INTRODUCTION
SUMMARY
von Willebrand factor (VWF) is a central component of hemostasis, serving both as an adhesive link between platelets and the injured blood vessel wall and as a carrier for clotting factor VIII (FVIII). Abnormalities in VWF function result in von Willebrand disease (VWD), the most common inherited bleeding disorder in humans. The overall prevalence of VWD has been estimated to be as high as 1 percent of the general population, although the prevalence of clinically significant disease is probably closer to 1:1000. VWD is associated with either quantitative deficiency (type 1 and type 3) or qualitative abnormalities of VWF (type 2). The uncommon type 3 variant is the most severe form of VWD and is characterized by very low or undetectable levels of VWF, a severe bleeding diathesis, and a generally autosomal recessive pattern of inheritance. Type 1 VWD, the most common variant, is characterized by VWF that is normal in structure and function but decreased in quantity (in the range of 20 to 50 percent of normal). In type 2 VWD, the VWF is abnormal in structure and/or function. Type 2A VWD is associated with selective loss of the largest and most functionally active VWF multimers. Type 2A is further subdivided into group 1, as a result of mutations that interfere with biosynthesis and secretion, and group 2, in which the mutant VWF exhibits an increased sensitivity to proteolysis in plasma. Type 2B VWD is caused by mutations clustered within the VWF A1 domain, in a segment critical for binding to the platelet glycoprotein Ib receptor. These mutations produce a “gain of function” resulting in spontaneous VWF binding to platelets and clearance of the resulting platelet complexes, leading to thrombocytopenia and loss of the most active (large) VWF multimers. Type 2N VWD is characterized by mutations within the FVIII binding domain of VWF, leading to disproportionately decreased factor VIII and a disorder resembling mild to moderate hemophilia A, but with autosomal rather than X-linked inheritance. Type 1 VWD can often be effectively managed by treatment with DDAVP (1-deamino-8-D-arginine vasopressin, desmopressin), which produces a two- to threefold increase in plasma VWF level due to release from endothelial storage sites in the vessel wall. Response to DDAVP is generally poor in type 3 and some type 2 VWD variants. These disorders often require treatment with factor replacement in the form of VWF/FVIII concentrates containing large quantities of intact VWF multimers.
Acronyms and Abbreviations
ADAMTS13, a disintegrin and metalloprotease with thrombospondin type 1 motifs; aPTT, activated partial thromboplastin time; DDAVP, 1-desamino-8-D-arginine vasopressin or desmopressin; ER, endoplasmic reticulum; GP, glycoprotein; HHT, hereditary hemorrhagic telangiectasia; PCR, polymerase chain reaction; RIPA, ristocetin-induced platelet aggregation; VWD, von Willebrand disease; VWF, von Willebrand factor.
DEFINITION AND HISTORY
In 1926, Eric von Willebrand described a bleeding disorder in 24 of 66 members of a family from the Åland Islands.1 Both sexes were afflicted, and the bleeding time was prolonged despite normal platelet counts and normal clot retraction. von Willebrand distinguished this condition from the other hemostatic diseases known at the time and recognized its genetic basis, calling the disorder “hereditary pseudohemophilia,” but incorrectly characterizing the inheritance as X-linked dominant. von Willebrand’s confusion about the inheritance pattern was probably the result of, at least in part, the greater recognition of bleeding symptoms in women because of the hemostatic stresses of menstruation and parturition. The proband in the original family, Hjördis, was 5 years old at the time of von Willebrand’s initial evaluation and ultimately died at age 13 years during her fourth menstrual cycle. Four of Hjördis’ sisters died between the ages of 2 and 4 years, and deaths in the family were also noted during childbirth.
An apparently similar disorder was independently reported in the United States by Minot and others in 1928. The original family in the Åland Islands was reexamined by von Willebrand and Jürgens in 1933, leading to the conclusion that the defect in this disorder was caused by an impairment of platelet function. It was not until 1953 that Alexander and Goldstein demonstrated reduced levels of coagulation factor VIII (FVIII) in von Willebrand disease (VWD) patients, along with prolonged bleeding time. This observation was confirmed by others, including studies of the original von Willebrand pedigree by Nilsson and coworkers. In the late 1950s, Nilsson and coworkers demonstrated that a fraction of plasma referred to as “I-O” could correct the FVIII deficiency and normalize the bleeding time, indicating that the defect in VWD was a result of the deficiency of a plasma factor rather than an intrinsic platelet abnormality. Infusion of fraction I-O promptly increased the FVIII level in a hemophilic patient, while in VWD the FVIII level rose gradually, peaking at 5 to 8 hours. Fraction I-O prepared from a hemophilia A patient was also shown to correct the defect in VWD, demonstrating that these disorders were caused by deficiencies of distinct plasma factors (reviewed in Refs. 2 and 3).
It was not until 1971 that Zimmerman, Ratnoff, and Powell prepared the first antibodies against what was thought to be a highly purified form of FVIII.4 This FVIII-related antigen was found to be normal in hemophilia A patients but decreased in VWD. This puzzle was finally resolved with the demonstration that von Willebrand factor (VWF) and FVIII are closely associated, with more than 98 percent of the mass of the complex made up of VWF (see section The Function of von Willebrand Factor below). Thus, antibodies raised against this complex predominantly recognize VWF. The first direct assay of VWF function was based on the observation that the antibiotic ristocetin induced thrombocytopenia and the demonstration by Howard and Firkin5 that ristocetin-induced platelet aggregation (RIPA) was absent in some VWD patients. Weiss and coworkers6 used this observation to develop a quantitative assay for VWF function that remains a mainstay of laboratory evaluation for VWD to this day. In 1973, several groups succeeded in dissociating VWF from FVIII procoagulant activity.7,8
Final proof that VWF and FVIII are independent proteins encoded by distinct genes came with the complementary DNA (cDNA) cloning of the two molecules in 1984 and 1985.9,10,11,12,13,14 These discoveries also marked the beginning of the molecular genetic era for the study of VWF and FVIII, leading to the identification of gene mutations in many patients with hemophilia and VWD, as well as considerable insight into the structure and function of these related proteins.
