (FIGURE 14.2B). The typical active site residues of the serine proteases, His57 and Ser195 (chymotrypsin numbering), are replaced with Lys and Asp, respectively, in the C-terminal domain.37
PS circulates as a single-chain molecule80 consisting of an N-terminus Gla domain (residues 1 to 41), a unique TSR (residues 42 to 75), four EGF-like domains (EGF1, residues 76 to 114; EGF2, residues 115 to 159; EGF3, residues 160 to 201; EGF4, residues 202 to 242), and a C-terminus sex hormone binding globulin (SHBG) domain (residues 243 to 635), which is completely different from other VKD proteins.80 The SHBG
region consists of two laminin G-like domains. PS functions as a cofactor for the anticoagulant APC and enhances the inactivation FVa and FVIIIa.27,81,82
region consists of two laminin G-like domains. PS functions as a cofactor for the anticoagulant APC and enhances the inactivation FVa and FVIIIa.27,81,82
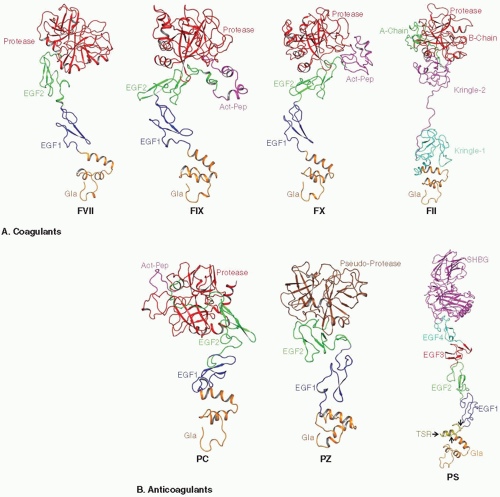 FIGURE 14.2 Modeled structures of the zymogen forms of coagulant (A) and anticoagulant (B) VKD proteins. The Gla domains are shown in orange, EGF1 domains in blue, EGF2 domains in green, and protease domains in FVII, FIX, FX, II, and PC in red. Kringle domains 1 and 2 of FII are colored deep teal and magenta, respectively, and the segments that become A and B chains in α-IIa are colored green and red, respectively. Activation peptide regions in FIX, FX, and PC are colored magenta. The pseudoprotease domain of PZ is colored brown. The thrombin-sensitive region (TSR), the EGF3, EGF4, and SHBG domains in PS are colored yellow, red, deep teal, and magenta, respectively; the SHBG domain in PS contains two laminin G-type binding subdomains. |
POSTTRANSLATIONAL PROTEIN PROCESSING
γ-Carboxylation and Propeptide Cleavage
The propeptide region contains the carboxylase recognition site. In FIX, site-specific mutagenesis of Phe(-16) to Ala and Ala(-10) to Glu inhibits γ-carboxylation.83,84 Naturally occurring Ala (-10) to Thr or Val and Asn(-9) to Lys mutations lead to warfarin hypersensitivity where severe depression of FIX occurs despite mild reductions in other VKD proteins.85,86,87 Synthetic FIX propeptides with Thr(-10) or Gly(-10) have reduced carboxylase affinity compared to Ala(-10),87 and mutation of Leu(-6) to Asp in FIX leads to impaired γ-carboxylation in tissue culture.88 Similar studies on FII show that the propeptide residues at -18, -17, -15, and -10 are important for γ-carboxylation.89 A DNA construct in which the propeptide region was placed adjacent to a region of thrombin rich in Glu residues resulted in γ-carboxylation of the Glu residues to Gla.90 Moreover, deletion mutants of proprotein C lacking residues -17 to -12 were also defective in γ-carboxylation.91 The NMR structure of synthetic FIX propeptide shows that the γ-carboxylase recognition site is likely within
an α–helical region that includes a hydrophobic surface similar to prothrombin.92,93 These data support a concept that the propeptide region itself is sufficient to support γ-carboxylation.
an α–helical region that includes a hydrophobic surface similar to prothrombin.92,93 These data support a concept that the propeptide region itself is sufficient to support γ-carboxylation.
Table 14.1 Properties of VKD zymogens | |||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
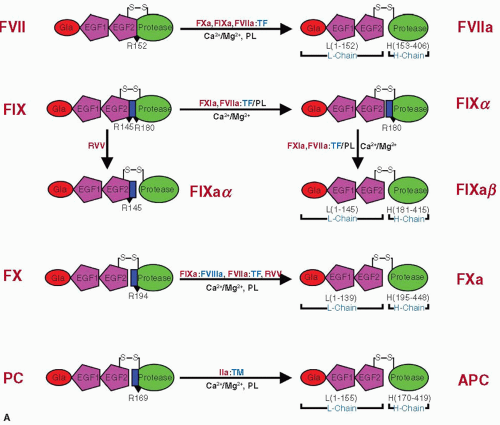 FIGURE 14.3 Schematic representation of zymogen activation. A: FVII, FX, and PC are converted to active forms by cleavage at a single residue at R152, R194, and R169, respectively. FIX is activated by FXIa and FVIIa/TF by sequential cleavages at R145 and R180 and by RVV by cleavage at R180. The activation peptide regions in FIX, FX, and PC are shown as blue rectangles, and the cleavage sites are marked with black triangles. |
Synthetic peptides with a complete propeptide sequence of FII were able to have their respective Glu residues γ-carboxylated efficiently with a Km of approximately 3 µM,94,95 whereas peptides with a shortened propeptide region or without the γ-carboxylase recognition site were poorly γ-carboxylated.94,95 Moreover, a synthetic peptide based upon the propeptide sequence of FX stimulates γ-carboxylation of a pentapeptide Phe-Leu-Glu-Glu-Leu approximately eightfold.96 Thus, it is feasible that the catalytic activity of carboxylase is regulated by binding to its normal substrate.
