Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
Maud B. Gorbet
Michael V. Sefton
Cardiovascular device manufacturers rely on elegant device designs and systemic pharmacologic agents to work wonders with existing materials despite their thrombogenicity. Adverse effects are minimized and existing devices are, if not risk free, providing sufficient benefit to outweigh the risks. The focus of research in biomaterials is to make better materials that pose fewer risks and offer greater benefits. Many strategies for lowering thrombogenicity have been identified, and all these strategies show a beneficial effect in at least one assay of thrombogenicity. However, the perfect material is yet to be identified.
Clinical manifestations of the bioincompatibility of cardiovascular devices are numerous1,2: Sudden and complete occlusion of stents3; acute and subacute thrombotic occlusion in medium-sized grafts (4 to 6 mm); embolic complications with artificial hearts,4 catheters,5 and prosthetic valves6; and thrombotic complications during cardiopulmonary bypass (CPB)7 and angioplasty.8 Large-diameter vascular grafts remain thrombogenic for many years, but fewer thrombotic complications are observed because high flows disperse the coagulation proteins. Even if the risk of thrombotic complication appears to be low (varying between 1% and 10% depending on the device), the complications may have fatal outcomes, and the cost associated with the follow-up intervention is not negligible. Furthermore, these thrombotic complications with cardiovascular devices occur, despite the use of antiplatelet and anticoagulant therapies. In the absence of nonthrombogenic materials, current devices rely on avoiding irregular flow geometries (i.e., sudden contractions, recirculation zones, and poor device streamlining) so that thrombi do not accumulate within the device.9,10 Coupled with this is the routine use of antithrombotic agents (see Spinler et al.11 for guidelines and Gorbet et al.12 for review). The inherent thrombogenicity of currently existing cardiovascular devices can be illustrated by the catastrophic impact that premature (prior to 6 months) discontinuation of dual antiplatelet therapy can have on the risk of thrombosis with drug-eluting stents (DES).13
This chapter focuses on biomaterials and their interactions with blood. We consider how each component, including leukocytes and complement, is thought to interact with biomaterials to induce thrombosis. The role that endothelial cells and diseases such as diabetes may play in exacerbating material-induced thrombosis is also discussed.
INTRODUCTION TO BIOMATERIAL-ASSOCIATED THROMBOSIS
Overview
Cardiovascular devices (except endothelial cell-seeded vascular grafts) expose a foreign surface lacking the properties of the endothelium to the circulation. This exposure triggers a complex series of events, including protein adsorption, platelet and leukocyte activation/adhesion, and the activation of complement and coagulation; the events are highly interlinked in this simplified picture (see FIGURE 96.1).
Criteria for Nonthrombogenicity
Much of the effort in biomaterials research over the last 30 years has been directed toward the development of inert materials that do not react with platelets and coagulation factors. A number of approaches, often conflicting, have been developed. For example, there is still no consensus as to whether a surface should be hydrophilic or hydrophobic.
Thrombogenicity is defined as the ability of a material to induce or promote the formation of thromboemboli.14 In the past, we have thought of a nonthrombogenic surface as one that prevented platelet adherence and did not shorten blood clotting time. These criteria are easily met by heparinized materials,15 but further studies have shown that some hydrogels, even with heparin, can activate platelets, leading to high platelet consumption.16 The latter materials promote the formation of microthrombi, although the thrombi do not attach to the materials. A clotting time measurement is a reasonable way of assessing the effect of a material on coagulation. However, this assay is likely unimportant for predicting thrombosis in vivo under normal and pathologic conditions.
The criteria that we use as hallmarks of nonthrombogenicity are given in Table 96.1. Rather than a clotting time, a specific thrombin production rate constant, kp, enables the extrapolation to indicate realistic situations. Experimental results suggest that kp is of the order of 10-3 per second for simple materials such as polyethylene (the same scale as mass transfer coefficients in flowing blood) but <10-4 per second for heparinized materials.17 According to a simple flow model (see FIGURE 96.2), only the latter low kp materials can be expected to minimize fibrin production. This is one of the reasons heparinization and other active methods of inhibiting thrombin formation are so popular as strategies for imparting low thrombogenicity. There are, however, other criteria that must also be met. It is a requirement that platelet interactions with the surface should not lead to thrombosis. Platelet activation and consumption may therefore be better indicators of platelet incompatibility than just deposition. Furthermore, some deposition may even be desirable since these deposits appear to passivate the surface to further activation. Although a nonthrombogenic surface should not support platelet adhesion, some ex vivo studies18,19 have demonstrated that even in the absence of adhesion, platelets can be “consumed.” That is, the platelets can be activated by the material and prematurely removed from the
circulation (e.g., shorter platelet lifespan, decrease in systemic platelet count). This suggests that low platelet adhesion is not a sufficient criterion of in vivo platelet compatibility. Reduced thrombogenicity is characterized by both low platelet adhesion and low platelet activation. Leukocyte activation (and complement activation) may also be key components in thrombogenicity (see subsequent text); with further research, these factors too may become crucial parameters that define the thrombogenicity of the surface.