Table 126–1 summarizes the current nomenclature and terminology for FVIII and VWF. VWD is a heterogeneous disorder with more than 20 variants described. The previous complex and confusing classification has been consolidated and simplified into six distinct types,15 as summarized in Table 126–2. Type 3 VWD is associated with very low or undetectable levels of VWF and severe bleeding. Type 1 VWD is characterized by concordant reductions in FVIII activity, VWF antigen, and ristocetin cofactor activity, generally to the range of 20 to 30 percent of normal, but sometimes up to 50 percent of normal, in association with a normal VWF multimer distribution. Type 2 VWD is heterogeneous and further divided into four subtypes (2A, 2B, 2M, and 2N) by the nature of the VWF qualitative dysfunction. Type 2A VWD results from abnormal VWF secretion or proteolysis and is characterized by a disproportionately low level of ristocetin cofactor activity relative to VWF antigen and absence of large and intermediate-sized multimers. Type 2B VWD results from an abnormal VWF molecule with increased affinity for platelet glycoprotein Ib (GPIb), and can also be associated with reduced high-molecular-weight VWF multimers and thrombocytopenia. Functional abnormalities in VWF can also result in defective interactions with platelets, as in type 2M VWD, or decreased FVIII binding to VWF, designated type 2N VWD and characterized by mild to moderate FVIII deficiency. Many other subtypes have been reported, including platelet-type (pseudo-) VWD, which is actually an intrinsic platelet disorder caused by mutations in GPIb (Chap. 120). Finally, acquired forms of VWD also occur, such as in patients with antibodies to VWF or thrombocytosis secondary to myeloproliferative neoplasms, resulting in accelerated loss of circulating VWF.
| Factor VIII |
| Antihemophilic factor, the protein that is reduced in plasma of patients with classic hemophilia A and most von Willebrand disease (VWD) and is measured in standard coagulation assays |
| Factor VIII activity (factor VIII:C) |
| The coagulant property of the factor VIII protein (this term is sometimes used interchangeably with factor VIII) |
| Factor VIII antigen (VIII:Ag) |
| The antigenic determinant(s) on factor VIII measured by immunoassays, which may employ polyclonal or monoclonal antibodies |
| von Willebrand factor (VWF) |
| The large multimeric glycoprotein that is necessary for normal platelet adhesion, a normal bleeding time, and stabilizing factor VIII |
| von Willebrand factor antigen (VWF:Ag) |
| The antigenic determinant(s) on VWF measured by immunoassays, which may employ polyclonal or monoclonal antibodies; inaccurate designations of historical interest only include factor VIII-related antigen (VIIIR:Ag), factor VIII antigen, AHF antigen, and AHF-like antigen |
| Ristocetin cofactor activity (VWF:RCo) |
| The property of VWF that supports ristocetin-induced agglutination of washed or fixed normal platelets |
| von Willebrand factor collagen-binding activity (VWF:CB) |
| The property of VWF that supports binding to collagen, measured by enzyme-linked immunosorbent assay (ELISA) |
| Type | Molecular Characteristics | Inheritance | Frequency | Factor VIII Activity | VWF Antigen | Ristocetin Cofactor Activity | RIPA | Plasma VWF Multimer Structure |
|---|---|---|---|---|---|---|---|---|
| Type 1 | Partial quantitative VWF deficiency | Autosomal dominant, incomplete penetrance | 1–30:1000; most common VWD variant (>70% of VWD) | Decreased | Decreased | Decreased | Decreased or normal | Normal distribution (mutant subunits permitted) |
| Type 3 | Severe quantitative reduction or absence of VWF | Autosomal recessive (or codominant) | 1–5:1,000,000 | Markedly decreased | Very low or absent | Very low or absent | Absent | Usually absent |
| Type 2A | Qualitative VWF defect; loss of large VWF multimers, decreased VWF-dependent platelet adhesion | Usually autosomal dominant | ~10–15% of clinically significant VWD | Decreased to normal | Usually low | Markedly decreased | Decreased | Largest and intermediate multimers absent |
| Type 2B | Qualitative VWF defect; increased VWF–platelet interaction (GPIb) | Autosomal dominant | Uncommon variant (<5% of clinical VWD) | Decreased to normal | Usually low | Decreased to normal | Increased to low concentrations of ristocetin | Largest multimers reduced/absent |
| Type 2M | Qualitative VWF defect; decreased VWF-platelet interaction, no loss of large VWF multimers | Usually autosomal dominant | Rare (case reports) | Variably decreased | Variably decreased | Decreased | Variably decreased | Normal and occasionally ultralarge forms |
| Type 2N | Qualitative VWF defect; decreased VWF-factor VIII binding capacity | Autosomal recessive | Uncommon; heterozygotes may be prevalent in some populations | Decreased | Normal | Normal | Normal | Normal |
| Platelet-type (pseudo-) | Platelet defect; decreased platelet-VWF interactions | Autosomal dominant | Rare | Decreased to normal | Decreased to normal | Decreased | Increased to low concentrations of ristocetin | Largest multimers absent |
ETIOLOGY AND PATHOGENESIS
VWF is synthesized exclusively in endothelial cells and megakaryocytes. The VWF monomer is assembled into higher-order multimers, a structure required for optimal adhesive function, and performs two major functions in hemostasis. First, VWF serves as the initial critical bridge between circulating platelets and the injured blood vessel wall, accounting for the apparent defect in platelet function and prolonged bleeding times historically observed in VWD patients. Second, VWF serves as the carrier in plasma for FVIII, ensuring its stability and localizing it to the initial platelet plug for participation in thrombin generation and fibrin clot formation (Chap. 113). This tight, noncovalent interaction between VWF and FVIII accounts for the copurification of these two molecules and the resulting initial confusion as to the origin of hemophilia and VWD. FVIII is encoded by the F8 gene on the X chromosome (Chaps. 113 and 123), while VWF is encoded by the VWF gene on human chromosome 12.
