Matthew K. Waldor, Edward T. Ryan
Vibrio cholerae
Cholera is an acute and often severe watery diarrheal disease that is among the most rapidly fatal infectious diseases of humans. Previously well individuals infected with Vibrio cholerae, the comma-shaped gram-negative rod that causes this disease, can die from dehydration in less than a day. Two epidemiologic features of cholera also distinguish it from other diarrheal diseases. First, cholera can appear in explosive epidemics, particularly among populations that lack access to clean water and adequate sanitation and who are immunologically naïve to the organism. Second, cholera occurs in pandemics, where one strain of V. cholerae spreads around the world in essentially clonal fashion. Currently, we are in the midst of the seventh pandemic of cholera, which began in 1961 on the Indonesian island of Sulawesi. Cholera is thought to have afflicted human populations for thousands of years, particularly on the Indian subcontinent.1 Throughout the centuries, cholera epidemics have influenced human history and killed millions. Studies of cholera and V. cholerae have had broad impact in several scientific fields, and rehydration strategies developed for the treatment of cholera have been extremely useful for treatment of other diarrheal diseases.2 Of note, John Snow’s studies in London in the 1840-1850s linking the spread of cholera to the water supply are considered central to the establishment of the field of epidemiology. Robert Koch isolated V. cholerae from cholera patients in Calcutta (Kolkata) in 1883; however, Filippo Pacini likely detected the organism in 1854 in the intestines of cholera victims in Florence.3
Classification and Genomics
V. cholerae are oxidase positive facultative anaerobes that can be identified using a variety of biochemical tests and selective media.4 V. cholerae are characteristically highly motile and bear a unipolar sheathed flagellum. V. cholerae is classified into more than 200 serogroups; differences in the composition of the O-specific polysaccharide (OSP) chains of lipopolysaccharide (LPS) molecules establish the chemical basis for the distinct antigenicity of the serogroups. The O1 serogroup of V. cholerae was, until 1992 with the emergence of V. cholerae O139, the only serogroup associated with epidemic cholera. However, some non-O1, non-O139 V. cholerae serogroups have given rise to gastroenteritis outbreaks, but not to large epidemics. In general, the majority of V. cholerae isolates from the environment, including those of V. cholerae O1, do not produce the virulence factors, such as cholera toxin, that are required to cause cholera. The O1 serogroup is divided into two principal serotypes, Inaba and Ogawa, which differ by the presence of a 2-O-methyl group in the nonreducing terminal sugar of the Ogawa OSP, which is absent from Inaba OSP.5 Cholera toxin-producing (toxigenic) O1 serogroup strains are also divided into two biotypes based on a number of microbiologic and biochemical differences.4 The classic biotype of V. cholerae O1 caused the second and sixth cholera pandemics and perhaps the other early pandemics as well, whereas the El Tor biotype of V. cholerae O1 is the cause of the ongoing seventh pandemic of cholera. The classic biotype of V. cholerae is now extinct.
V. cholerae, like many other bacterial enteric pathogens, is classified as a γ-proteobacteria. Unlike most γ-proteobacteria, such as E. coli, whose genomes consist of single circular chromosomes, the genome of V. cholerae is multipartite. The V. cholerae genome (and that of all Vibrio species) is unequally divided between two circular chromosomes.6 Its large chromosome (≈3 kb) contains the vast majority of the essential genes as well as genes implicated in pathogenicity, whereas the small chromosome (≈1kb) is enriched for genes of unknown function. A plausible explanation for the evolution of the bipartite V. cholerae genome is that a remote ancestor of V. cholerae (and all vibrios), acquired a large plasmid that subsequently obtained essential genes and other chromosome-like features.7 Acquisition of virulence-linked genes from other (unknown) donor organisms via lateral gene transfer has also played a central role in the evolution of pathogenic V. cholerae.8
Pathogenicity
V. cholerae has no known natural vertebrate host besides humans. However, the pathogen is not dependent on humans for its propagation; V. cholerae grows in brackish estuaries and coastal seawaters, often in close association with copepods or other zooplankton. V. cholerae can also grow in water of lower salinity when it is warm and adequate organic material is available.9 Because patients with severe cholera can excrete up to 20 L of “rice water” stool laden with ≈109 V. cholerae cells/mL per day, humans can greatly facilitate the propagation and dissemination of the pathogen. Humans usually become infected with V. cholerae after ingestion of contaminated water or food, although there may be direct fecal-oral spread between people that does not involve water or food. The infectious dose in volunteer studies carried out in the United States is relatively high.10 Low stomach pH is known to lower the infectious dose in volunteers, and additional factors likely impact the infectious dose as well.10 Because the frequency of hypochlorhydria (caused by chronic H. pylori infection) in cholera endemic regions is often high, the V. cholerae infectious dose may be considerably lower in these regions.