Importantly, the Gla domains also play a role in γ-carboxylase binding. In FIX, linkage of the propeptide to the Gla domain increased its affinity approximately 20-fold for γ-carboxylase.97 Moreover, the Gla domain was found to play an important role in the rate of release of FIX from γ-carboxylase as the koff of fully
γ-carboxylated FIX Gla domain was approximately threefold faster than the koff of noncarboxylated Gla domain.97 Absence of γ-carboxylation at two or more sites results in a prothrombin molecule that is an ineffective substrate for the prothrombinase complex.98 Thus, the prothrombin deficient in γ-carboxylation cannot bind Ca2+ and fails to undergo conformational changes that occur with Ca2+ binding. Moreover, the molecules are defective in binding to the PL surface on which the prothrombinase complex assembles.
γ-carboxylated FIX Gla domain was approximately threefold faster than the koff of noncarboxylated Gla domain.97 Absence of γ-carboxylation at two or more sites results in a prothrombin molecule that is an ineffective substrate for the prothrombinase complex.98 Thus, the prothrombin deficient in γ-carboxylation cannot bind Ca2+ and fails to undergo conformational changes that occur with Ca2+ binding. Moreover, the molecules are defective in binding to the PL surface on which the prothrombinase complex assembles.
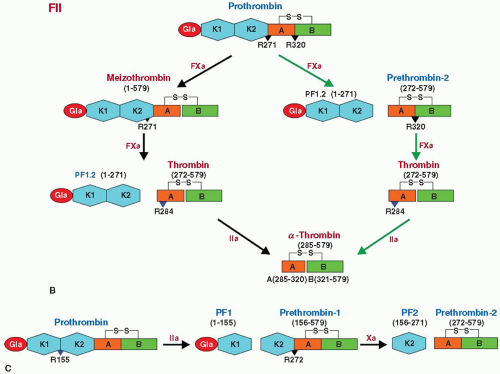 FIGURE 14.3 (Continued) B: Physiologic generation of thrombin from prothrombin. FXa converts prothrombin to thrombin, by proteolytic cleavages at R272 and R320, which occurs by two pathways. In one pathway (black arrows), cleavage at R320 yields meizothrombin, which is then cleaved at R271 to yield thrombin and PF1.2. In the second pathway (green arrows), cleavage at R271 yields PT2 and PF1.2. The generated PT2 is cleaved at R320 by FXa to yield thrombin. Thrombin generated by either pathway is further cleaved at R284 by thrombin itself to yield stable α-thrombin. FXa cleavage sites are indicated by black triangles and the thrombin cleavage site by the blue triangle. C: Thrombin-initiated conversion of prothrombin to thrombin starts with cleavage at R155, yielding PF1 and PT1. FXa cleavage at R272 in PT1 yields PF2 and PT2, which is converted to thrombin. Gla, γ-carboxyglutamic acid-rich domain; EGF1, epidermal growth factor-like domain 1; EGF2, epidermal growth factor-like domain 2; K, kringle domain; PF1, prothrombin fragment-1; PF2, prothrombin fragment-2; PF1.2, prothrombin fragment-1 and fragment-2. |
Table 14.2 Cofactors for VKD enzymes | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
The propeptide is cleaved from each VKD protein by PACE/furin,99 which recognizes a consensus sequence Arg-X-Arg/Lys-Arg at positions -4 to -1 and cleaves after the -1 position Arg residue. In FIX, this sequence includes three basic residues,
Arg(-4)-Pro-Lys-Arg(-1)-Tyr(+1). Hemophilic mutations at the -4, -2, and -1 positions (P4, P2, P1) cause a severe bleeding tendency due to dysfunctional FIX.100,101,102 FIX of hemophiliacs with Arg(-4) to Trp, Gln, or Leu react normally to antibodies that bind in the presence of Mg2+, indicating that γ-carboxylation has occurred, but the FIX mutant proteins require more than 5 mM Ca2+ for binding to the PL surfaces.103 Similarly, site-specific mutagenesis of Arg(-1) to Thr reduced propeptide cleavage by more than 99%, and nonconservative mutations at Pro(-3) and Leu(-6) also reduced propeptide cleavage in FIX.104 Recombinant profactor IX with105 full γ-carboxylation does not bind to PL membrane and is not activated by FXIa unless the propeptide is cleaved by PACE/furin. Thus, a free N-terminus is essential for proper conformational folding of the Gla domain, and acylation of Tyr(+1) is sufficient to prevent FIX binding to PL.103
Arg(-4)-Pro-Lys-Arg(-1)-Tyr(+1). Hemophilic mutations at the -4, -2, and -1 positions (P4, P2, P1) cause a severe bleeding tendency due to dysfunctional FIX.100,101,102 FIX of hemophiliacs with Arg(-4) to Trp, Gln, or Leu react normally to antibodies that bind in the presence of Mg2+, indicating that γ-carboxylation has occurred, but the FIX mutant proteins require more than 5 mM Ca2+ for binding to the PL surfaces.103 Similarly, site-specific mutagenesis of Arg(-1) to Thr reduced propeptide cleavage by more than 99%, and nonconservative mutations at Pro(-3) and Leu(-6) also reduced propeptide cleavage in FIX.104 Recombinant profactor IX with105 full γ-carboxylation does not bind to PL membrane and is not activated by FXIa unless the propeptide is cleaved by PACE/furin. Thus, a free N-terminus is essential for proper conformational folding of the Gla domain, and acylation of Tyr(+1) is sufficient to prevent FIX binding to PL.103
β-Hydroxylation
The VKD proteins undergo posttranslational modification via hydroxylation of Asp and Asn residues by aspartyl β-hydroxylase.106,107 FIX has β-hydroxylation at Asp64 in the EGF1 domain.108 However, only one-third of the Asp64 residues are hydroxylated,109 inhibition of hydroxylation in recombinant FIX does not decrease activity,110 and the hydroxyl group is not necessary for Ca2+ binding.111,112 Bovine FVII contains a β-hydroxyaspartic acid in the EGF1 domain at Asp63; however, Asp63 in human FVII is not β-hydroxylated.113 PS contains a β-hydroxyaspartic acid at residue 95 in the first EGF domain, and the other three EGF domains contain β-hydroxylated Asn residues, which are incompletely hydroxylated.114,115 Asp63 is hydroxylated in FX,116 and Asp71 in PC,117 whereas Asp64 is β-hydroxylated in PZ.118
Signal Peptide and Its Cleavage
In FIX, the hydrophobic signal peptide residue Ile30 is important as the hemophilic mutation to Asn results in no detectable FIX antigen.100 FIX signal peptide cleavage occurs between Thr18 and Cys19 and mutation of Cys19 to Gly, Arg, Trp, or Tyr is associated with hemophilia B.100 When the signal and propeptide sequences of FIX are substituted for those of PC, full γ-carboxylation occurs,119 indicating that the signal and propeptide sequences are not specific among homologous VKD proteins (see Chapter 9).