The VWF cDNA was initially cloned from endothelial cells,11,12,13,14 and the corresponding gene mapped to the short arm of chromosome 12 (12p13.3).11 The VWF mRNA is approximately 9 kb in length, encoding a primary translation product of 2813 amino acid residues with an estimated Mr of 310,000. Comparison of the primary peptide sequence obtained from plasma VWF16 with the VWF cDNA sequence established the prepropolypeptide nature of VWF.17 Prepropolypeptide VWF is composed of a 22-amino-acid signal peptide, a 741-amino-acid precursor polypeptide known as the VWF propeptide (VWFpp), and the mature subunit.11,17,18,19,20 Cleavage of the 741-amino-acid propeptide from the amino terminus produces the mature VWF monomer subunit of 2050 amino acids (Fig. 126–1).
Figure 126–1.
Schematic of the human VWF gene, mRNA, and protein. The VWF gene and VWFP1 pseudogene are depicted at the top, with boxes representing exons and the solid black line representing introns. Schematics of the VWF mRNA encoding the full prepro-VWF subunit are depicted in the middle as bar and lettered boxes. The upper schematic denotes commonly annotated regions of internally repeated sequences; the lower schematic illustrates the multiple repeating motifs of VWF. The locations of signal peptide (sp) and VWF propeptide (VWFpp) cleavage sites are indicated by arrowheads. The approximate localizations for known VWF functional domains within the mature VWF subunit are indicated at the bottom. Numbers underneath the domains refer to amino acid residues numbered from the ATG start site; numbers in parentheses indicate the amino acid residue position in the mature VWF subunit. aa, Amino acids; chr, chromosome. (Adapted with permission from Ginsburg D, Bowie EJW. Molecular genetics of von Willebrand disease. Blood 79(10):2507–2519, 1992.)
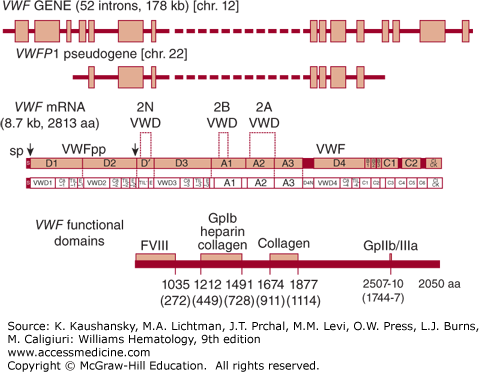
Analysis of the VWF sequence identifies four distinct types of repeated domains: three A domains, three B domains, two C domains, and four D domains,18,21 within which appear additional repeating motifs (schematic in Ref. 22). The first pair of D domains is tandemly arranged in the VWFpp, followed by a partial and full D domain at the N terminus of the mature subunit. The final complete D domain is separated by a segment of more than 600 amino acids containing the triplicated A domains. The repeated domain structure of VWF suggests that the gene may have evolved via a complex series of partial duplications, although exon structure is not highly conserved between homologous domains.
Comparison of the VWF amino acid sequence to other proteins identifies a superfamily of related proteins that share sequence similarity with the VWF A domains.23 The common theme among these potentially evolutionarily related genes is a role in extracellular matrix or adhesive function. Consistent with this notion, VWF functional domains for binding to the platelet receptor GPIb and specific ligands within the extracellular matrix have been localized to the VWF A repeats. A potential relationship between the VWF C domains and portions of thrombospondin and procollagen has also been proposed.24
The VWF gene spans 178 kb and is divided into 52 exons.25 Exons range in size from 40 bases to 1.4 kb (exon 28). Exon 28 is unusually large, encoding the entire Al and A2 domains and containing most of the known type 2A and all of the type 2B VWD mutations. The concentration of these defects within one exon has facilitated the identification of human mutations responsible for these VWD variants (see “Molecular Genetics of von Willebrand Disease,” below). A partial, nonfunctional duplication of the VWF gene, termed a pseudogene, is located on human chromosome 22.26 The pseudogene, known as VWFP1, duplicates the middle portion of the VWF structural gene, spanning exons 23 to 34 and the intervening noncoding sequences. VWFP1 is approximately 97 percent identical in sequence to the authentic VWF gene, indicating that it is of fairly recent evolutionary origin.27 Gene conversion involving the VWFP1 pseudogene, possibly through recombination with the large homologous exon 28 sequence, has been proposed as a mechanism for introducing mutations into the VWF gene.28,29,30,31
VWF is synthesized exclusively in megakaryocytes and endothelial cells and, as a result, has frequently been used as a specific histochemical marker to identify cells of endothelial cell origin. Although generally assumed to mark all endothelial cells, VWF is expressed at widely varying levels among endothelial cells, depending on the size and location of the associated blood vessel.32,33 A careful survey in the mouse identified wide differences in the level of VWF mRNA, with 5 to 50 times higher concentrations in the lung and brain, particularly in small vessels, than in comparable vessels in the liver and kidney. In general, the higher levels of VWF mRNA and antigen were found in the endothelial cells of large vessels rather than in microvasculature, and in venous rather than arterial endothelial cells.33
Specific DNA sequences within or near the proximal promoter of the VWF gene appear to be required for endothelial-specific gene expression,34,35,36,37,38,39 although it is likely that additional important regulatory elements exist outside of this region, some of which may lie at a great distance.40 VWF is expressed in most, but not all, endothelial cells,41 and this vascular-bed specific gene expression program is likely a result of the concerted action of multiple regulatory elements. Endothelial VWF gene expression also appears to be upregulated by exposure to shear stress. The length of a polymorphic GT repeat in the proximal VWF promoter correlates with the magnitude of this response, and several other more distal DNA sequences are predicted to be involved in a shear stress response.42 However, this GT repeat does not appear to influence circulating VWF levels.43
The processing steps involved in the biosynthesis of VWF are similar in megakaryocytes and endothelial cells (reviewed in Refs. 46 and 47). VWF is first synthesized as a large precursor monomer polypeptide, depicted schematically in Fig. 126–1. VWF is unusually rich in cysteine, which accounts for 8.3 percent of its amino acid content. All cysteines in the mature VWF molecule are thought to be involved in disulfide bonds,48 although these bonds may be exposed in circulating mature VWF by shear stress.49 Pro-VWF monomers are assembled into dimers through disulfide bonds at both C termini, and only dimers are exported from the endoplasmic reticulum (ER).48,50,51
Glycosylation begins in the ER, with 12 potential N-linked glycosylation sites present on the mature subunit and three on the propeptide. Extensive additional posttranslational modification of VWF occurs in the Golgi apparatus, including the addition of multiple O-linked carbohydrate structures, sulfation, and multimerization through the formation of disulfide bonds at the N termini of adjacent dimers. It is unusual for a protein to undergo extensive disulfide bond formation at this late stage, and this process appears to be catalyzed by disulfide isomerase activity present within the VWFpp.52 Mutations at either of two specific cysteines within the propeptide that are thought to be critical for disulfide isomerase activity, or a shift in the spacing between them, results in loss of multimer formation.52 An intermediate species with disulfide bonds between the propeptide and VWF D’D3 domain appears briefly in either the late ER or early Golgi,53 which may position these domains for subsequent multimerization. The multimerization process appears to require the slightly acidic environment of the distal Golgi.54 The VWFpp self-associates and may also serve to align VWF subunits for multimer assembly.55 However, the propeptide facilitates multimer assembly even when coexpressed as a separate molecule from the mature VWF monomer.56,57
Propeptide cleavage occurs late in VWF synthesis or just prior to secretion. Cleavage occurs adjacent to two basic amino acids, Lys-Arg at positions –2 and –1. An Arg at position –4 is also required for recognition by the intracellular protease responsible for propeptide cleavage.58 Multimerization and propeptide cleavage are not linked to each other. The multimers secreted by cultured endothelial cells contain both pro-VWF and mature subunits,59,60 and recombinant VWF with a point mutation inhibiting propeptide cleavage is still assembled into normal multimer structures.61 Although propeptide cleavage appears to occur primarily intracellularly, cleavage may also occur after secretion.