After passage through the stomach, V. cholerae has the unusual capacity to survive and multiply in the small intestine. Although many processes and gene products contribute to the organism’s ability to colonize the small intestine,11 TCP, the toxin coregulated pili, are the most critical identified factors primarily dedicated to mediating colonization.12 The major subunit of these pili is encoded by tcpA, the first gene of the tcp operon; with the exception of tcpF, the other genes of the operon encode the machinery for the biogenesis of TCP.13 The TCP assembly apparatus also secretes TcpF, an unusual secreted colonization factor. TCP are thought to promote V. cholerae intestinal colonization in at least three ways: (1) the pili mediate bacterium–bacterium interactions, enabling microcolony formation within the small intestine; (2) pili confer protection against toxic factors produced in the intestine; and (3) the pili promote attachment to the intestinal epithelium.13 The tcp operon is found within the TCP pathogenicity island, an ≈41 kb chromosome segment that also encodes additional virulence-associated factors,14,15 which appears to have been acquired via horizontal gene transfer by a nonpathogenic ancestor of contemporary V. cholerae.
While colonizing the small intestine, V. cholerae secretes cholera toxin (CT), the AB5 subunit type protein toxin that causes the secretory diarrhea that is characteristic of cholera. The sufficiency of CT to elicit cholera-like diarrhea was demonstrated experimentally; volunteers fed as little as 5 µg of purified CT developed severe diarrhea indistinguishable from cholera.16 Thus, cholera is a toxin-mediated disease, and V. cholerae is a noninvasive mucosal pathogen. The pentameric B subunit of CT binds to GM1, a glycosphingolipid on the surface of epithelial cells. The CT-GM1 interaction targets the toxin through a retrograde trafficking pathway from the plasma membrane to the Golgi apparatus and endoplasmic reticulum and then to the cytoplasm.17 In the cytoplasm, the enzymatic A subunit of cholera toxin mediates the transfer of adenosine diphosphate (ADP)-ribose from nicotinamide adenine dinucleotide (NAD) to the G protein that regulates adenylyl (adenylate) cyclase activity, leading to elevation in the intracellular cyclic adenine monophosphate (cAMP) concentration.18 Increased cAMP levels promote chloride ion (Cl−) secretion by intestinal crypt cells and decreased absorption by villous cells. Water moves from epithelial cells into the bowel lumen to maintain osmolality, and diarrhea results when the resorptive capacity of the remainder of the gut is exceeded. Additional actions of CT (besides dysregulation of adenylyl cyclase) likely contribute to the secretory response underlying cholera.18 Also, other factors besides cholera toxin contribute to the diarrheal response because volunteers who ingest V. cholerae deficient for ctxA often develop mild diarrhea.19
The genes encoding cholera toxin, ctxAB, are not part of the ancestral V. cholerae genome; instead, these genes are embedded in the genome of CTXΦ, a filamentous bacteriophage that infected V. cholerae, rendering it toxigenic.20 Of note, the receptor for this phage is TCP, suggesting that a TCP+, ctxAB− strain was the precursor of contemporary pathogenic V. cholerae. The intricate interdependence of these two mobile elements—the TCP pathogenicity island and the CTX prophage—extends even further; ToxT, a key transcriptional activator of ctxAB expression, similar to the tcp operon, is encoded within the TCP island. Mobile genetic elements, such as phages, plasmids, integrative conjugative elements (ICEs), and pathogenicity islands have played critical roles in the evolution of most bacterial pathogens.
