INTRODUCTION
SUMMARY
Blood vessels, especially their endothelial lining, play a critical role in the maintenance of vascular fluidity, arrest of hemorrhage (hemostasis), prevention of occlusive vascular phenomena (thrombosis), and regulation of inflammatory cell processes.* The endothelium extends to all recesses of the body and maintains an intimate association with flowing blood and blood cells. However, endothelial cell morphologies, gene-expression profiles, and functions vary among different vascular beds. For example, in straight arterial segments, but not at branch points or curvatures of the arteries or veins, endothelial cells align themselves in parallel to the direction of blood flow. Similarly, endothelial cells in post capillary venules are primarily responsible for mediating adhesion and transmigration of leukocytes, whereas arteriolar endothelium is important for regulation of vasomotor tone. Proteomic studies have revealed that endothelial cells have the unique capacity to express and elaborate thromboregulatory molecules, which can be classified according to their chronologic appearance following vascular injury. Early thromboregulators appear prior to thrombin formation and late thromboregulators arrive after thrombin has formed. This chapter reviews some of the mechanisms by which the vascular wall regulates hemostasis, and discuss their implications for vascular health and disease (Table 115–1).
Acronyms and Abbreviations
APC, activated protein C; Apo, apolipoprotein; APS, antiphospholipid syndrome; C5a, complement factor 5a; CAM, cell adhesion molecule; COX, cyclooxygenase; DAG, diacylglycerol; DDAVP, deamino D-arginine vasopressin; EPCR, endothelial protein C receptor; GMP, guanosine monophosphate; IL, interleukin; IP3, inositol triphosphate; Lp(a), lipoprotein(a); NFκB, nuclear factor kappa B; NO, nitric oxide; NOS, nitric oxide synthase; PAF, platelet-activating factor; PDGF, platelet-derived growth factor; PECAM, platelet endothelial cell adhesion molecule; PGI2, prostacyclin; PGIS, prostacyclin synthase; PSGL, P-selectin glycoprotein ligand; scu-PA, single-chain urokinase-type plasminogen activator; TAFI, thrombin-activatable fibrinolysis inhibitor; TF, tissue factor; TFPI, tissue factor pathway inhibitor; TM, thrombomodulin; TNF, tumor necrosis factor; t-PA, tissue-type plasminogen activator; VWF, von Willebrand factor.
Early thromboregulators
Late thromboregulators
|
| Class | Type | Site of Action | Aspirin Sensitivity | Mode of Action |
|---|---|---|---|---|
| Eicosanoids | PGI2, PGD2 | Fluid phase autacoid | Sensitive | Elevation of platelet cAMP |
| Nitrovasodilators | EDRF/NO | Fluid phase autacoid | Insensitive | Elevation of platelet cGMP |
| Ectonucleotidases | CD39/ENTPD1 | Endothelial cell surface | Insensitive | Enzymatic removal of secreted ADP |
| Thromboxane | TXA2 | Fluid phase vasoconstrictor | Sensitive | Lowers platelet cAMP and platelet agonist |
| Endothelins | ET-1, ET-2 | Fluid phase vasoconstrictor | Insensitive | Direct vasoconstrictor peptide |
*Dr. Aaron Marcus died on May 6, 2015
VASCULAR FUNCTION IN HEMOSTASIS: INTRODUCTION
The endothelium represents a dynamic interface between flowing blood and the vessel wall, and produces a variety of factors that regulate blood fluidity (Fig. 115–1). Endothelial cells are subject to unique shear stress forces, to soluble factors in the blood, and to signals emanating from cells in the circulation, vascular wall, and tissues, all of which create region-specific phenotypes.1,2,3 In addition to modulating vascular permeability and fragility, the endothelium regulates the fluid state of blood through its thromboresistant nature, profibrinolytic properties, and antiinflammatory potential. These activities maintain vascular patency.4
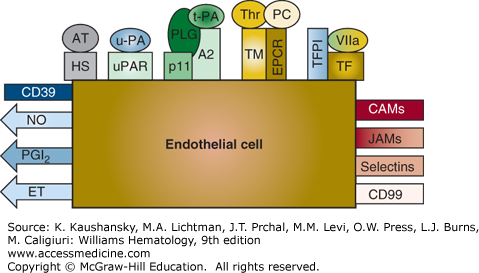
Figure 115–1.
Schematic of endothelial cell thromboregulatory molecules. Products that are secreted and exert their effects in the fluid phase are represented by arrows. Cell-surface–associated molecules are shown as rectangles. Metabolites synthesized by endothelial cells are indicated. Thromboregulators that modulate platelet activation, recruitment, and blood vessel contractility are shown on the left. Agents that regulate components of the coagulation cascade and/or fibrinolytic system are located at the top. Inflammatory molecules whose expression or activity is directed by inflammatory mediators are shown at the right. A2, annexin 2; AT, antithrombin; CAMs, cellular adhesion molecules; CD39, endothelial cell ecto-ADPase/CD39; EPCR, endothelial cell protein C receptor; ET, endothelin; FVIIa, factor VIIa; HS, heparan sulfate; JAMs, junctional adhesion molecules; NO, nitric oxide; PC, protein C; PGI2, prostacyclin; PLG, plasminogen; TF, tissue factor; TFPI, tissue factor pathway inhibitor; TM, thrombomodulin; t-PA, tissue plasminogen activator; u-PA, urokinase plasminogen activator; uPAR, urokinase plasminogen activator receptor. These components are discussed further in the text.