VWF is stored in tubular structures within the α-granules of platelets and within the Weibel-Palade bodies in endothelial cells62,63 (reviewed in Ref. 64). These large VWF structures form by tubular packing of the VWF N-terminal domains within the secretory granules.65 Weibel-Palade bodies are derived from the Golgi apparatus and are found in most endothelial cells, although the number varies considerably between endothelial cell beds. It has been shown that VWF and FVIII colocalize in storage granules. Although VWF is not required to traffic FVIII to platelets,66 VWF appears to play a role in trafficking FVIII to Weibel-Palade bodies in endothelial cells.67,68 Weibel-Palade bodies mature as they move to the periphery of the cell in an ordered process dependent on Rab proteins and Rab effector proteins, which act as chaperones and organizers of the various stages of Weibel-Palade body maturity and subsequent exocytosis (reviewed in Ref. 69). The transmembrane glycoprotein P-selectin is also found in the membranes of both the α-granule and the Weibel-Palade body.70 The VWF D’D3 domain has been shown in vitro to associate with P-selectin and to be necessary for the recruitment of P-selectin to Weibel-Palade bodies.71 There appears to be heterogeneity within Weibel-Palade body populations both in relative content of VWF and P-selectin and in response to regulated secretion by different stimuli.72 In addition to VWF and P-selectin, the Weibel-Palade body also contains tissue-type plasminogen activator (t-PA), a thrombolytic secreted protein that also may be released distinctly from VWF,73 and several other proteins that are known to participate in inflammation or angiogenesis (for a complete list of Weibel-Palade contents see Ref. 74).
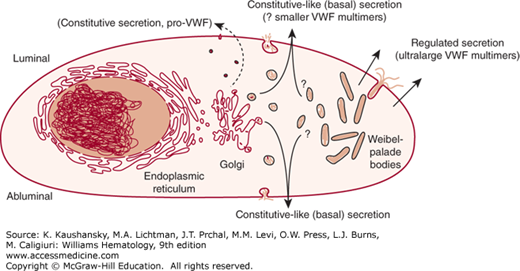
VWF is secreted from endothelial cells continuously via constitutive and constitutive-like (or basal) pathways and upon stimulated release of storage granules via a classic regulated pathway.46,75 Regulated secretion of VWF from its storage site in the Weibel-Palade body is triggered by a number of secretagogues, including thrombin,76 fibrin,77 histamine,78 the C5b-9 complement complex,79 and several inflammatory cytokines.80 Recent in vitro data suggests that there may also be suppression of regulated VWF secretion by statins.81,82 The secretagogue desmopressin acetate (DDAVP), a vasopressin analogue, is used clinically for its capacity to cause a marked release of VWF and FVIII in vivo by acting through type 2 vasopressin receptors to induce secretion from the Weibel-Palade bodies in endothelial cells.83 Constitutive-like secretion of VWF occurs evenly at the luminal and abluminal surface, while regulated secretion from the Weibel-Palade body is highly polarized in the luminal direction (Fig. 126–2).75,84 While constitutively secreted multimers are of relatively small size, the multimers stored within the Weibel-Palade body are the largest, most biologically potent form.85,86 The VWF stored in platelet α-granules is also enriched for large multimers.87 The N-terminal D domains appear to be required for VWF storage, with deletion of any of the individual domains resulting in constitutive secretion.88,89 It also appears that cleavage of the VWFpp is required for efficient formation of storage granules.90
Figure 126–2.
Schematic of von Willebrand factor (VWF) processing and secretion from endothelial cells. VWF dimers are formed in the endoplasmic reticulum, where VWF begins to be glycosylated. VWF dimers are transported to the Golgi, where the VWF undergoes further glycosylation and sulfation. Multimerization begins in the Golgi and continues within the secretory granules (Weibel-Palade bodies). A small amount of immature VWF is released constitutively (i.e., without regulation or storage) from endothelial cells as dimers or very small multimers. VWF is also released continuously from both the luminal and abluminal endothelial cell surfaces by constitutive-like (or basal) secretion. This VWF has been processed in the Golgi and may be transiently stored in an intermediate secretory granule or Weibel-Palade bodies. Mature VWF is packaged and stored as ultralarge multimers in Weibel-Palade bodies. This ultralarge VWF is released from the luminal surface of stimulated endothelial cells by regulated secretion. Once in circulation, VWF multimers undergo proteolysis by ADAMTS13 (a disintegrin and metalloprotease with a thrombospondin type 1 motif member 13) under moderate to high shear conditions. (Adapted with permission from Johnsen J, Lopez JA. VWF secretion: What’s in a name? Blood 112(4):926–927, 2008.)