circulation (e.g., shorter platelet lifespan, decrease in systemic platelet count). This suggests that low platelet adhesion is not a sufficient criterion of in vivo platelet compatibility. Reduced thrombogenicity is characterized by both low platelet adhesion and low platelet activation. Leukocyte activation (and complement activation) may also be key components in thrombogenicity (see subsequent text); with further research, these factors too may become crucial parameters that define the thrombogenicity of the surface.
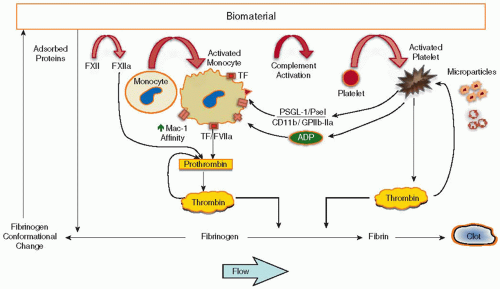 FIGURE 96.1 On or in the adsorbed protein layer, coagulation factors change their conformation or are converted into active enzymes and result in the generation of thrombin and fibrin. In parallel, platelets adhere to the surface (or adsorbed protein), become activated, change their shape, and form platelet aggregates. Both processes are linked most notably through thrombin generated on the activated platelet surface. Complement is activated on many biomaterials within the protein adsorbate and leukocytes are activated to express TF and other procoagulant activities. Flow (shear rate) is a crucial factor affecting both transport coefficients (from bulk to the surface and reverse) and the activation of leukocytes and platelets. (From Gaamangwe T, Gorbet MB, unpublished work, with permission.) |
Table 96.1 Hallmarks of nonthrombogenicity | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||
MECHANISTIC DETAILS
The failure to create nonthrombogenic biomaterials is directly attributable to our limited understanding of the complexity of
biomaterial-associated thrombosis. The variety of mechanisms that are involved are summarized here.
biomaterial-associated thrombosis. The variety of mechanisms that are involved are summarized here.
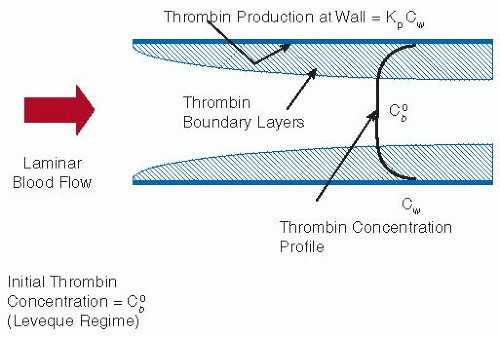 FIGURE 96.2 Model illustrating thrombin production at the surface of a tube as a balance between autoaccelerative production and mass transfer away from the wall. (From Rollasson G, Sefton MV. Measurement of the rate of thrombin production in human plasma in contact with different materials. J Biomed Mater Res 1992;26(5):675-693, with permission.) |
Coagulation
In the past, the discussion of coagulation at the biomaterialblood interface has focused on the intrinsic coagulation system and the effects of the adsorbed protein layer. Protein adsorption is certainly the first event in blood-material interactions, and adsorption of the contact-phase proteins may result in activation of the intrinsic cascade. Recent studies, however, demonstrate that tissue factor (TF) and the extrinsic coagulation system are also involved.