The heterogeneity of endothelial cells is mediated by two mechanisms.5,6 First, extracellular biochemical and biomechanical signals trigger posttranscriptional and/or posttranslational changes that vary across the vascular tree. Second, certain site-specific properties of the endothelium are genetically programmed, and therefore, independent of the extracellular milieu. This phenotypic variability serves at least two important purposes: (1) It allows endothelial cells to meet the specific metabolic needs of the surrounding tissue. For example, the tight junctions of the blood–brain barrier protect neurons from fluctuations in composition of the aqueous blood supply, whereas the fenestrated discontinuous endothelium of hepatic sinusoids allows ready access of nutrient-rich portal venous blood for the metabolic systems in hepatocytes; and (2) phenotypic variability provides endothelial cells with site-specific mechanisms for thriving within many different microenvironments. For example, endothelial cells in the inner medulla of the kidney must survive the relatively hypoxic and hyperosmolar local environment, whereas endothelial cells in the pulmonary capillary bed have adapted to an oxygen-rich environment.
A rapid endothelial cell response is required for sudden environmental perturbations. Translational control mechanisms, which are more immediate than transcriptional changes, provide regulatory responses for up to 10 percent of genes expressed in endothelial cells.7 Because of their close association with both flowing blood and solid tissues, endothelial cells are subject to a broad spectrum of agonistic and inhibitory external signals that frequently require rapid functional and phenotypic responses. Clinically, such stimuli are associated with sepsis, inflammation, ischemia–reperfusion injury, and direct mechanical vascular trauma induced clinically by stents, balloon catheters, and graft procedures.
Thromboregulatory compounds, such as eicosanoids, nitric oxide, and the ecto-ATP/Dase-1/CD39, control platelet and vascular reactivity during the early stages of thrombus formation (Table 115–2).8 Eicosanoids are hydrocarbon compounds derived from essential fatty acids in the diet. The most important endothelial eicosanoid is prostacyclin (PGI2), which blocks platelet reactivity, induces vascular relaxation, and stimulates cytokine production.9 Nitric oxide (NO) is a naturally occurring gas released from vascular endothelial cells in response to binding of vasodilators to endothelial cell membrane receptors. Thus, it is a short-lived vasodilator and inhibitor of platelet reactivity. By activating guanylate cyclase, the resulting increase in cyclic guanylate monophosphate (GMP) inhibits platelet function and induces vascular relaxation.10,11 Endothelial cell ecto-ATP/Dase-1/CD39 is a membrane-associated apyrase that metabolizes adenosine diphosphate (ADP) in the primary platelet releasate, preventing further platelet activation and recruitment.12,13
Late thromboregulators produced by endothelial cells act either to prevent excessive thrombin generation or to promote lysis of intravascular thrombi (see Table 115–1). Antithrombin, a natural anticoagulant, acts as an inhibitor of thrombin and factor Xa in the circulation. Endothelial cell heparan proteoglycans act as cofactors for antithrombin. The tissue factor pathway inhibitor (TFPI) inhibits the complex between factor VIIa and tissue factor (TF). The thrombomodulin/endothelial cell protein C receptor (EPCR)/protein C system in the vascular wall regulates hemostasis through inactivation of procoagulant cofactors, and antiinflammatory activity.14 The fibrinolytic system is intimately involved with the vascular endothelium because endothelial cells not only synthesize and secrete tissue plasminogen activator (t-PA), but also regulate formation of plasmin from its precursor, plasminogen, through the expression of receptors.15 Impairment of fibrinolytic potential can play a central role in the etiology of occlusive vascular disease.16 Finally, endothelial cell adhesion molecules, including the cell adhesion molecules (CAMs: mucosal addressin cell adhesion molecule [MAdCAM]-1, intercellular adhesion molecule [ICAM]-1, vascular cell adhesion molecule [VCAM]-1, and platelet endothelial cell adhesion molecule [PECAM]-1) and the selectins (P- and E-selectin), are glycoproteins that modulate multiple interactions between the endothelium and various classes of circulating leukocytes, thereby modulating vascular patency.17 Together, these mechanisms define thromboregulation, the processes by which blood cells and cells of the vessel wall, through their close proximity, interact to facilitate or inhibit thrombus formation.18
The physiologic defense systems that render endothelial surfaces and blood cells antithrombotic can be overwhelmed by excessive shear stress, increased turbulence, injury, inflammation, and severe atherosclerosis.19 These events transform the endothelial cells into a prothrombotic and antifibrinolytic phenotype,20 which is accompanied by upregulation of leukocyte and endothelial CAMs, increased expression of TF, and accumulation of monocytes/macrophages in the vessel wall.21 These events commonly occur at the site of fissured atherosclerotic plaques in the coronary and cerebrovascular circulation.22 Because the eicosanoids such as PGI2 (prostaglandin [PG]I2), as well as NO, and the ecto-ATP/Dase-1/CD39 group reach peak activity very early in the hemostatic/thrombotic cascade (Figs. 115–2, 115–3, 115–4), they represent potential targets for therapeutic intervention in the sequence of events beginning with platelet activation, and leading to coagulation, thrombosis, and atherogenesis.21,22 Finally, functional and physical contacts between platelets and endothelial cells are of critical importance for the maintenance of vascular integrity and cell permeability.1,23
Figure 115–2.