The concentration of VWF in plasma is approximately 10 mcg/mL, with approximately 15 percent of circulating VWF localized to the platelet compartment.91 Marrow transplants between normal and VWD pigs demonstrate that platelet VWF is derived entirely from synthesis within marrow megakaryocytes and does not contribute to the normal plasma VWF pool.92,93,94 These studies also demonstrate that both the plasma and the platelet VWF pools are required for full hemostasis, although the plasma pool appears to be more critical.
Plasma VWF is further processed in the circulation through cleavage by a specific protease, ADAMTS13 (a disintegrin and metalloprotease with thrombospondin type 1 motifs–13), resulting in reduction in the size of the largest multimers (reviewed in Ref. 95). After regulated secretion in vitro, ultralarge VWF multimers may anchor to the endothelial cell surface via P-selectin96,97 resulting in shear stress and VWF cleavage by ADAMTS13. The major proteolytic cleavage site maps to the peptide bond between Tyr1605 and Met1606 in the VWF A2 domain,98 and recombinant VWF missing the A2 domain is resistant to proteolysis.99 VWF carrying a subgroup of type 2A VWD mutations exhibits increased susceptibility to cleavage by this protease,100 and this is the proposed mechanism for the selective loss of large VWF multimers in this group of patients (see “Molecular Genetics of von Willebrand Disease,” below). Increased VWF susceptibility to proteolysis by ADAMTS13 has also been described in a subset of type 1 VWD patients, but the clinical significance of this is unclear as increased proteolysis appears to only occur under certain conditions.101 Decreased ADAMTS13 activity, either because of congenital deficiency or acquired inhibitors, plays a central role in the pathophysiology of thrombotic thrombocytopenic purpura (Chap. 132).
VWF is a large multivalent adhesive protein that plays an important role in platelet attachment to subendothelial surfaces, platelet aggregation at sites of vessel injury, and stabilization of coagulation FVIII in the circulation. Not only is the interaction of VWF and FVIII important for the protection of FVIII from inactivation or degradation, FVIII bound to VWF may localize to cells and/or sites where it can more readily participate in the promotion of blood coagulation and/or thrombus formation.
VWF is required for the adhesion of platelets to the subendothelium, particularly at moderate to high shear force. VWF performs this bridging function by binding to two platelet receptors, GPIb and GPIIb/IIIa, as well as to specific ligands within the exposed subendothelium at sites of vascular injury (reviewed in Ref. 103). Binding of VWF to its platelet receptors generally does not occur in the circulation under normal conditions. However, the interaction of VWF with exposed ligands in the vessel wall, combined with high shear stress conditions, facilitates VWF binding to platelet GPIb and subsequent platelet adhesion and activation. Activation of platelets leads to the exposure of the GPIIb/IIIa complex, an integrin receptor that can bind to fibrinogen, VWF, and other ligands, to form the platelet–platelet bridges required for thrombus propagation. Platelet adhesion to VWF immobilized at a site of injury appears to be a two-step process, with the initial tethering of the rapidly moving platelet dependent on the VWF–GPIb interaction and subsequent firm adhesion occurring through GPIIb/IIIa after platelet activation.104 VWF may also play a role in inflammation by directly interacting with leukocytes,105 but the clinical significance of this observation is not clear.
VWF binds to the vessel wall at sites of vascular endothelial injury (reviewed in Ref. 106). VWF binds to several different types of collagens, including types I through VI. Distinct binding domains for the fibrillar collagens, types I and III, have been localized to specific segments within the VWF A1 and A3 repeats (see Fig. 126–1),107,108 and a potential third domain has been identified in the VWFpp.109 Studies of recombinant VWF suggest that the A3 collagen-binding domain may be the most important.110,111 VWF has also been shown to bind to the nonfibrillar collagen type VI, which is resistant to collagenase112 and colocalizes with VWF in the subendothelium.113 Type VI collagen supports the binding of VWF under high shear through cooperative interactions between binding domains within the VWF A1 and A3 repeat.114 Although VWF binding has also been demonstrated to a number of other potential components of the subendothelium, including glycosaminoglycans,115,116 sulfatides,117 and VWF itself,118 the biologic significance of these interactions remains to be demonstrated.
VWF interacts with platelets both to mediate platelet aggregation and platelet localization to sites of vascular injury (reviewed in Ref. 106). Circulating VWF does not spontaneously interact with platelets, but once bound to an injured vessel wall VWF is subjected to higher shear stresses and a platelet-binding site in the VWF A1 domain is uncovered. VWF interacts with a receptor complex on the surface of platelets composed of the disulfide-linked GPIbα and GPIbβ chains noncovalently associated with GPIX and GPV. The binding site for VWF is within a 293-amino-acid segment at the N-terminus of GPIb and requires sulfation of several key tyrosine residues for optimal binding.119 The GPIb binding domain within VWF lies within the A1 segment, within the disulfide loop formed between the cysteine residues at 1272 (amino acid 509 in mature VWF) and 1458 (695) (see Fig. 126–1).120,121 GPIb binding to the A1 domain enhances proteolysis of recombinant VWF fragments by ADAMTS13 and suggests a feedback mechanism for limiting thrombus propagation in vivo.122 Scanning mutagenesis studies of recombinant VWF characterized a number of amino acid residues within the VWF A1 domain that are critical for binding to GPIb and for interaction with botrocetin.123 Several mutations were also identified that increase platelet binding, an effect similar to that of mutations associated with type 2B VWD (see “Molecular Genetics of von Willebrand Disease,” below). These natural and synthetic mutations cluster in a small area on the surface of the VWF A1 domain structure, as revealed by x-ray crystallographic studies.124 The complexity of the VWF A1–GPIb interaction is evidenced by the ability of gain-of-function and loss-of-function VWF A1 mutants to counterbalance each other in mice.125 The structure of the A1 domain closely resembles that of other previously studied A domains, including the VWF A3 domain.126,127,128 The structure of GPIb in complex with the VWF A1 domain provides insight into the structural basis for the gain of function mutations associated with type 2B VWD.129 The abundant plasma protein β2-glycoprotein I can bind the VWF A1 domain when VWF is structurally open to GPIbα binding. This may result in biologically relevant inhibition of the VWF-platelet interaction, as inhibitory anti–β2-glycoprotein I autoantibodies found in some patients with antiphospholipid antibody syndrome are associated with thrombosis.130
Ristocetin binds to both VWF and platelets, but the mechanism by which it enhances the VWF/GPIb interaction is still poorly understood.131,132 The snake venom botrocetin appears to induce GPIb binding through a different alteration in the VWF A1 domain and is also used to study this interaction.128 Heparin binds the VWF A1 domain within the loop formed by the disulfide bond formed between the residues Cys1272 and Cys1458,133 where it appears to competitively inhibit VWF binding to GPIb134,135 and enhance VWF proteolysis by ADAMTS13 in vitro.136 Although it has been suggested that this may account for hemorrhage not predicted by conventional heparin monitoring, the clinical significance of the VWF-heparin interaction is not clear.