Glycosylation, Sulfation, and Phosphorylation
Glycosylation occurs either via Asn or Ser/Thr residues, forming amide linked (N-linked) or O-glycosidically linked (O-linked) carbohydrate chains. Human FVII contains two N-linked carbohydrate chains attached to the residues Asn145 and Asn322113 and two O-linked carbohydrate chains attached to the residues Ser52 and Ser60 in the EGF1 domain. Ser52 is linked to disaccharide (Xyl-Glc) or trisaccharide (Xyl2-Glc),120 whereas Ser60 is linked to a fucose.121 Alanine mutations at Ser52/Ser60 revealed that these O-glycosylation sites do not affect TF binding or the proteolytic activity of FVIIa/TF complex toward FX or FIX, even though the mutant FVIIa had reduced activity.122
Bovine FIX contains 26% carbohydrate123 and human FIX contains 17%,124 oligosaccharides primarily N-linked to consensus Asn-Xxx-Thr/Ser sequences (four in bovine and two in human). The two sites of N-linked glycosylation in FIX are Asn157 and Asn167, both within the activation peptide region.33 FIX from all species, all besides humans have an N-linked glycosylation at Asn260, all have Asn157, all but porcine have Asn167, and the sequences of ovine, bovine, and porcine predict an Asn172 site.125 Notably, FIX activity is preserved following sialic acid removal with neuraminidase.126 In the first EGF domain of FIX, Ser53 has O-linked di- and trisaccharides [Xyl1– or Xyl2-Glc-O-Ser]120 and Ser61 contains a tetrasaccharide [NeuAc-Gal-GlcNAc-Fuc-O-Ser].127 Of note is the fact that FIX proteins from other species such as porcine do not have homologous Ser residues, so O-linked carbohydrate moieties in the EGF1 domain are unlikely to have functional importance. In the FIX activation peptide, the hydroxyl groups of Thr159 and Thr169 are glycosylated.128 Plasma-derived FIX has an O-linked oligosaccharide at Thr172, sulfation of Tyr155, and phosphorylation of Ser158.129 Importantly, of all the activation peptide modifications in FIX, only the equivalent of Ser158 phosphorylation and N-linked glycosylation at Asn157 are conserved in other species. Lastly, the catalytic serine protease domain has no posttranslational modifications.129,130
Human FX contains two N-linked carbohydrate chains attached to residues Asn181 and Asn19135 and two O-linked carbohydrate chains attached to Thr159 and Thr171.131 The O-glycosylation sites are found in the activation peptide region, which is removed during activation to FXa. Human FX contains neutral bi-, tri-, and tetra-antennary complex type oligosaccharides without a fucose residue at the N-glycosylation sites and disialylated Galβ1-3GlaNAc sequences at the O-linked sites.132 The removal of the activation peptide or the Asn191 to Ala mutation affects the activation of FX by FVIIa/TF133,134 and FIXa/FVIIIa133 marginally. However, in another study, the carbohydrates contribute to the cofactor-dependent recognition of the zymogen by both the extrinsic and intrinsic Xase complexes.135 Thus, the physiologic role of carbohydrate chains in FX remains unclear.133,134,135 Human FII contains approximately 10% carbohydrates attached through the three N-linked glycosylation sites at Asn78 and Asn100 in the kringle-1 domain of PF1 and Asn102 of the thrombin B chain.136 Partial deglycosylation at Asn102 using neuraminidase, β-galactosidase, and β-N-acetylglucosaminidase suggests that the carbohydrates at this site had no effect on thrombin functions.137
Similar to prothrombin, human PC contains only N-linked glycosylation sites. The N-linked glycosylation in PC is highly variable with several glycosylation variants present in human plasma that contain bi-, tri-, or tetra-N-linked carbohydrates attached to Asn97, Asn248, Asn313, and Asn329.68,138 Mutational studies carried out at each glycosylation site of PC indicate that Asn97 glycosylation is essential for the efficient secretion of PC and affects the degree of core glycosylation at Asn329.139 All glycosylation variants of PC can be activated; however, they have different rates of activation as well as different anticoagulant activities. Elimination of all the glycosylation sites in PC increases the anticoagulant activity two- to threefold.140 Elimination of only the N-linked site at Asn313 yields a 2.5-fold increase in the rate of PC activation due to a decrease in Km.139 A polylactosamine on PC has been implicated in inhibiting cell adhesion through E selectin and may represent a potential anti-inflammatory function.140
Human PZ contains nine potential carbohydrate sites, of which five are N-linked sites and the other four are O-linked sites. The potential N-linked oligosaccharide are Asn59, Asn185, Asn193, Asn266, and Asn292, and the potential O-linked oligosaccharide sites are Ser53, Ser72, Ser196, and Ser275.38 Ser53 is linked to either Xyl-Glc or the Xyl2-Glc.120 The functional role of
carbohydrate chains in PZ is unknown. PS contains three potential N-linked glycosylation sites at residues Asn458, Asn468, and Asn489 in the SHBG domain.27,141 Asn to Gln mutagenesis revealed that Asn458 mutant affects cofactor activity while the Asn468 mutant affects γ-carboxylation.142,143