Following injury to the blood vessel wall, platelets adhere to the damaged surface of the endothelial cell. Concomitant with adhesion platelets and endothelial cells become activated. P-selectin is expressed on the endothelial cell surface. Platelet surface receptors glycosylphosphatidylinositol (GPI)bα and P-selectin glycoprotein ligand (PSGL)-1 interact with endothelial P-selectin, thereby mediating platelet rolling. Firm adhesion is mediated by the integrin αIIbβ3. In parallel with these intercellular events, platelet activation and release occur. The enzyme CD39 on the endothelial surface modulates the ambient concentration of adenosine diphosphate (ADP) by metabolizing it.7,8 5-HT, 5-hydroxytryptamine; TXA2, thromboxane A2. (Adapted with permission from Gawaz M, Langer H, May AE: Platelets in inflammation and atherogenesis. J Clin Invest 11(12):3378–3384, 2005.)
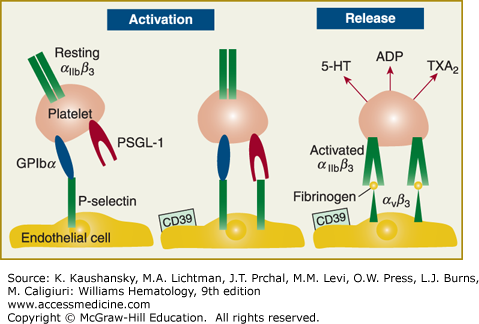
Figure 115–3.
Adherent activated platelets induce an inflammatory response in endothelial cells. Platelet adhesion involving αIIbβ3 induces exposure of P-selectin (CD62P) and release of platelet CD40 ligand (CD40L) and interleukin (IL)-1β which then stimulate endothelial cells to respond with an inflammatory reaction that supports prothrombotic and proatherogenic alterations in the endothelium. IL-8 and MCP-1 (monocyte chemoattractant protein-1) are the principal chemoattractants for neutrophils and monocytes. ICAM, intercellular adhesion molecule; MMP, matrix metalloproteinase; u-PA, urokinase plasminogen activator; uPAR, urokinase plasminogen activator receptor; VCAM, vascular cell adhesion molecule. (Adapted with permission from Gawaz M, Langer H, May AE: Platelets in inflammation and atherogenesis. J Clin Invest 11(12): 3378–3384, 2005.)
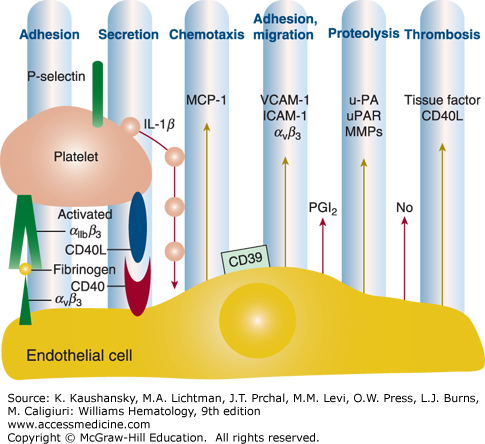
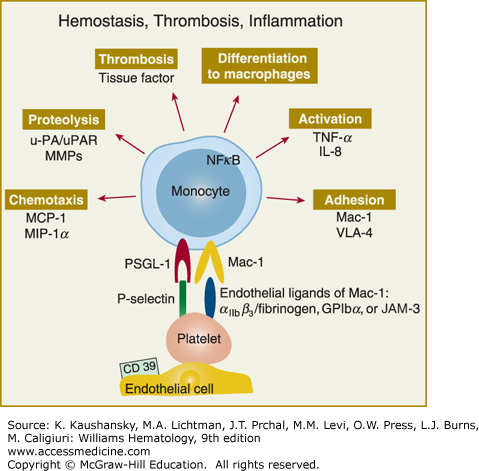
Figure 115–4.
Adherent/activated platelets promote an inflammatory response in monocytes. The platelets mainly interact with monocyte P-selectin glycoprotein ligand (PSGL)-1 with monocytic PSGL-1 via P-selectin and with monocyte Mac-1 (αMβ2) via αIIbβ3 (and fibrinogen bridging) or glycosylphosphatidylinositol (GPI)bα. Through this mechanism platelets initiate monocyte secretion of chemokines, cytokines, and procoagulant tissue factor. These serve to upregulate and activate adhesion receptors and proteases. In parallel, they induce monocyte differentiation into macrophages. Therefore platelet-monocyte interactions provide a prothrombotic and atherogenic milieu at the vascular wall, which can eventually support plaque formation. IL, interleukin; JAM, junctional adhesion molecule; MCP, monocyte chemoattractant protein; MIP, macrophage inhibitory protein; MMP, matrix metalloproteinase; NFκB, nuclear factor kappa B; u-PA, urokinase plasminogen activator; uPAR, urokinase plasminogen activator receptor; TNF, tumor necrosis factor; VLA, very late antigen. (Adapted with permission from Gawaz M, Langer H, May AE: Platelets in inflammation and atherogenesis. J Clin Invest 11(12):3378–3384, 2005.)
THE EICOSANOID PATHWAY IN BIOLOGY AND MEDICINE: CELL–CELL INTERACTIONS AND TRANSCELLULAR METABOLISM
Dietary fatty acids give rise to arachidonic acid, the starting point for synthesis of other eicosanoids. Originating from different cells, intermediates in the arachidonic acid pathway can interact with each other to produce new products with new biologic activities. Oxygenation and further enzymatic transformation of arachidonic acid gives rise to eicosanoids (formerly classified as PGs) and hydroxy acids, such as the leukotrienes. Eicosanoids are autocoids, an important group of transient, physiologically active endogenous substances, that act within the immediate environment of the cell, where they promote or inhibit a biologic function.24 These autacoids have a very short life span and may act within a few seconds, a phenomenon that is clinically important but difficult to study experimentally.