The arg-gly-asp-ser (RGDS) sequence at amino acids 2507 to 2510 of the mature VWF subunit serves as the binding site within VWF for GPIIb/IIIa. The GPIIb/IIIa complex, also known as integrin αIIbβ3, is a member of the integrin family of cell surface receptors. GPIIb/IIIa undergoes a conformational change to a high-affinity ligand-binding state following platelet activation which, in addition to VWF, can bind a number of other adhesive proteins, including fibrinogen. Although VWF is present in blood at much lower concentrations than is fibrinogen, evidence suggests that VWF may be a critical ligand. VWF participates in platelet tethering and adhesion to fibrin under flow conditions,104,137 where the C1C2 domains of VWF are required for fibrin binding.138 An RGD sequence is also present in the VWFpp, although its functional significance is unknown.
The noncovalent interaction between FVIII and VWF is required for the stability of FVIII in the circulation, as is evident from the FVIII levels of less than 10 percent that are observed in most severe VWD patients. Although each VWF subunit appears to carry a binding site for FVIII, the stoichiometry for the VWF–FVIII complex found in normal plasma is approximately one to two FVIII molecules per 100 VWF monomers.139 FVIII bound to VWF is also protected from proteolytic degradation by activated protein C (reviewed in Ref. 140). FVIII also appears to increase the susceptibility of VWF to proteolysis by ADAMTS13 under shear conditions.141
The FVIII binding domain within VWF has been localized to the first 272 N-terminal amino acids of the mature subunit.142 In mice, expression of the VWF D’-D3 domains alone has been shown to be sufficient to stabilize FVIII levels.143 Antibody studies suggest a particularly critical role for amino acids 841 to 859.144,145 The mutations identified in patients with type 2N VWD, in which VWF binding to FVIII is specifically affected (see “Molecular Genetics of von Willebrand Disease,” below), are all clustered in this region, including the most common type 2N mutation Arg854Gln.146 The corresponding binding site for VWF on FVIII includes an acidic region at the N-terminus of the light chain (residues 1669 to 1689)147 and requires sulfation of Tyr1680 for optimal binding.148 Overlay of VWD 2N mutations with the crystal structure of the VWF A1–FVIII domains shows the 2N mutations clustered in a dynamic VWF TIL’ domain (shown in Fig. 126–3), implicating a need for flexibility in this domain for normal FVIII binding.149 Thrombin cleavage after Arg1689 in FVIII activates and releases FVIII from VWF. Thus, VWF may serve to efficiently deliver FVIII to the sites of clot formation, where it can complex with factor IXa on the platelet surface.
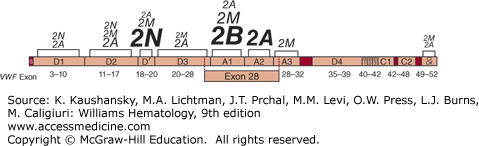
Figure 126–3.
von Willebrand disease (VWD) mutations. The von Willebrand factor domain locations of all reported mutations associated with type 2A, 2B, 2M, and 2N VWD. Lettering size represents the proportion of total mutations reported within the designated VWF domain for that subtype, with larger letters indicating more mutations. Shown below are the relative positions of the VWF gene exons. Type 1 and type 3 VWD associated mutations have been reported throughout the VWF gene. (Mutation data from Nichols WC and Ginsburg D: von Willebrand disease. Medicine (Baltimore) 76(1):1–20, 1997 and the VWD mutation database at www.vwf.group.shef.ac.uk.)
VWD is an extremely heterogeneous and complex disorder, with more than 20 distinct subtypes reported (referenced in Ref. 150). A large number of mutations within the VWF gene have been identified (see Fig. 126–3). However, because of both the genetic complexity of VWD and the practical considerations of VWF gene sequencing in most clinical settings, a VWF gene mutation is not required for the diagnosis of VWD.151 A list is maintained by a consortium of VWD investigators and can be accessed through the Internet at http://www.vwf.group.shef.ac.uk/. These findings form the basis for the simplified classification of VWD outlined in Table 126–2151 and used throughout this chapter. Types 1 and 3 VWD are defined as pure quantitative deficiencies of VWF that are either partial (type 1) or complete (type 3). Type 2 VWD is characterized by qualitative abnormalities of VWF structure and/or function. The quantity of VWF found in type 2 VWD may be normal, but it is usually mildly to moderately decreased (see Table 126–2).