carbohydrate chains in PZ is unknown. PS contains three potential N-linked glycosylation sites at residues Asn458, Asn468, and Asn489 in the SHBG domain.27,141 Asn to Gln mutagenesis revealed that Asn458 mutant affects cofactor activity while the Asn468 mutant affects γ-carboxylation.142,143
VKD CRYSTAL STRUCTURES
The coagulation process is regulated by VKD proteins that appeared to be evolved from the common ancestor. These proteins share high degree of sequence and structural homology; however, each VKD protein performs its function in a precise, controlled manner. Among the VKD proteins, FVIIa is the only one whose complete structure has been determined.144,145 FVIIa adopts an extended conformation, where the Gla domain is on one end of the molecule and the catalytic domain is on the other. The binding of FVII/FVIIa to TF makes FVII/FVIIa somewhat more rigid and less flexible.146 The sequence similarity and domain organization of FIXa, FXa, and APC indicate that these proteins may assume similar conformations.147,148,149,150,151,152,153,154,155
Crystal structures of IIa and its different forms as well as of FII fragments have been determined; these include PF1,156,157 PT1,158 meizothrombin des-F1,159,160 PF2,161 PT2,162,163,164 and native and mutant thrombins.165,166,167,168,169,170,171,172,173,174,175 The EGF2-protease segment of PZ in complex with ZPI has been reported,176,177 but for PS, only the NMR structures of EGF3-EGF4 domains have been reported.178
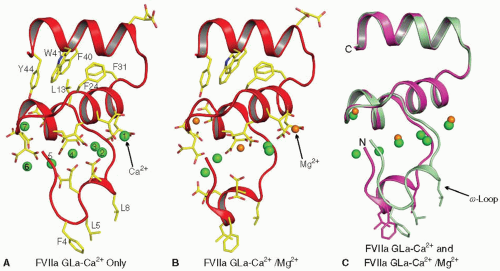 FIGURE 14.4 Structure of the FVIIa Gla domain in the presence of Ca2+ only, and with Ca2+ and Mg2+. Left: Structure of the Gla domain in the presence of Ca2+ only.144 The Gla domain of FVIIa contains seven metal binding sites, and all are occupied by Ca2+ in this structure. Center: Folding of the Gla domain in the presence of Ca2+/Mg2+.145 In this structure, the external (numbers 1 and 7) and the central (number 4) metal sites are occupied by Mg2+ while the other sites are occupied by Ca2+. Right: The superimposed structures shown in “A” and “B.” Although the divalent metal ions occupy similar positions in the two structures, the fold of the ω-loop is different between the two structures. The membrane binding residues Phe4, Leu5, and Leu8 in the ω-loop as well as the Gla residues and the hydrophobic residues in the C-terminal helix of the Gla domain are shown in stick representation. The Ca2+ (green) and the Mg2+ (orange) are shown as spheres. The numbering system used is that of Tulinsky et al.156 |
Gla Domains
The common structural element among all VKD proteins is their N-terminal Gla domain, the glutamic acid residues within the first 40 residues being converted to Gla residues by carboxylase. The binding of Ca2+ and Mg2+ divalent metal ions to the Gla domain induces a structural transition from an unfolded, non-functional form to a folded functional form. In its functional form, the Gla domain binds to phosphatidylserine-exposed PL membranes with the aid of solvent-exposed hydrophobic residues in the ω-loop. Prothrombin, FVII, APC, and PS each contain seven divalent metal binding sites in their Gla domains, whereas FIX, FX, and PZ contain more than seven sites. In addition to membrane binding, Gla domains serve as cofactor and/or substrate binding recognition sites.
FVII/VIIa. Seven divalent metal binding sites144 have been identified in the Gla domain of FVII/FVIIa and are necessary for stabilizing its functional fold. Among the 10 Gla residues present in FVII/FVIIa, 9 are associated with the metal ions. In the crystal structure of FVIIa/TF solved in the presence of Ca2+ only (PDB id 1DAN), all seven sites in the Gla domain are occupied by Ca2+ ions,144 whereas in the presence of Ca2+/Mg2+ (PDB id 2A2Q,145), three of the seven Ca2+ ions at positions 1, 4, and 7 are replaced by Mg2+ (compare FIGURE 14.4A,B). The fold of the Gla domain ω-loop in FVIIa is different between these two structures (FIGURE 14.4C). Folding of the ω-loop under near physiologic concentrations of Ca2+/Mg2+ is similar to 2A2Q
structure, with identical positions for four Ca2+ and three Mg2+ (PDB id 3TH2,179). Further, the folding of the ω-loop under 45 mM Ca2+/5 mM Mg2+ is similar to that reported in the presence of Ca2+ only (PDB id 3TH4,179); however, positions 1 and 7 are occupied by Mg2+ (PDB id 3TH4). Thus, it would appear that four Ca2+ and three Mg2+ ions are bound to the Gla domain in circulating FVII/FVIIa.
structure, with identical positions for four Ca2+ and three Mg2+ (PDB id 3TH2,179). Further, the folding of the ω-loop under 45 mM Ca2+/5 mM Mg2+ is similar to that reported in the presence of Ca2+ only (PDB id 3TH4,179); however, positions 1 and 7 are occupied by Mg2+ (PDB id 3TH4). Thus, it would appear that four Ca2+ and three Mg2+ ions are bound to the Gla domain in circulating FVII/FVIIa.