Early studies that were focused on protein adsorption on glass or biomaterial surfaces showed how fibrinogen is replaced over time by high molecular weight kininogen (the Vroman effect), suggesting a possible role of the intrinsic pathway in material thrombosis.20 On the other hand, factor XII (FXII) adsorption has been observed in moderate amounts on materials used in vascular grafts21 and hemodialysers22 but has not been found in its activated form.23 In some instances, in vitro activation of FXII and kallikrein has been reported with biomaterials;24,25 whether this activation results in any significant coagulation is not known. Other studies have actually shown that only minute amounts of thrombin or thrombin-antithrombin complex (TAT) are generated when biomaterials are incubated with undiluted plasma alone.26,27,28 Furthermore, the levels of adsorbed kallikrein and FXII on biomaterials do not correlate with TAT formation,26 suggesting that the contact-phase proteins, by themselves, play little role in the activation of coagulation. In fact, two studies27,29 suggested that the presence of leukocytes and/or erythrocytes is required for activation of the coagulation cascade. TAT formation on polyvinyl chloride (PVC)27 and titanium29 was found to be negligible in both plasma and platelet-rich plasma, whereas significant levels of TAT were observed in whole blood. In vivo, a small increase of FXIIa is observed during CPB,30,31 but it appears to be in response to the surgical intervention. The establishment of extracorporeal circulation (i.e., exposure to the biomaterials of the circuit) does not further increase FXIIa levels.31 Furthermore, no significant correlation has been observed between FXIIa and thrombin generation.31 In vivo results with hemodialysis also failed to show any significant increase of FXIIa.32 That a patient with a severe FXII deficiency showed levels of thrombin generation during CPB similar to normal patients33 casts further doubt on the role of FXII in the initiation of coagulation with biomaterials. Taken together, these studies do not support the view that activation of the contact-phase proteins is important in the activation of coagulation by biomaterials. Rather a TF-dependent pathway of initiation for coagulation may be more important, and this is discussed in the context of leukocyte activation in subsequent text.
Platelets
Platelet activation and adhesion is known to occur during CPB and hemodialysis, as well as in vascular grafts, stents, and catheters.2,34 The thrombotic complications associated with cardiovascular devices are clearly linked to their ability to activate platelets. Adherent platelets35 and circulating platelet microparticles (PMPs) generated by material contact36,37 have been shown to be procoagulant in nature. Association between platelets and leukocytes through P-selectin occurs in the presence of cardiovascular devices,38 and such associations may directly or indirectly contribute to thrombin generation (by monocyte TF) or may participate in the removal of platelets from the circulation because several platelets can be bound to each neutrophil or monocyte. Generation of PMPs is also induced by biomaterials; when compared to balloon angioplasty alone, higher levels of PMPs have been observed within 15 minutes following coronary stenting.39
Following contact with the layer of adsorbed proteins on the artificial surface, platelets will either adhere or “bounce off,”40 most likely depending on their state of activation and the ligands present at the interface.41 Many investigators have studied the influence of surface characteristics of biomaterials (i.e., hydrophilicity/hydrophobicity, surface free energy, chemistry, charge, and roughness) on platelet adhesion in vitro, but the results have been contradictory and adsorbed proteins remain the crucial factor in platelet adhesion. Few platelets are found on biomaterials that preferentially adsorb albumin. Preadsorbed fibrinogen, fibronectin, vitronectin, and von Willebrand factor (vWF) support platelet adhesion in vitro on biomaterials. However, initial platelet adhesion on surfaces in vitro is mediated mainly by fibrinogen and glycoprotein (GP) IIb/IIIa.42 No initial platelet activation is required, and binding of GP IIb/IIIa occurs through the aminoterminal of the γchain in fibrinogen.43 A very small amount of fibrinogen appears to be necessary to support platelet adhesion (<10 ng/cm2),42 suggesting that a biomaterial has to be extremely resistant to protein adsorption to prevent adhesion. Such biomaterials can be characterized as “ultralow” fibrinogen-adsorbing surfaces, whereby <5 ng/cm2 of adsorbed fibrinogen is required. Despite surface modification, such a design criterion appears difficult to meet, especially under in vitro conditions mimicking the in vivo environment (i.e., 100% plasma).44 The role of protein in platelet adhesion on biomaterial surfaces is also highly dependent on shear rate; at low shear, platelet adhesion was reported to be dependent only on fibrinogen while at high shear both fibrinogen and vWF were reported to affect platelet adhesion.44 Interaction with vWF
(by GP Ib/IX/V) and fibronectin (by GP IIb/IIIa) is believed to improve platelet adhesion once adherent platelets have been activated.45,46,47 Adherent platelets on biomaterials become activated and undergo morphologic changes; the extent of platelet spreading or flattening is directly related to the increase in activation.48 The state of activation of adherent platelets is also dependent on the biomaterial.49,50 Activation of adherent platelets may result in degranulation, with the release of activation products such as β-thromboglobulin, thromboxane B2, adenosine 5′-diphosphate (ADP) or serotonin (which can activate bulk platelets), and neutrophil-activating protein-2. In addition, adherent platelets express P-selectin (translocated to the surface during release from the α-granulae), which promotes the binding of monocytes and neutrophils. The activated adherent platelet surface supports thrombin generation (i.e., prothrombinase and tenase formation) and can also shed procoagulant microparticles (MPs) from their plasma membrane.