The diagnosis of VWD, particularly type 1 VWD, can be confounded by the incomplete penetrance of the disease and the wide range of VWF levels in normal populations (see “Laboratory Features” below). Nonpathogenic variation can impact laboratory assays in vitro, as is the case with Asp1472His, which alters VWF-ristocetin interactions but has no impact on hemostasis in vivo.152 Ethnicity should also be considered. European ancestries were overrepresented in the studies which informed laboratory cutoffs, while African Americans exhibit generally higher VWF and FVIII levels. Additionally, there are numerous VWF gene “mutations” previously thought to be causative of VWD which are now known to be common in African Americans153,154 who have normal VWF and FVIII levels,153 including the Asp1472His variant.155
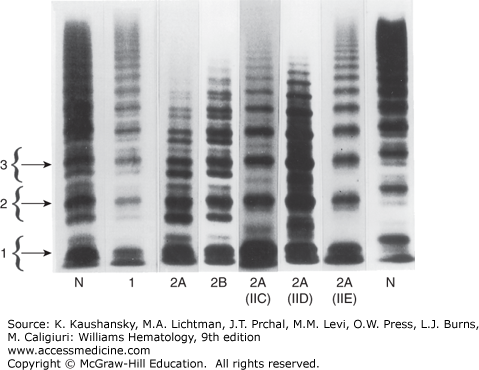
Type 1 VWD is the most common form, accounting for approximately 70 percent of patients with the disease. Type 1 VWD is generally autosomal dominant in inheritance and is associated with coordinate reductions in FVIII, ristocetin cofactor activity, and VWF antigen with maintenance of the full complement of multimers (Fig. 126–4). Subgroups within type 1 VWD have been proposed based on the relative levels of VWF present in the plasma and platelet pools,156,157,158,159 but with the exception in some circumstances for an accelerated VWF clearance phenotype (unofficially termed VWF type 1C160), subtype distinctions in type 1 VWD are not generally used in clinical practice.
Figure 126–4.
Agarose gel electrophoresis of plasma von Willebrand factor (VWF). VWF multimers from plasma of patients with various subtypes of von Willebrand disease (VWD) are shown. The brackets to the left encompass three individual multimer subunits, including the main band and its associate satellite bands. N indicates normal control lanes. Lanes 5 through 7 are rare variants of type 2A VWD. The former designations for these variants are indicated in parentheses below the lanes (IIC through E). (Adapted with permission from Zimmerman TS, Dent JA, Ruggeri ZM, Nannini LH: Subunit composition of plasma von Willebrand factor. Cleavage is present in normal individuals, increased in IIA and IIB von Willebrand disease, but minimal in variants with aberrant structure of individual oligomers (types IIC, IID, and IIE). J Clin Invest 1986 Mar;77(3):947–951.)
Type 1 VWF was previously assumed to simply represent the heterozygous form of type 3 VWD. However, in a large Canadian study, 48 percent of heterozygous carriers of type 3 VWD gene mutations carried a diagnosis of type 1 VWD, while the remainder were asymptomatic.161 Furthermore, in two large studies of type 1 VWD families, numerous putative VWF mutations were identified, but very few were predictive of VWF null alleles.31,162 Thus, some, but probably not all (see section Clinical Features below), type 1 VWD is the result of defects within the VWF gene. Studies of type 1 VWD mutations in patients, in vitro, and in animal models have characterized diverse mechanisms underlying type 1 VWD, including decreased VWF production,163 retention of VWF in the ER,164,165 impaired VWF secretion,165,166 and decreased VWF survival.163,166,167 Mutations that give rise to defective VWF subunits that interfere in a dominant negative way with the normal allele may be particularly likely to cause symptomatic VWD in the heterozygote.168 For example, mutations at several cysteine residues in the VWF D3 domain and in the VWFpp of patients with moderately severe type 1 VWD. VWF carrying one of these mutations is retained in the ER, where it is proposed to exert a dominant negative effect on VWF derived from the normal allele via heterodimerization and degradation.169,170
To date, most mutation studies and genetic linkage analysis of type 1 VWD have been consistent with defects within the VWF gene. Although no single mutation can explain the majority of type 1 VWD, common VWF founder mutations can occur within populations, such as Tyr1584Cys identified in 14 percent of Canadian type 1 VWD patients, and possibly a similar proportion of patients in Europe.162,171 The Tyr1584Cys mutation is associated with decreased VWF survival, likely a result of increased susceptibility to proteolysis by ADAMTS13.172,173,174,175 These large multicenter studies of type 1 VWD families found candidate VWF mutations in 63 to 70 percent of families with type 1 VWD, leaving 37 to 40 percent of type 1 VWD index cases without a putative mutation in VWF.162,171 Cases with VWF gene mutations tend to be more severe and highly heritable, while cases without an identifiable VWF mutation generally have higher VWF antigen (VWF:Ag) levels (>30 IU/dL).31 In a large, multicenter European study of 150 type 1 VWD families, about one-third of cases historically diagnosed to have type 1 VWD were found to have abnormal multimers, and of these nearly all (95 percent) had a putative VWF gene mutation and significantly lower VWF:Ag, VWF ristocetin cofactor activity (VWF:RCo), assay of FVIII activity (FVIII:C), and VWF collagen-binding assay (VWF:CB) levels. Conversely, index cases with normal multimers had higher laboratory VWF values and fewer identifiable VWF mutations (55 percent), suggesting that the pathogenic mechanism(s) underlying this cohort of “true” type 1 VWD patients is more genetically complex.162 Given the complex biosynthesis and processing of VWF, defects at a number of other loci could also be expected to result in quantitative VWF abnormalities (reviewed in Ref. 176). This concept is supported by families with type 1 VWD in which bleeding histories and low ristocetin cofactor activities do not always cosegregate with genetic markers at the VWF locus,31,177 while one or more genetic factors outside the VWF locus may be associated with the variation in bleeding severity observed within VWD pedigrees.178,179 It is interesting to note that a spontaneous mouse model of type 1 VWD exhibits up to 20-fold reductions in plasma VWF as a result of an unusual mutation in a glycosyltransferase gene, leading to aberrant posttranslational processing of VWF and accelerated clearance from plasma.180 Similar mechanisms affecting VWF survival, perhaps combined with altered proteolysis,181,182,183 may explain the observed modifying effect of the ABO blood group glycosyltransferases on plasma VWF survival.184 Additional genetic factors have been implicated to influence VWF via altered survival, including the clearance receptors CLEC4M and LRP1 (CD91) (reviewed in Ref. 185). The biologic consequences of VWF modifiers identified in normal populations are unclear, and studies are needed to determine their significance in VWD.