The portion of the Gla domain located at the bottom of FVIIa (FIGURE 14.5A) contains three solvent-exposed hydrophobic residues (Phe4, Leu5, and Leu8) present in the ω-loop that are hypothesized to be inserted into the PL membrane.180,181 The carboxy terminus of sTF is also located near the bottom of the structure; in intact TF, this part of the molecule is linked to the membrane anchor by a short peptide sequence. Therefore, the membrane surface should be located at the bottom of the Gla domain, with TF linked to the membrane by its membraneanchoring domain and the FVIIa interacting with PL head groups through the Gla domain. Fluorescence resonance energy transfer experiments indicate that the FVIIa/TF complex is oriented almost perpendicular to the membrane, with the active site of FVIIa located approximately 78 Å above the membrane surface.182
In addition to membrane binding, FVIIa Gla domain is involved in binding to the C-terminal region of TF, a mainly hydrophobic interaction involving the hydrophobic residues present in the Gla C-terminal helix. The FVIIa Gla domain also plays a role in FIX and FX substrate interaction where it serves the exosite recognition sites for macromolecular substrates. Biochemical and modeling data suggest that residues Asp33, Ala34, Gla35, Arg36, Leu39, Trp41, Ile42, and Lys48 in the FVIIa-Gla domain are involved in substrate interaction.183,184,185,186,187
FIX/IXa. The Gla domain of human FIX contains 12 Gla residues,188 the first 10 of which are at homologous positions of other VKD proteins. The FIXa Gla domain undergoes a transition to an ordered structure on addition of Ca2+ or Ca2+/Mg2, as seen in x-ray diffracted crystals189,190; NMR studies indicate a very similar structure for FIX Gla domain in solution.191,192 The coordination of the Ca2+ within the FIX Gla domain differs slightly from those in FVII, FX, and FII (FIGURE 14.5A).193,194 The FIX Gla domain binds to eight Ca2+, five of which are internal and three are on the surface and contribute to surface charge.189,190 The three surface sites bind to Mg2+ as well.189 Of the Gla residues, 8 of the first 10 that are homologous to the other VKD proteins are essential for activity. Of the 10, hemophilia B is associated with mutations in all but Gla15,100 which coordinates to Ca2+-7.189,190 Gla21 mutations result in an inactive protein.193 This is interesting because Gla21 coordinates to Ca2+-6189 and Ca2+-6 coordinates to lysophosphatidylserine, displacing a water molecule from the coordination sphere in PF1.190 In contrast, the nonhomologous Gla36 and Gla40 do not contribute to activity194; however, they provide all four ligands in coordinating to Ca2+-8 or Ca2+-9193, indicating that Ca2+-8 or Mg2+-8 (FIGURE 14.5A) is not necessary for FIXa activity.
Antibodies that bind to the FIX Gla domain in the presence of divalent metal ions indicate the presence of both Ca2+ and Ca2+/Mg2+-dependent epitopes, and the latter are localized to the carboxy terminus of the domain.195 Mg2+ binding to the Gla domain are important for FIX activity.196,197 Comparison of immunoreactivity of FIX molecules substituted with portions of the FVII Gla domain indicates that Gla33, Thr39, and Gla40 are involved in a high affinity where Mg2+ can bind.198 The ω-loop in FIXa is important in binding to negatively charged PL surface, involving Lys5 (FIGURE 14.5A) binding to the glycerol phosphate backbone and the carboxyl group of phosphatidylserine coordinating to Ca2+-5 and Ca2+-6 as has been shown for PF1.157,199 Further, Mg2+ binding to the Gla domain enhances binding of FIX to PL at physiologic Ca2+.200
Gla20 and Gla21 are in a conserved 4-amino acid disulfide loop that is buried with hydrophobic interactions to an aromatic stack composed of Phe41, Trp42, and Tyr45 near the carboxy terminus of the domain. Hemophilic or recombinant mutations of the Cys residues inactivate the protein.201 Recombinant FIX without the aromatic stack can be activated by FXIa but has low specific activity; mutagenesis of Phe41 to Val or Asp similarly affects activity whereas Tyr41 retains 64% of FIX activity.202 The Gla domain of FIX is important for activation by both FVIIa/TF203 as well as by FXIa.204,205 The Gla domain of FIXa has been implicated in binding to TF in the FVIIa/TF:FIXa ternary complex187,206 and binds directly to FXIa.205 The Gla domain of FIXa also binds to the FVIIIa C2 domain (A3-C1-C2),207,208 most likely involving Phe25 and Val46. Mutation of Gly12 to Arg disrupts FVIIIa binding209; however, this is most likely due to a structural impairment rather than from a direct binding effect. More studies are needed to elucidate the exosites on FIXa important in binding to FVIIIa.
Cultured endothelial cells have specific receptors that bind FIX and FIXa,210,211 allowing intrinsic FX activation without the need for platelets; this binding may also occur in vivo.212 As is the case with the PL, binding of the Gla domain of FIXa involves residues 3 through 11213 although collagen may account for some of the binding.214 Using protein fragments to inhibit 125I-labeled FIX binding to cultured endothelial cells, determinants in the first EGF-like domain of FIX were demonstrated, whereas binding to PL was only competed with the Gla domain peptides.215 From domain exchange experiments with FIX and PC, optimal endothelial cell binding appears to involve the entire FIX Gla domain including its aromatic stack.216 In kinetic studies, FVIIa activated FIX, but not FX, directly on the endothelial surface, and FIX activation was about one-sixth as rapid as compared to observed with stimulated platelets.217
In vivo, there is a basal low level of FIX activation by FVIIa,218 presumably at the endothelial cell surface. Mutation of Lys5 to Ala or Val10 to Lys considerably reduces the affinity of FIX for endothelial cells, whereas Lys5 to Ala increases FIX affinity for bovine vascular endothelial cells threefold.213 The endothelial cell-binding site for FIX involves two sites on collagen IV that are 50 and 98 nm from the C-terminus of collagen IV.214,219 Further, FIX binds in vivo to the endothelial cell collagen IV surface,220 and this binding may play an important role in regulating the concentration of FIX in the blood.