In 1975, Hamberg and colleagues discovered that a new eicosanoid, PGI2, was derived from arachidonic acid in endothelial cells.25 Soon thereafter, Moncada realized that the effects of PGI2 opposed those of thromboxane, namely vasodilation and inhibition of platelet aggregation.26,27 The biologic half-life of PGI2 was found to be 10 to 20 seconds. In addition, it was determined that the first step in arachidonic acid oxidation and conversion, which is carried out by cyclooxygenase (COX)-1, is inhibited by aspirin (acetylsalicylic acid), which donates an acetyl group that inactivates COX-1 and inhibits platelet function.28
PGI2 is the major and most important eicosanoid produced by endothelial cells. A broad range of stimuli, including hormones, biochemicals, or physical forces such as shear stress can elicit release of PGI2. Kinetic studies revealed two distinct patterns of PGI2 production: (1) rapid release, independent of new COX-1 mRNA or protein synthesis, and (2) slower production reflecting increased COX-2 expression.
In the case of rapid stimulation of PGI2 production, as induced by thrombin, histamine, bradykinin, and ionophore, the response plateaus at 10 minutes.29 These agonists activate phospholipase C which generates inositol trisphosphate (IP3) and diacylglycerol (DAG). The released IP3 induces an elevation of intracellular calcium, which translocates phospholipase A to the outer portion of the nuclear envelope and endoplasmic reticulum. Phospholipase A then couples functionally to COX-1, which is located on the luminal membrane. Prostacyclin synthase (PGIS) colocalizes with COX-1 in endothelial cells. Activated phospholipase A2 (cPLA2) catalyzes the release of arachidonic acid from membrane phospholipids, and the free arachidonate interacts with COX-1 and is converted to the endoperoxide PGH2. PGIS converts prostaglandin H2 (PGH2) to PGI2. The half-life of COX-1 is approximately 10 minutes, whereupon it autoinactivates.
Stimulation of PGI2 production by proinflammatory cytokines and growth factors, such as lipopolysaccharide (LPS), interleukin (IL)-1β, tumor necrosis factor (TNF)-α, and platelet-derived growth factor (PDGF), is a slower, more sustained process.29 In response to these agonists, PGI2 production occurs within 30 to 60 minutes, and parallels the time course of production induced by COX-2, but not COX-1.
The recognition that there was a constitutive and an inducible cyclooxygenase (COX-1 and COX-2, respectively), was a major advance.30 Cloning studies of an immediate to early response gene from 3T3 fibroblasts revealed that the COX-2 complementary DNA was highly homologous to that of COX-1.30,31,32,33,34 COX-2 is inducible in endothelial cells by prothrombotic, inflammatory, or mitogenic stimuli, and in neutrophils by inflammatory stimuli.35,36
Within a specific species, there is approximately 60 percent homology between deduced amino acid sequences of COX-1 (576 residues) and COX-2 (587 residues). The C-terminal sequence of 18 amino acids in COX-2 is absent in COX-1. Therefore, antibodies directed at this C-terminal sequence can identify COX-2 in tissues by immunoblot. The catalytic activity of both COX enzymes are similar and all amino acids critical for COX-1 activity are conserved in COX-2. The active site in COX-1 is slightly larger than that of COX-2, a fact that has impacted design of COX inhibitors. COX-2 contains mannose, and an N-glycosylation site within the 18-amino acid C-terminal sequence. An N-glycosylation site at Asn410 is required for COX-1 to fold into its active conformation.
The gene for COX-1 is located on chromosome 9 and spans 22 kb of genomic DNA, while the gene for COX-2 is located on chromosome 1 and spans 8 kb of DNA. Transcription of COX-2 proceeds via several signaling mechanisms initiated by cyclic adenosine monophosphate (cAMP)/protein kinase A, protein kinase C, tyrosine kinases, and pathways activated by growth factors, endotoxin, and cytokines.33,37,38,39 The discoveries of COX-1 and COX-2 were of great importance and have led to new concepts concerning the structure and function of COX-induced autacoids.40
PGI2 is released from stimulated endothelial cells by a broad range of agonists, and plays a critical role in the maintenance of vascular integrity by promoting thromboresistance and inhibiting inflammatory responses in the vasculature. Production of PGI2 is dynamically regulated to meet the challenges arising from frequent prothrombotic and proinflammatory events.29 As an autacoid, PGI2 has a half-life of 3 minutes, whereupon it undergoes chemical hydrolysis to 6-keto-PGF1α. It acts on the type I platelet PG receptor (IP) by increasing cAMP levels in a paracrine manner.41 IP is a 7-transmembrane, G-protein– and adenylyl cyclase–coupled receptor. The latter binds to and activates protein kinase A (PKA), resulting in inhibition of platelet activation and recruitment.42 Physical or chemical perturbation of endothelial cells results in enhanced PGI2 production, which increases platelet cAMP resulting in abolition of platelet shape change, inhibition of platelet secretion and recruitment, and impaired binding of von Willebrand factor (VWF) and fibrinogen to the platelet surface. PGI2 also inhibits platelet adhesion to subendothelium, especially at high shear rates.43
The discovery of PGI2 revealed that the vascular endothelium had a protective effect on blood fluidity.2,8 It also meant that PGI2 released from endothelial cells could counteract the effect of excessive thromboxane formation. In addition, it was appreciated that intermediates in the synthesis of PGI2 from arachidonic acid could interact with other cells and tissues. Thus, PGI2 could be synthesized from platelet-derived endoperoxides by cultured human endothelial cells.44 Because of a low threshold for toxicity (hypotension and diarrhea), PGI2 does not display a satisfactory therapeutic window. An interesting compendium of eicosanoid-related disorders is described in a review on eicosanoids in health and disease.45