Patients with type 3 VWD account for 1 to 5 percent of clinically significant VWD, have very low or undetectable levels of plasma and platelet VWF:Ag and VWF:RCo, and generally present early in life with severe bleeding.186 FVIII coagulant activity is markedly reduced but usually detectable at levels of 3 to 10 percent of normal. Type 3 VWD has generally been considered an autosomal recessive disorder, but in a recent Canadian study of 100 individuals in 34 families, 48 percent of “carriers” had a diagnosis of type 1 VWD,161 suggesting the dominant type 1 VWD pattern of inheritance is common in type 3 VWD families.
Mutations associated with type 3 VWD have been reported throughout the VWF gene (http://www.vwf.group.shef.ac.uk/). Gross VWF gene deletion detectable by Southern blot26,187,188,189,190 or multiple ligation-probe amplification161,191 is the molecular mechanism for type 3 VWD in only a small subset of families. However, large deletions may confer an increased risk for the development of alloantibodies against VWF.26,189 A similar correlation between gene deletion and risk for alloantibody formation has been observed in hemophilia (Chap. 123). Comparative analysis of VWF genomic DNA and platelet VWF mRNA has identified nondeletion defects resulting in complete loss of VWF mRNA expression as a molecular mechanism in some patients with type 3 VWD.192,193 A number of nonsense and frameshift mutations that would be predicted to result in loss of VWF protein expression or in expression of a markedly truncated or disrupted protein have been identified in some type 3 VWD families.168,194,195,196 A frameshift mutation in exon 18 appears to be a particularly common cause of type 3 VWD in the Swedish population and has been shown to be the defect responsible for VWD in the original Åland Island pedigree.197,198 This mutation results in a stable mRNA encoding a truncated protein that is rapidly degraded in the cell.199 This mutation also appears to be common among type 3 VWD patients in Germany,200 but not in the United States.201
Type 2A is the most common qualitative variant of VWD and is generally associated with autosomal dominant inheritance and selective loss of the large and intermediate VWF multimers from plasma (see Fig. 126–4). A 176-kDa proteolytic fragment present in normal individuals is markedly increased in quantity in many type 2A VWD patients. This fragment is consistent with proteolytic cleavage of the peptide bond between Tyr1605 and Met1606.98,202 Based on this observation, initial DNA sequence analysis in patients centered on VWF exon 28, in the region encoding this segment of the VWF protein, leading to the identification of the first point mutations responsible for VWD.203 Since that time, a large number of mutations have been identified, accounting for the majority of type 2A VWD patients.194 Many of these mutations are clustered within a 134-amino-acid segment of the VWF A2 domain (between Gly1505 and Glu1638; see Fig. 126–3), and the most common, Arg1597Trp, appears to account for about one-third of type 2A VWD patients.194,195,204
Type 2A VWD mutations have been grouped by two distinct molecular mechanisms. In the first subset, classified as group 1, the type 2A VWD mutation has been commonly considered a defect in intracellular transport, with retention of mutant VWF in the ER. In addition to retention or degradation of mutant VWF in the ER, type 2A mutations can also disrupt intracellular processing and secretion via defective multimerization and/or loss of regulated storage.205 In the second subset, or group 2, mutant VWF is normally processed and secreted in vitro, and thus loss of multimers in vivo is presumed to occur based on increased susceptibility to proteolysis in plasma98,206,207,208,209 at the Tyr1605-Met1606 site cleaved by ADAMTS13.101,210 The susceptibility of type 2A VWD mutations to proteolysis by ADAMTS13 in vitro supports accelerated proteolysis as a mechanism for the loss of high-molecular-weight VWF multimers in these patients.204
The multimer structure of platelet VWF correlates well with the underlying type 2A mechanisms. Group 1 patients show loss of large VWF multimers within platelets as a result of defective synthesis, while group 2 patients have normal VWF multimers within the protected environment of the α granule.206 These observations confirm the earlier subclassification of type 2A VWD based on platelet multimers.156 Subclassification into group 1 or 2 might be expected to predict response to DDAVP therapy, although this remains to be demonstrated.
In addition to the major classes of type 2A VWD described above, a number of rare variants historically classified as types IIC to IIH, type IB, and “platelet discordant” are included in the more general type 2A category. Most of these rare variants were distinguished on the basis of subtle differences in the multimer pattern (see Fig. 126–4; multimer changes relative to the location of type 2 mutations is reviewed in Ref. 211). The IIC variant is usually inherited as an autosomal recessive trait and is associated with loss of large multimers and a prominent dimer band. Several mutations have been identified in the VWFpp of these patients,212,213,214 presumably interfering with multimer assembly and/or trafficking to storage granules. A mutation at the C terminus of VWF, interfering with dimer formation, was described in a patient with the IID variant.215 Most of the other reported variants of type 2A VWD are quite rare, often limited to single case reports.
Type 2B VWD is usually inherited as an autosomal dominant disorder and is characterized by thrombocytopenia and loss of large VWF multimers. The plasma VWF in type 2B VWD binds to normal platelets in the presence of lower concentrations of ristocetin than does normal VWF and can aggregate platelets spontaneously. Accelerated clearance of the resulting complexes between platelets and the large, most adhesive forms of VWF accounts for the thrombocytopenia and the characteristic multimer pattern (see Fig. 126–4).
The peculiar functional abnormality characteristic of type 2B VWD suggested a molecular defect within the GPIb binding domain of VWF. For this reason, initial DNA sequence analysis focused on the corresponding portion of VWF exon 28.216,217 Type 2B mutations are located within the VWF A1 domain at one surface of the described crystallographic structure.124,129 The four most common mutations are clustered within a 36-amino-acid stretch between Arg1306 and Arg1341 (see Fig. 126–3); together, these account for more than 80 percent of type 2B VWD patients.195 Functional analysis of mutant recombinant VWF218,219,220,221,222 confirms that these single-amino-acid substitutions are sufficient to account for increased GPIb binding and the resulting characteristic type 2B VWD phenotype. Structural studies of type 2B VWD mutations show that these residues interact with the leucine rich repeats of GPIb thought to be critical to the VWF A1–GPIb interactions under shear.223 Type 2B mutations have now been modeled extensively in mice, all of which exhibited accelerated VWF clearance, as expected.224 Type 2B VWD mice also had short-lived platelets, with evidence of macrophage-mediated platelet clearance.225 In these models, platelets were observed to be coated by type 2B VWF,225
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