Many of V. cholerae‘s key virulence factors, such as CT and TCP, are not constitutively expressed. Instead, fairly shortly after V. cholerae is ingested, as yet undefined host signal(s) trigger the coordinated expression of the principal virulence factors of V. cholerae.12 A single membrane-embedded transcription activator, ToxR, sits at the top of a signaling cascade that promotes expression of many of the pathogen’s virulence associated genes.21 There may be a specific temporal order of V. cholerae gene expression during expression; ctxAB expression occurs after and depends on tcpA expression.22 Furthermore, as the pathogen passes to more distal parts of the intestine, expression of “late” genes promotes V. cholerae survival outside the human host and engenders a transient “hyperinfectious” state, which promotes the organism’s transmission to new hosts.23,24
Epidemiology
Clinical descriptions of cholera-like illness exist in ancient Sanskrit texts, and cholera has likely been endemic to the Ganges delta regions for centuries, if not longer. Seven cholera pandemics have been recorded since 1817. All of the pandemics are thought to have originated in Asia, particularly from the Indian subcontinent. The El Tor O1 V. cholerae strain causing the ongoing seventh pandemic is evolving. Polymorphisms in the genome of the pandemic strain have been reported to have arisen in the Bay of Bengal and then to have independently spread to other continents.25 All seven pandemics were likely caused by V. cholerae O1, but in 1992, V. cholerae O139 emerged in India and Bangladesh to become the first non-O1 serogroup to give rise to epidemic cholera. Although V. cholerae O139 initially caused large cholera outbreaks, at present, this serogroup only causes a small fraction of cholera cases in Asia; furthermore, V. cholerae O139 has not spread outside of Asia. Horizontal gene exchange appears to explain the molecular origins of V. cholerae O139; the genome of this novel V. cholerae serogroup is essentially identical to that of El Tor O1 V. cholerae, except that the genes encoding O1 antigen biosynthesis have been replaced by genes encoding the O139 O-antigen.26
Cholera had been absent from the Americas for nearly a century until 1991, when a large outbreak began in Peru and rapidly spread throughout much of South and Central America. During the ensuing decade, more than one million cases were reported. The source of the El Tor O1 strain that gave rise to this epidemic is not certain; it has been proposed that contaminated ship ballast water was the source, but this idea has been questioned.27 Cholera did not reach the Caribbean during this outbreak. However, in October 2010, a severe cholera epidemic began in Haiti on the Caribbean island of Hispaniola, just 10 months after a devastating earthquake had compromised that nation’s already tenuous infrastructures for sanitation and water. In the 2 years after the onset of the epidemic, more than 600,000 cases and 7400 deaths were recorded in Haiti, and at least 6.1% of the population had been infected (Fig. 216-1).28 In Hispaniola, cholera spread from Haiti to the Dominican Republic, and from Hispaniola to Cuba, and imported cholera cases from Hispaniola have been reported in several countries, including the United States.29 Several lines of evidence indicate that cholera may have been introduced to Haiti by United Nations security forces from Nepal,30,31,32 suggesting that asymptomatic carriers of V. cholerae can play an important role in transmitting cholera over long distances.