NITRIC OXIDE: AN ENDOTHELIAL VASODILATOR AND INHIBITOR OF PLATELET ACTIVATION AND RECRUITMENT
In vascular endothelial cells, NO synthase (NOS) catalyzes formation of NO from L-arginine, in the presence of nicotinamide adenine dinucleotide phosphate (NADPH) and oxygen.46 The L-arginine is subsequently converted to citrulline and NO. The endothelial cell isoform of NO synthase (eNOS or the NOS3 gene product) functions constitutively, and is further activated by receptor-agonists that elevate intracellular calcium. Major stimuli include ADP, thrombin, bradykinin, and shear stress.43 Shear forces induce transcriptional activation of the eNOS gene because its promoter contains a shear response consensus sequence (GAGACC). The NO that forms activates guanylate cyclase, thereby generating cyclic GMP. NO becomes oxidized to nitrite and then to nitrate, which is measurable in blood samples. NO in the circulation is rapidly inactivated by erythrocytes.11,47,48 NO has a vasodilatory effect on the pulmonary vasculature, and, in patients with congestive heart failure, its inhalation decreases pulmonary hypertension and increases pulmonary ventilation.10,11,47,48,49,50,51,52,53,54 Acetylcholine released by activated nerve terminals in the vessel wall activate the endothelial cell to produce and release NO. This NO effect also explains the action of nitroglycerin, which has long been used to treat patients with angina resulting from coronary artery disease.54
Importantly, production of NO by endothelial cells is impaired in the presence of the thiol-containing amino acid, homocysteine. Cynomolgus monkeys with diet-induced hyperhomocysteinemia demonstrated reduced blood flow in the lower extremity and an impaired response to endothelial cell-dependent vasodilators.51 Similarly, production of NO by endothelial cells in vitro is significantly inhibited in the presence of homocysteine, possibly by a mechanism involving impairment of the enzyme glutathione peroxidase.52,53
There are two isoforms of NOS, the constitutive form (eNOS), synthesized by the endothelial cell and regulated by Ca2+ and calmodulin, and the cytokine-inducible, posttranscriptionally regulated form (iNOS).47 Both constitutive and inducible forms are mainly cytosolic, although a membrane-bound constitutive NOS isoform containing a myristoylation consensus sequence has been isolated from bovine aortic endothelial cells.43 eNOS has a molecular mass of 144 kDa and shares 57percent amino acid sequence identity with neuronal NOS. The cofactor (6R-tetrahydro-L-biopterin [H4B]) participates in inducible and constitutive NOS isoform reactions. It is thought that H4B stabilizes the enzyme in a manner allowing for maximum activity of the NOS subunit to which the pterin binds.10,11,47,54
Platelet activation and recruitment in response to all agonists, such as ADP, collagen, epinephrine, and thrombin, is blocked by NO. Blockade also occurs in vivo via formation of NO from endothelium.10 Importantly, the inhibitory action of NO is not affected by aspirin either in vivo or ex vivo. Therefore, NO production is not caused by participation of endothelial cell eicosanoids.
In addition to eNOS, the NOS3 gene product, endothelial cells stimulated by agonists such as cytokines express the inducible form of NO synthase, iNOS, the NOS2 gene product. Through this mechanism, NO can further inhibit platelet reactivity and reduce basal vessel tone by inducing relaxation of vascular smooth muscle. The biochemical basis for the reaction is that NO binds to the heme prosthetic group of guanylyl cyclase. The inhibitory effect of NO on platelet activation can be monitored by measuring surface expression of P-selectin. The ability of NO to inhibit mobilization of intracellular platelet calcium results in reduction of the conformational changes in platelet membrane glycoprotein (GP)IIb/IIIa, an absolute requirement for fibrinogen binding and subsequent platelet aggregation. There is a broad spectrum of other effects of NO, including inhibition of leukocyte adhesion to endothelial cell surfaces, inhibition of smooth muscle migration, and reduction of smooth muscle cell proliferation. These phenomena suggest that secretion of NO into the microenvironment is a major component of the response to vascular injury43
INHIBITION OF PLATELET ACTIVATION AND RECRUITMENT BY ECTO-ATP/DASE1-CD39
In addition to the platelet inhibition by PGI2 and NO, endothelial cells inhibit platelet function via the action of endothelial cell ecto-ATP/Dase-1/CD39, an ecto-apyrase with ADPase and adenosine triphosphatase (ATPase) activities. The cluster designation symbol for this compound is CD39, the product of the ENTPD1, ectonucleotide triphosphate diphosphohydrolase gene.55 CD39 is localized mainly in endothelial cells and leukocytes. In endothelial cells, CD39 is located on the cell surface with the major portion of the molecule facing the vessel lumen.12,13,56 The enzyme has both N- and C-terminal transmembrane regions with small cytosolic portions anchoring the molecule.57 In addition to CD39, CD73 (5’-nucleotidase) is present on vascular cells and converts the adenosine monophosphate (AMP) generated from CD39 metabolism to adenosine (Fig. 115–5). In contrast to all other known platelet inhibitors, acting in concert with CD73, CD39 can convert the local environment from a prothrombotic ADP/ATP-rich entity to an antithrombotic adenosine-rich environment.58 This phenomenon was evident from observations that platelets became unresponsive to all agonists when in motion or in proximity to endothelial cells, even when eicosanoid and NO production were blocked.2 Importantly, CD39 and CD73 do not exert their action on the platelet per se but act in series to metabolize ATP and ADP secreted from activated platelets to AMP and hence to adenosine.13,59 ADP released from activated platelets is metabolized by CD39, thereby inhibiting ADP-induced platelet activation, release and aggregation (Fig. 115–5).