The distance from a rhodamine-labeled lipid surface to a fluorescein-labeled FIXa active site is at least 70 Å,221 a value consistent with the molecular dimensions of the crystallographic structure if lipid binding involves the Gla domain and the balance of the protein extends vertically above the surface.147 Electron crystallographic studies of two-dimensional (2-D) crystals of human FIX zymogen bound to a lipid layer support this orientation.222
FX/Xa. The human FX Gla domain contains 11 Gla residues and binds 8 Ca2+ ions (FIGURE 14.5A),223 7 of which have been observed in the crystal structure of the Gla domain of bovine FX. The Ca2+-8 was not seen in the crystal structure and is
modeled based upon the structure of the Sr2+ bound Gla domain of bovine PF1.224 An additional Ca2+ site is in the Gla domain of bovine FX involving Gla residues 35 and 40; residue 35 is Asp in human FX Gla domain, which precludes binding of Ca2+ at this site. Ca2+ sites at positions 1, 4, and 7 were occupied by Mg2+ in the crystal structure of Gla domain of human FX in the presence of Mg2+ only.152 Mutation of Gly11 to Val observed in FX deficient patients disrupts the activation of FX by either FVIIa/TF or FIXa/FVIIIa225; this mutation could partially impair folding of the ω-loop. Another mutation (Gla7 to Gly) may impair binding of Ca2+ at sites 2 and 3 and proper folding of the Gla domain.226 The Gla domain of FXa is also important in the activation of FVII/TF to FVIIa/TF.227
modeled based upon the structure of the Sr2+ bound Gla domain of bovine PF1.224 An additional Ca2+ site is in the Gla domain of bovine FX involving Gla residues 35 and 40; residue 35 is Asp in human FX Gla domain, which precludes binding of Ca2+ at this site. Ca2+ sites at positions 1, 4, and 7 were occupied by Mg2+ in the crystal structure of Gla domain of human FX in the presence of Mg2+ only.152 Mutation of Gly11 to Val observed in FX deficient patients disrupts the activation of FX by either FVIIa/TF or FIXa/FVIIIa225; this mutation could partially impair folding of the ω-loop. Another mutation (Gla7 to Gly) may impair binding of Ca2+ at sites 2 and 3 and proper folding of the Gla domain.226 The Gla domain of FXa is also important in the activation of FVII/TF to FVIIa/TF.227
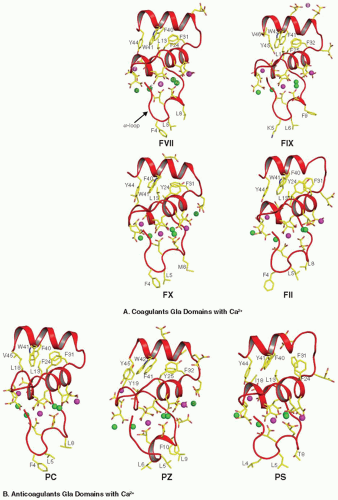 FIGURE 14.5 Structures of the Gla domains of coagulant (A) and anticoagulant (B) VKD proteins in the presence of Ca2+. FVII, FII, PC, and PS contain seven metal binding sites, while FIX, FX, and PZ contain more than seven sites in their respective Gla domains. The Gla residues and the membrane binding hydrophobic residues at the C-terminal helix of the domain are shown in stick representation. The Ca2+ metal sites, which cannot be replaced by Mg2+ are shown as green spheres and those which could be replaced by Mg2+ are shown in magenta. In circulation, the depicted Mg2+ sites are most likely not occupied by Ca2+ and are likely specific for Mg2+. The metal binding sites in the Gla domain of PZ and of PS are modeled based upon other VKD proteins. |
Prothrombin (II). The first x-ray structure of the Gla domain was obtained for bovine PF1.156 The Ca2+ ions are necessary to stabilize the Gla domain fold of FII.156,228 Human FII contains 10 Gla residues and binds 7 Ca2+ in the absence of Mg2+ and 4 in the presence of Mg2+.145,229 Based upon the crystal structures of the FVIIa and FIXa Gla domains,145,189 positions 1, 4, and 7 can be occupied by Mg2+ (FIGURE 14.5A). All Gla residues except Gla6 are important for proper function in FII.230 Ca2+/Mg2+ binding to the Gla domain are required for insertion of designated hydrophobic amino acids from the ω-loop into the PL surface.112,156
PC/APC. The Gla domain of PC/APC contains nine Gla residues and binds seven Ca2+ ions (FIGURE 14.5B)68,187 in the absence of Mg2+, and four in the presence of Mg2+. Mutations of Gla residues 7, 20, 26, and 29 results in nearly complete loss of APC anticoagulant activity. Mutation of Gla25 decreases activity by 75%, but Gla6, 14, and 19 are not critical for APC functions.231,232,233,234,235,236 Mutations at the hydrophobic residues near the N-terminus, particularly Leu5,233 disrupt PL binding, and Arg15 is important in Ca2+ binding, as is the disulfide bond for function.236 His10 is largely responsible for the 10-fold higher membrane binding affinity of human PC over bovine PC, which has a Pro at this position.237 Endothelial protein C receptor (EPCR) can substitute for PL in promoting PC activation by binding to the Gla domain, but such binding requires near complete γ-carboxylation.238,239,240 Binding of PC to EPCR increases PC activation via decreasing the Km.241,242 Soluble EPCR inhibits PC activation by blocking PC binding to cellular EPCR.242 Human, but not bovine243 FVa can potentiate PC activation on endothelium,244,245 and human FVa can accelerate PC activation in solution.245