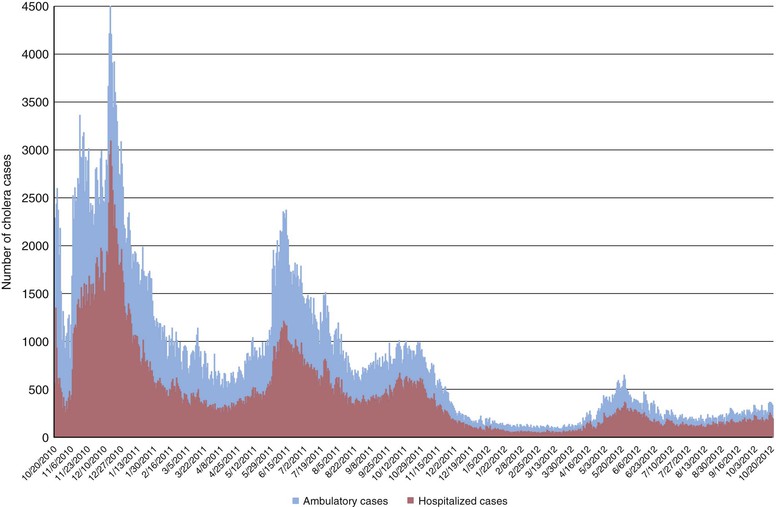
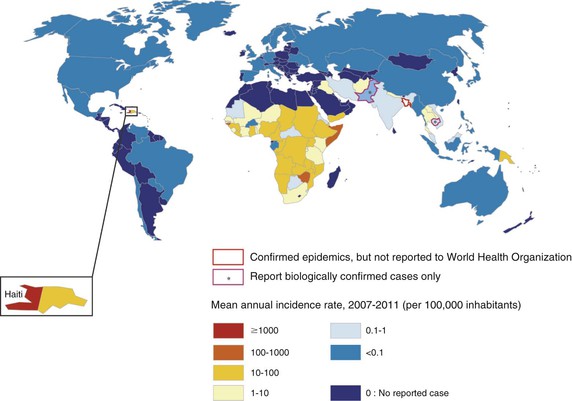
In 2011, there were approximately 600,000 cases of cholera and nearly 8000 cholera-related deaths reported to the World Health Organization (WHO) from 58 countries (Fig. 216-2).33 These estimates of cholera morbidity and mortality significantly underrepresent the true burden of this disease because many cases are not diagnosed or reported. Recent estimates suggest that approximately three to five million cholera cases occur annually worldwide, with 100,000 deaths.34 In the decade preceding the outbreak in Haiti, more than 90% of reported cholera cases were reported from Africa, although the burden of disease in Asia is significantly underreported.
Clinical Manifestations
Cholera manifests as an acute and often severe watery diarrhea that can lead to death from dehydration within hours of onset. The spectrum of disease in persons infected with V. cholerae ranges from asymptomatic intestinal colonization of V. cholerae, observed in individuals with preexisting immunity, through mild, moderate, and severe diarrhea. In severe cholera, the volume of watery diarrhea can exceed 1 L/hr. Vomiting is common. In the initial stages, diarrhea may contain intestinal contents; however, as diarrhea progresses, it becomes more watery, eventually becoming clear, with flecks of white mucus (rice water stool) with a fishy odor. Individuals with cholera are usually afebrile and may be hypothermic because of severe dehydration. The extensive fluid secretion into the intestinal lumen can lead to abdominal cramping and discomfort, and the severe dehydration and perturbations in calcium and potassium levels can lead to ileus, muscle pain and spasm, and even tetany. In severely dehydrated patients, the skin can become mottled or darkened, reflecting the hyperviscosity and poor circulation of blood.
The clinical manifestations of severe cholera (referred to as cholera gravis) reflect the degree of dehydration. Individuals may be lethargic or unresponsive, have sunken eyes, and markedly decreased skin turgor, often leading to skin “tenting” (when skin is pulled back, it takes a long time for it to fall back into place). Individuals may have cold and clammy skin, dry mucous membranes, and may be anuric. Extensive loss of bicarbonate in stool and lactic acidosis secondary to compromised circulation and perfusion can lead to Kussmaul breathing.35 The peripheral pulse is often rapid in the initial stages of dehydration; however, it can become progressively fainter, eventually becoming nonpalpable as dehydration progresses. Hypoglycemia can be severe, especially in children with limited glycogen stores, and can contribute to lethargy, seizures, and coma. Paradoxically, stool volume can decrease as the degree of dehydration increases, giving a false impression of improvement before total circulatory collapse. Cholera is an acute illness, and the complications of cholera reflect severe hypoperfusion and can include stroke, acute tubular necrosis with renal dysfunction, and aspiration pneumonia from the extensive vomiting.36 Diarrhea is usually most prominent for 1 to 3 days.