Figure 115–5.
Released platelet adenosine diphosphate (ADP) is a major control system for hemostasis: ADP → adenosine monophosphate (AMP) → adenosine. Perturbation of endothelial cells, as a consequence of vascular injury, initiates the release of newly synthesized prostacyclin as well as nitric oxide, both of which inhibit platelet reactivity in the fluid phase. The apyrase CD39 is a cell-associated inhibitory thromboregulator. CD39 is substrate-activated and, in concert with CD39, CD73 brings the reaction to completion with the formation of adenosine.309,310 The early metabolic deletion of ADP from the system may serve as a biologic safeguard to avoid excessive platelet accumulation, which would result in thrombosis.21,22,309,310 NO, nitric oxide; PGI2, prostacyclin.
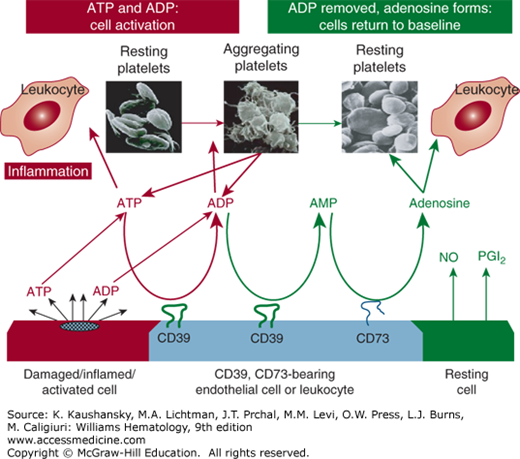
Most platelet agonists initiate secretion of dense granule contents within 15 to 20 seconds. The enhanced metabolism of ATP and ADP by therapeutically administered soluble CD39 would also reduce secondary autoamplification and recruitment, and, consequently, thrombus formation.9,27,60 Because CD39 and CD73 are probably acting together, they will theoretically increase levels of endogenous adenosine and elevate the threshold for platelet activation in the local microenvironment. In a murine model, soluble CD39 administration ameliorates the extent of stroke and reverses excessive platelet reactivity without bleeding complications, even if administered 3 hours following stroke induction.61 Therapeutic benefit of soluble CD39 has also been demonstrated in animal models of cardiac ischemia,62 in the development of atherosclerosis,63 regulation of leukocyte proinflammatory activity,64 inhibition of metastasis,65 and in transplantation medicine.66 That the preclinical therapeutic use of soluble CD39 could abrogate thrombosis without inducing the hemorrhage seen with the use of existing antiplatelet therapies67 could provide a therapeutic advantage over existing therapies for thrombotic disorders, including those who are resistant to existing therapeutic paradigms.9 CD39 represents a major control system for blood fluidity.68
THE PROTEIN C PATHWAY
The protein C pathway69 plays a critical role in the prevention of thrombosis and is an integral part of the host inflammatory response. This pathway is initiated on the endothelial cell surface when thrombin combines with the endothelial receptor protein thrombomodulin (TM). Although thrombin is capable of slowly activating protein C, this reaction is markedly inhibited in the presence of physiologic concentrations of calcium ions. Upon binding of thrombin to TM, the rate of protein C activation is dramatically enhanced and becomes dependent on the presence of calcium. The detailed biochemistry of this activation reaction has been reviewed elsewhere.70 Another protein found predominantly in large vessels, the EPCR, can bind protein C and further augment its activation by the thrombin–TM complex.70 Activated protein C (APC) can dissociate from EPCR and interact with protein S on either the endothelial cell or other membrane surface to exert its anticoagulant function. The function of APC can be found in several reviews.14,71,72,73
By far, the best known function of TM is its role in protein C activation. When thrombin is bound to TM, it is no longer able to clot fibrinogen, activate platelets, activate factors V and VIII,74 or interact with the protease-activated receptors.75,76 Instead, thrombin-TM acts as a direct anticoagulant. TM also promotes the activation by thrombin of the plasma thrombin-activatable fibrinolysis inhibitor (TAFI).77 TAFI inhibits plasmin-mediated fibrinolysis by removing carboxy-terminal lysine residues from fibrin, thereby reducing available binding sites for plasminogen and t-PA. In addition, TAFI is the major enzyme responsible for the removal of a C-terminal arginine from complement factor 5a (C5a),78,79 leading to the inactivation of this potent anaphylotoxin generated during complement activation. Other vasoactive substances may also be inactivated by this enzyme. TM also accelerates the proteolytic inactivation of prourokinase (also called single-chain urokinase-type plasminogen activator [scu-PA]) by thrombin,80,81 which may affect both fibrinolysis and tissue remodeling.82 Despite these antifibrinolytic effects of TM, many in vivo experiments have demonstrated that soluble TM infusion results in a net antithrombotic and/or antiinflammatory effect.83
Independent of its effect on hemostasis, TM is essential to normal fetal development. When the TM gene is deleted by homologous recombination in mice, embryos die on day 8.5, prior to the development of a functional cardiovascular system,84 implying that TM has functions in addition to its anticoagulant and antifibrinolytic properties. Both TM85 and EPCR86 are highly expressed on the giant trophoblast cells of the placenta. If TM expression is maintained on these cells, the TM null embryos survive past this blockade point.87,88
The EPCR is a 220-amino-acid, type 1 transmembrane protein.89,90,91,92 EPCR has two extracellular domains that show structural homology with the α and β domains of major histocompatibility complex (MHC) class 1 molecules, most notably the CD1d family. Because there are three Cys residues in the extracellular domain, the possibility of crosslinking with another protein exists. The cytoplasmic domain of human EPCR is only three amino acids long, Arg-Arg-Cys. The terminal Cys can be acylated with palmitate, which may have functional consequences.93 Both protein C and APC bind to EPCR with similar affinity, approximately 30 nM.89 Binding requires the presence of calcium and is enhanced in the presence of magnesium ions. In addition, a soluble form of EPCR found normally in plasma94 is also capable of binding both protein C and APC with equivalent affinity.