(by GP Ib/IX/V) and fibronectin (by GP IIb/IIIa) is believed to improve platelet adhesion once adherent platelets have been activated.45,46,47 Adherent platelets on biomaterials become activated and undergo morphologic changes; the extent of platelet spreading or flattening is directly related to the increase in activation.48 The state of activation of adherent platelets is also dependent on the biomaterial.49,50 Activation of adherent platelets may result in degranulation, with the release of activation products such as β-thromboglobulin, thromboxane B2, adenosine 5′-diphosphate (ADP) or serotonin (which can activate bulk platelets), and neutrophil-activating protein-2. In addition, adherent platelets express P-selectin (translocated to the surface during release from the α-granulae), which promotes the binding of monocytes and neutrophils. The activated adherent platelet surface supports thrombin generation (i.e., prothrombinase and tenase formation) and can also shed procoagulant microparticles (MPs) from their plasma membrane.
The absence of significant platelet adhesion does not preclude platelet activation in the bulk as shown by the generation of platelet MPs with polyvinyl alcohol (PVA) hydrogel both in vitro51 and ex vivo in a canine shunt model.52 In a baboon arteriovenous (AV) shunt model, the rate of platelet consumption was directly linked to the water content of hydrogels,18 despite the absence of significant adhesion. All materials that were tested,53 not just hydrogels, were observed to activate platelets, resulting in their premature removal from the circulation.
It is not clear whether inhibition of platelet adhesion will reduce platelet consumption or inhibit the generation of platelet MPs. It appears that the effects of antiplatelet agents vary, depending on the surface characteristics as shown by Spijker et al.54 For example, abciximab is less effective in reducing platelet adhesion on hydrophilic surfaces than on hydrophobic surfaces. The strategies available to minimize platelet adhesion (e.g., polyethylene glycol immobilization) have not been sufficient to prevent platelet consumption.55 The mechanism of material-induced platelet activation is often presumed to be through the generation of thrombin due to activation of the intrinsic coagulation cascade or the release of ADP from damaged red blood cells or platelets. Even in the presence of heparin, small amounts of thrombin are generated and may activate platelets. However, the inability of thrombin and kallikrein inhibitors to reduce platelet activation suggests that platelet activation is at least mediated in part by other agonists.56 Classical pathway complement inhibitors can have some beneficial effects on platelets: pentamidine (12 mg/kg, daily, intra muscular) but neither abciximab nor low molecular weight heparin reduced PVA-induced platelet consumption in the canine AV shunt (unpublished study, Gemell CH & Sefton MV). A correlation between complement activation and thrombocytopenia has also been noted during dialysis.57,58
Complement activation can lead to platelet activation in many ways. Platelets possess a receptor for C1q that induces GP IIb/IIIa activation, P-selectin expression, and procoagulant activity.59 Insertion of the complement assembly C5b-9 in platelets has also been associated with increased platelet procoagulant activity.60
Leukocytes
Contact with biomaterials in vivo activates both neutrophils and monocytes. Indicators of leukocyte activation such as L-selectin shedding and CD11b upregulation on leukocytes have been widely observed following angioplasty,61 hemodialysis,62 and CPB63 (for review, see Asimakopoulos and Taylor64). Degranulation with the release of elastase and lactoferrin and the presence of cytokines, such as IL-1 and TNF-α, have been associated with extracorporeal circulation65 and further demonstrate leukocyte activation. Activation of the respiratory burst is also a common response to hemodialysis.66
Both in vitro67 and in vivo68 studies have found that material-induced leukocyte activation is dependent on the exposed surface area. Monocytes express TF after coming into contact with a biomaterial.67,69,70 Material-induced monocyte TF expression in vitro depends on complement activation and platelets.71 On the other hand, the removal of platelets or the blocking of GP IIb/IIIa by monoclonal antibody (7E3) has no effect on CD11b upregulation. Contact with materials such as PVA hydrogel72 or pellethane73 leads to a twofold upregulation of CD11b, typical of the degree of leukocyte activation induced by phorbol esters.
Material-induced leukocyte activation results in increased adhesion to artificial surfaces. Because the biomaterial is larger than a microorganism and cannot therefore be engulfed by leukocytes, adherent neutrophils and monocytes respond with a frustrated attempt at phagocytosis, whereby these cells release their array of potent oxygen metabolites, growth factors, cytokines, and proteolytic enzymes.74 The state of adherent leukocyte activation depends on the type of surface, the protein ligands present at the interface,75,76,77 and the presence of adherent platelets.71,78 Hydrophobic surfaces tend to promote leukocyte adhesion and activation, whereas less activation is observed in leukocytes adherent to hydrophilic surfaces.79 In vivo studies have demonstrated activated leukocytes adhering to stents,80 oxygenators,81 and hemodialysis membranes.82,83
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