PZ. The human PZ Gla domain contains 13 Gla residues, which is the highest for any VKD protein.37,38 Similar to FIX Gla domain, PZ has an additional residue at position 4 (FIGURE 14.5B).37 The ω-loop of PZ contains four hydrophobic residues, unusual for the VKD proteins, while the others contain only three hydrophobic residues. Ca2+ binding studies performed with PZ and Des-Gla PZ revealed that only the Gla domain has Ca2+ binding sites, whereas the EGF-like domains and the pseudoprotease domain do not.246 Direct Ca2+ binding studies performed from 0 to 0.8 mM Ca2+ indicate that the Gla domain of PZ contains only four sites247; however, from the positions of the Gla residues and the similarity with FIX and PF1, PZ might contain more than seven metal binding sites (FIGURE 14.5B). Two point mutations (Gln or Lys) have been reported in patients at residue Glu30, affecting PZ secretion from transfected cells.247,248,249 PZ functions as a cofactor for PZI inhibition of FXa by binding to the PL through its Gla domain.24 PZ has nearly 100-fold slower membrane binding and dissociation kinetics as compared to PS and PC because of the unique Gla residue at position 11.250
PS. Human PS contains 11 Gla residues and may contain seven metal binding sites (FIGURE 14.5B) and binds to PL membrane with higher affinity than PZ or PC.251 Chemical modifications in the Gla residues of PS affect PL binding and also self association.252 The Gla domain of PS binds to PL membrane in a Ca2+ dependent manner, but high Ca2+ (10 mM) disassociates the Gla domain from PL.253 The TSR region in PS may affect Ca2+ binding and disrupt the structure of the Gla domain.254 The Gly11Asp and Thr37Met missense mutations are reported in patients with PS deficiency, and mutagenesis studies reveal that Gly11 to Asp mutation affects its cofactor activity, while the Thr37 to Met mutation affects PS secretion.255 Mutation at Gla36 affects its cofactor activity by 89% for FVa inactivation and completely for FVIIIa.256 PS enhances the binding of APC to PL membrane,257 and the Gla domain of PS is implicated in plasma clotting assays.257
EGF-Like Domains
EGF domains are mainly involved in protein-protein interactions and include human EGF receptor family ligands such as EGF and Transforming Growth Factor-α (TGF-α), low-density lipoprotein receptor, matrix proteins, the transmembrane receptor Notch, and proteins involved in blood coagulation and fibrinolysis.107 The EGF domain contains approximately 45 amino acids and the fold is primarily stabilized by the three signature intradisulfide bond patterns as 1-3, 2-4, and 5-6. The EGF domain has two antiparallel β-sheets, which are referred to as major (N-terminal) and minor sheets (C-terminal). FVII, FIX, FX, PC, and PZ each contain two EGF-like domains, whereas PS contains four EGF-like domains in tandem. The first EGF domain in FVII, FIX, FX, and PC is a Ca2+ binding type domain, with functional significance, restricting Gla domain movement, which is necessary for the biologic activity. The first EGF domain in PZ or PS lacks the Ca2+ binding site and may be more flexible.
FVII/VIIa. The EGF domains present in FVIIa make strong interactions with TF. The main sites of contact on FVIIa are located in the first EGF domain and the protease domain, with additional points of contact within the second EGF domain and the aromatic stack region of the Gla domain. The sites of contact on TF are located on both fibronectin type III domains and the interfacial region between them,144,145,258,259 Binding studies with proteolytic fragments and domain swapping experiments indicate that FVII EGF domains are important in binding to TF.259,260 The N-terminus of the EGF1 domain contains a strong Ca2+ binding site, which is important for cofactor interaction and proteolytic function.261,262 The Ca2+-bound structure of EGF1 (FIGURE 14.6) shows Ca2+ coordinated to the main chain carbonyl oxygen of Gly47 and Gln64 as well as to the side chain carboxyl group oxygen of Asp46 and Asp63. Site-directed fluorescence experiments reveal that complex formation between sTF and FVIIa is initiated upon Ca2+ binding to EGF1.263 The structural similarity between the Ca2+-bound and Ca2+-free structures suggest that Ca2+ binding is responsible for the increased affinity of FVII/FVIIa for TF.264 Site-directed mutagenesis experiments in which Ca2+ binding residues Asp46 and Asp63 are replaced with Asn abolished Ca2+ binding and reduced FVIIa proteolytic activity.265 The Asn57 to Asp
mutation in EGF1 affects the FVIIa secretion as well as its activity.265 Two other EGF2 mutations (Arg110 to Cys and Asp123 to Tyr) reduce FVII secretion.266 FVII EGF domains also play a role in FX activation.267 A peptide sequence from the EGF2 domain of FVII inhibits TF-dependent FX activation, most pronounced for a constrained sequence corresponding to residues 91 to 102.267 These results suggest a direct role for the EGF2 domain of FVII, and in particular its loop I, in the formation and function of the FVII/TF/FX complex.267
mutation in EGF1 affects the FVIIa secretion as well as its activity.265 Two other EGF2 mutations (Arg110 to Cys and Asp123 to Tyr) reduce FVII secretion.266 FVII EGF domains also play a role in FX activation.267 A peptide sequence from the EGF2 domain of FVII inhibits TF-dependent FX activation, most pronounced for a constrained sequence corresponding to residues 91 to 102.267 These results suggest a direct role for the EGF2 domain of FVII, and in particular its loop I, in the formation and function of the FVII/TF/FX complex.267