EPCR augments protein C activation by the thrombin–TM complex in vitro and in vivo, primarily by decreasing the Km (Michaelis-Menten dissociation constant) for protein C.70,95,96 Just as thrombin changes its function from procoagulant to anticoagulant when it binds to TM, it appears that APC bound to EPCR undergoes a similar switch from anticoagulant to antiinflammatory molecule.97,98 Unfortunately, however, early studies that suggested a possible therapeutic role for APC in human sepsis have not been borne out in clinical trials.99 Deletion of the EPCR gene by homologous recombination leads to early embryonic lethality around day 9.5,100 at which time EPCR is highly expressed in the giant trophoblasts of the placenta, but not in the embryo itself.86 In contrast to TM knockout animals,101 the placentas of EPCR knockout embryos show significant fibrin deposition at the fetal maternal interface.
VASCULAR FIBRINOLYSIS
Plasmin, the major clot-dissolving protease in humans, is formed upon the cleavage of a single peptide bond within the zymogen plasminogen (Chap. 135). This tightly regulated reaction is strongly influenced by cells of the blood vessel wall, including endothelial cells, smooth muscles cells, and macrophages, which express plasminogen activators, plasminogen activator inhibitors, and fibrinolytic receptors.
In 1958, Todd demonstrated that human blood vessels possess fibrinolytic activity that is dependent upon an intact endothelium.102,103 We now know that the endothelium is the principal source of t-PA in vivo where it appears to be highly restricted to small blood vessels in specific anatomic locations, a pattern that likely reflects the heterogeneity of endothelial cells as they respond to a myriad of tissue-specific cues.104,105 In the baboon, for example, sites of t-PA production include 7 to 30 μm precapillary arterioles and postcapillary venules, but not large arteries and veins.106 In the mouse lung, similarly, bronchial, but not pulmonary, endothelial cells express t-PA.107 Moreover, enhanced expression of t-PA at branch points of pulmonary blood vessels may reflect stimulation by laminar shear stress.108 In addition, peripheral sympathetic neurons that invest the walls of small arteries may represent a significant source of circulating t-PA.109
Although in vitro studies suggest that t-PA expression in cultured endothelial cells is regulated by a wide array of factors, only a few of these pathways have been confirmed in vivo. Thrombin,110 histamine,111,112 oxygen radicals,113 phorbol myristate acetate,114 DDAVP (deamino D-arginine vasopressin),115 and butyric acid liberated from dibutyryl cAMP116 all increase t-PA mRNA in cultured endothelial cells. Both thrombin and histamine appear to act via receptor-mediated activation of the protein kinase C pathway.105 Laminar shear stress stimulates both t-PA secretion117 and steady-state mRNA levels.118 Hyperosmotic stress and repetitive stretch also enhance t-PA expression.119,120 In addition, differentiating agents, such as retinoids,121,122 stimulate transcription of t-PA in endothelial cells in vitro.
In vivo, the circulating half-life of t-PA is approximately 5 minutes. Infusion of DDAVP, bradykinin, platelet-activating factor (PAF), endothelin, or thrombin is associated with an acute release of t-PA, and a burst of fibrinolytic activity can be detected within minutes.123 In the mouse lung, exposure to hyperoxia leads to 4.5-fold upregulation of t-PA mRNA in small-vessel endothelial cells.107 In humans, infusion of TNF into patients with malignancy is associated with an increase in plasma t-PA.123 Deficient release of t-PA in response to venous occlusion in humans is associated with deep venous thrombosis,124 as well as atrophie blanche and other cutaneous vasculitides.125
In vivo, urokinase plasminogen activator (u-PA) is not a product of resting endothelium,126 but is produced primarily by renal tubular epithelium.127 Expression of u-PA mRNA in endothelium, however, is strongly stimulated during wound repair and physiologic angiogenesis within ovarian follicles, corpus luteum, and maternal decidua.128 Endothelial cells passaged in culture do synthesize u-PA,129 and expression of its mRNA is stimulated by TNF-α by 5- to 30-fold.130 Small increases in u-PA have also been observed in vitro in response to IL-1 and LPS.131,132,133
The association of u-PA with the blood vessel wall appears to reflect its association with the u-PA receptor (uPAR) which may fulfill a variety of nonproteolytic functions ranging from directed cell migration to cellular adhesion, differentiation, and proliferation (Fig. 115–6).134 In the adult mouse, uPAR mRNA is not normally detected by in situ hybridization in the endothelium of either large or small blood vessels.135 However, upon stimulation with endotoxin, expression is detected in endothelium lining aorta, as well as arteries, veins, and capillaries in heart, kidney, brain, and liver,135 and in renal tubular epithelial cells.127
Figure 115–6.