The clinical manifestations of cholera caused by V. cholerae O1 and O139 are indistinguishable. Similarly, the disease caused by the Ogawa and Inaba serotypes of V. cholerae O1 is indistinguishable. At the population level, disease caused by classic biotype V. cholerae O1 is more severe than that caused by O1 El Tor biotype strains. Differences in the amino acid sequences of the CT produced by the two biotypes may account for the heightened severity of diarrhea associated with the classic biotype. Recently identified so-called hybrid (or altered) strains of V. cholerae O1 El Tor that express classic biotype cholera toxin may be associated with more severe clinical manifestations than traditional El Tor strains.37
Host factors are also likely to contribute to clinical manifestations of cholera. For example, when V. cholerae is first introduced into an immunologically naïve population (e.g., into Haiti in 2010), individuals of all ages can be equally affected, and disease is often severe. In comparison, in regions where cholera is an endemic, many older children and adults have preexisting immunity, and these individuals can be asymptomatically colonized or present with mild-to-moderate diarrhea.38 Individuals with blood group O are more likely to present with severe cholera than individuals with non-O blood groups.39 Hypochlorhydria increases the likelihood and severity of disease.40,41 Retinol deficiency is associated with an increased risk for cholera,42,43 and concomitant intestinal parasitic and bacterial infection can modulate anti–V. cholerae immune responses after cholera.44,45 First-degree relatives of cholera patients also have greater odds of becoming infected than non–first-degree relatives in the same household, independent of blood group,42 suggesting that other genetic factors may contribute to susceptibility, and in a candidate gene study, a variant in the promoter region of BPIFB1 (also known as LPLUNC1) involved in innate immune responses is associated with cholera.46,47 In a genome-wide study, host susceptibility to cholera was also associated with potassium channel genes involved in cyclic adenosine monophosphate-mediated chloride secretion and for components of the innate immune system involved in nuclear factor κB signaling.
Diagnosis
Cholera should be considered in any area of the world when an individual aged 5 years or older develops severe dehydration or dies from acute watery diarrhea, and when an individual aged 2 years or older develops acute watery diarrhea in an area of the world known to be endemic for cholera.48 Microbiologic analyses of stool samples permit confirmation of V. cholerae as well as determination of antimicrobial susceptibility profiles; however, once cholera is known to be endemic in an area and the antimicrobial susceptibility profile of the outbreak strain is known, routine stool culture from each individual is not indicated. V. cholerae can be isolated from stool by using selective media, such as taurocholate tellurite gelatin agar (TTGA) and thiosulfate citrate bile sucrose (TCBS) agar, and recovery of organisms can be enhanced by enrichment of stool in alkaline peptone water.49 Serogroup and serotype identification are determined with specific antibodies.50 Darkfield microscopy can be used to rapidly evaluate whether watery stools contain the characteristic “shooting star” motility of vibrios, which can be inhibited by the action of specific antibodies.51 A number of immunoassays that can detect V. cholerae LPS and/or cholera toxin directly in stool have been developed.52 These assays are particularly helpful when included in surveillance systems in areas of the world at risk for cholera. During laboratory evaluation and under field conditions, the sensitivity and specificity of these assays have ranged from 60% to 100%.52
Treatment
The primary focus of treating patients with cholera is fluid management. Individuals with suspected cholera should be rapidly restored to euvolemia, and ongoing fluid losses need to be matched. Algorithms for rapid assessment of fluid status in any setting, including in sites lacking electricity or laboratory support, are shown in Table 216-1. Although individuals with severe cholera or who are unresponsive require administration of intravenous fluids in the initial stages of treatment, many individuals with less severe cholera and who are alert can be managed with oral rehydration therapy alone.
TABLE 216-1
How to Approach Rehydration in Patients with Suspected Cholera48
| DEGREE OF DEHYDRATION | ||||
| None (<5%) | Some (5%-10%) | Severe (>10%) | ||
| Assess degree of dehydration | Mentation | Alert | Restless, irritable | Lethargic or unconscious |
| Eyes | Normal | Sunken | Sunken | |
| Skin turgor | Normal recoil | Slow recoil | Very slow recoil (>2 sec) | |
| Pulse | Normal | Rapid, low volume | Weak or absent | |
| Thirst | Drinks normally | Thirsty, drinks eagerly | Drinks poorly or unable to drink | |
| Approach to rehydration* | Fluid replacement | Ongoing losses only | 75 mL/kg in addition to ongoing losses | >100 mL/kg in addition to ongoing losses |
| Preferred route of administration | Oral† | Oral or intravenous | Intravenous | |
| Timing | Usually guided by thirst | Replace fluids over 3-4 hr | As rapidly as possible until circulation is restored; complete the remainder of fluids within 3 hr | |
| Monitoring | Observe until assured ongoing losses can be adequately replaced by ORS | Observe every 1-2 hr until all signs of dehydration resolve and patient urinates | Once circulation is established, monitor every 1-2 hr | |






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