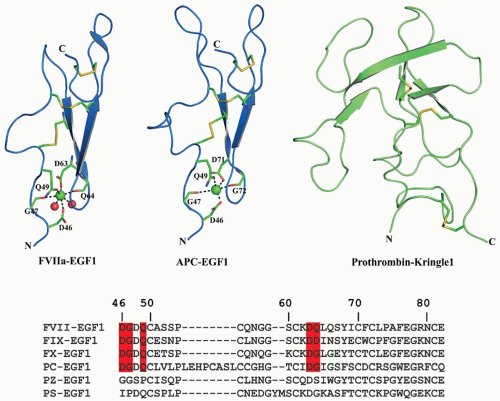 FIGURE 14.6 Cartoon representation of the EGF1 domains and the prothrombin kringle domain-1 (K1). The FVIIa EGF1 domain is on the left, APC EGF1 domain at the center, and K1 domain of prothrombin fragment-1 (PF1) is on the right. The Cys residues which form disulfide bridges are shown in stick representation. Ca2+ ions are shown as green spheres. The Ca2+ binding loops are similar in FVII, FIX, and FX, whereas APC has an eight-residue insertion in its Ca2+ binding loop. The multiple sequence alignment of EGF domains of FVII, FIX, FX, PC, PZ, and PS are shown at the bottom. The residues that serve as ligands for Ca2+ binding are highlighted in red; Mg2+ cannot replace Ca2+ in these domains. PZ or PS does not contain a Ca2+ binding site in the EGF1 domain. |
FIX/IXa. The FIX EGF1 domain extends from the aromatic stack to EGF2, which lies under the catalytic domain.147,148 The two FIX EGF domains interact through a salt bridge between Glu78 and Arg94 and by a hydrophobic patch.268,269 There is more restriction of rotation between the two hydrophobic patches in FIXa than in FXa.270 The interface between the EGF2 domain and the serine protease domain has extensive contacts involving aromatic residues.270 The structure of the EGF fragment by 2-D-NMR271 indicates that antiparallel β-pleated sheets observed in crystals also exist in solution. The first EGF domain of FIX was crystallized with Ca2+ bound to the Ca2+ site.272 Similar to FVII, the first EGF domain in FIX binds a single Ca2+ with high affinity,111,273,274,275,276 whereas the second EGF domain does not bind Ca2+. Ca2+ in the EGF1 domain of FIX coordinates to the side chains of residues Asp47, Gln50, and Asp64 and to the backbone carbonyls of residues Gly48 and Asp65273; Val46Glu or Gln50Glu mutations do not affect binding of Ca2+ to the EGF1 domain.276 Tyr69 to Cys produces severe hemophilia B100 and site-specific mutagenesis indicates that this residue is important for activity but not for Ca2+ binding or β-hydroxylation.277 Gly60 to Ser is a mild hemophilic mutation,100 and modeling predicts misfolding
due to steric clash with the EGF1 Cys73-Cys82 disulfide bond.278 Pro55 to Ser or Leu also causes mild hemophilia, caused by impaired FX activation.279
due to steric clash with the EGF1 Cys73-Cys82 disulfide bond.278 Pro55 to Ser or Leu also causes mild hemophilia, caused by impaired FX activation.279
Domain exchange among homologous VKD factors has provided further evidence for key structural elements of the EGF-like domains. Lin et al.280 found that replacing the first EGF-like domain of FX into FIX had little effect on activity and that substitution of both EGF-like domains with those of FX retained only 4% FIX activity. Conversely, replacing both the Gla and first EGF-like domain of FIX into FX led to a requirement of higher concentrations of FVa for FX activity281; therefore, cofactor binding was suboptimal but did not require specific FX sequences within the replaced segment. When the EGF-like domains of FIX were substituted with those of PC, the resulting APC had only 10% activity.282 The first EGF-like domain of FVII has also been substituted for that of FIX in a recombinant protein283; this species is of particular interest because FVIIa/TF activates FIX, and this portion of FVII participates in TF binding. Curiously, the chimeric protein had twice the specific activity of wild-type FIX, enhanced binding to FVIIIa, and even enhanced activity in canine hemophilia B. Monoclonal antibody that recognizes the C-terminal part of the EGF1 domain, particularly Trp72 and Lys80, blocks FIX activation by FXIa, marginally decreases activation by the FVIIa/tTF complex, and slightly decreases the kcat for FX activation.284 Further, the Fab fragment of this EGF1 monoclonal antibody has 10-fold higher affinity for FIXa as compared to FIX,285 indicating that the conformational changes that occur in the protease domain during activation and removal of the activation peptide are linked to the EGF1 domain and that the FIXa EGF1 domain is not involved in binding to FVIIIa.
The FIX EGF1 domain is required for activation by the FVIIa/TF complex but not by FXIa.286,287 Further studies by Zhong et al.288 revealed that the EGF1 domain of FIX interacts with TF in the FVIIa/TF. Recently, Ndonwi et al.227 reported that EGF1 domain of FIXa is important for the feedback activation of FVII/TF to FVIIa/TF. Several studies have emphasized the importance of Ca2+ binding to the FIXa EGF1 domain for FVIIIa binding on PL.111,268 However, Gln50 to Pro mutation only reduced affinity for FVIIIa in the presence of PL,289 and replacement of the FIX EGF1 domain with that of PC resulted in approximately 80-fold decreased affinity for FVIIIa in the presence of PL and a minor difference without PL.289 Monoclonal antibody to the EGF1 domain showed that the antibody does not affect FVIIIa binding to FIXa.285 This study and others question whether the hydrogen bond between Glu78 in the EGF1 domain and Arg94 in the EGF2 domain is crucial for the interaction of FIXa with the light chain of FVIIIa, as hypothesized by Christophe et al.268 Therefore, it appears that the EGF1 domain does not directly bind to FVIIIa but rather correctly positions the EGF2 and protease domains for proper interactions.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