Schematic of principal endothelial cell fibrinolytic receptors. A. The annexin A2/S100A10 heterotetrameric complex. Annexin A2 consists of a hydrophilic aminoterminal tail domain (A-Tail, approximately 3 kDa), and a membrane-oriented carboxyl terminal core domain (approximately 33 kDa).311,312 The tail domain contains residues required for tissue-type plasminogen activator (t-PA) binding. The core domain is composed of four homologous annexin repeats (A1, A2, A3, and A4), each consisting of five α-helical regions that contribute to calcium-dependent phospholipid binding sites. Repeat 2 appears to be most important for the interaction of annexin A2 with the endothelial cell surface. Plasminogen (PLG) binding requires lysine residue 307 within helix C of repeat 4. B. Urokinase plasminogen activator receptor (uPAR) is a 55- to 60-kDa, glycosylphosphatidylinositol-linked protein that consists of three disulfide-linked domains (U1, U2, U3).314 Domain 1 contains sequences required for urokinase plasminogen activator (u-PA) binding, while domains 2 and 3 mediate the receptor’s interaction with matrix proteins such as vitronectin. Domain 3 contains glycosylphosphatidylinositol-linked membrane anchor. (A, adapted with permission from Gerke V, Creutz CE, Moss SE: Annexins: linking Ca2+ signalling to membrane dynamics. Nat Rev Mol Cell Biol 6(6):449–461.)
Plasminogen activator inhibitor (PAI)-1 is likely to function as a major regulator of plasmin generation in the vicinity of the endothelial cell. Thrombin, IL-1, transforming growth factor β, TNF, lipoprotein(a) (Lp[a]), and LPS all induce dramatic increases in steady state PAI-1 message levels.110,131,132,136,137 Heparin-binding growth factor 1 reduces PAI-1 mRNA production by cultured endothelial cells, but has no effect on t-PA.138 Thus, synthesis and secretion of PAI-1 by the endothelial cell in vitro appears to be regulated independently of t-PA.
In vivo, elevated levels of circulating PAI-1 have been linked epidemiologically to risk for myocardial infarction.124 Although the liver is the major source of plasma PAI-1, endothelial expression of PAI-1 is detected near neovascular sprouts during decidual neovascularization in the ovary.128 In addition, inflammatory cytokines are powerful stimuli for induction of PAI-1 in a variety of tissues including liver, as injection of TNF in both rats and humans with active malignancy results in a striking increase plasma concentrations of PAI-1.105,123
The endothelial cell coreceptor for t-PA and plasminogen, the annexin A2/S100A10 complex (see Fig. 115–6), appears to be expressed constitutively in vivo by endothelial cells in a wide variety of tissues in the chicken,139 mouse,140 rat,141 and human.142 Annexin A2 is upregulated transcriptionally by hypoxia both in vivo and in endothelial cells in vitro,143 and by nerve growth factor in neuronal-like PC12 cells.144 In addition, the in vitro transition of human monocyte to macrophage is associated with a severalfold increase in both annexin A2 protein and steady state mRNA expression.145
The evidence that the annexin A2 system plays a role in maintaining vascular patency includes the findings that (1) overexpression of annexin A2 in blast cells in acute promyelocytic leukemia blast cells increases plasmin production and contributes to hyperfibrinolytic bleeding,146,147,148,149 (2) systemic injection of annexin A2 diminishes thrombotic vascular occlusion resulting from vascular injury in experimental animals,150 (3) annexin A2–deficient mice display fibrin deposition on microvessels and impaired clearance of arterial thrombi following vascular injury,151 (4) high titer antibodies directed against annexin A2 are associated with thrombosis in antiphospholipid syndrome and in individuals with cerebral venous thrombosis,152,153 and (5) that polymorphisms in the ANXA2 gene are associated with cerebral vascular occlusion and osteonecrosis of bone in patients with sickle cell disease.154,155,156 Whether defects in S100A10, which could serve either as a chaperone for annexin A2 or as a direct binding site for plasminogen,157 might also be associated with these clinical entities remains to be determined.
Although not yet demonstrated in vivo
Related posts:
 Hematopoietic Stem Cells, Progenitors, and Cytokines
Examination of the Marrow
Therapeutic Apheresis: Indications, Efficacy, and Complications
Hemolytic Anemia Resulting from Infections with Microorganisms
Structure and Composition of the Erythrocyte
Disorders of Hemoglobin Structure: Sickle Cell Anemia and Related Abnormalities
Hematopoietic Stem Cells, Progenitors, and Cytokines
Examination of the Marrow
Therapeutic Apheresis: Indications, Efficacy, and Complications
Hemolytic Anemia Resulting from Infections with Microorganisms
Structure and Composition of the Erythrocyte
Disorders of Hemoglobin Structure: Sickle Cell Anemia and Related Abnormalities
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


