Unusual Nonepithelial Tumors of the Head and Neck
 GLOMUS TUMORS
GLOMUS TUMORS
Anatomy
Glomus bodies are found in the jugular bulb and along the tympanic (Jacobson) and auricular (Arnold) branches of the tenth nerve in the middle ear or in other anatomic sites (Fig. 48.1). Depending on the location, glomus tumors (chemodectoma or paraganglioma) are classified as tympanic (middle ear), jugulare, or carotid vagal or designated as originating from other locations, such as the larynx, adventitia of thoracic aorta, abdominal aorta, or the surface of the lungs (Fig. 48.2).1
Glomus tumors (GT) or chemodectomas consist of large epithelioid (smooth muscle) cells with fine granular cytoplasm embedded in a rich capillary network and fibrous stroma with reticulin fibers, which derive from embryonic neural crest cells. Although histologically benign, they may extend along the lumen of the vein to regional lymph nodes, but rarely to distant sites. These tissues are responsive to changes in oxygen and carbon dioxide tensions and pH.
FIGURE 48.1. Anatomy of the region of the glomus jugulare. (From Hatfield PM, James AE, Schulz MN. Chemodectomas of the glomus jugulare. Cancer 1972;30:1165–1168, with permission.)

FIGURE 48.2. Distribution of paragangliomas of the head and neck region. Laterality was not specified in three patients with carotid body paragangliomas. The diagram does not include one left carotid body paraganglioma that was found incidentally at autopsy and a left vagal body paraganglioma that presented in a patient who had two other paragangliomas. (From Lack EE, Cubilla AL, Woodruff JM, et al. Paragangliomas of the head and neck region. Cancer 1977;39:3997–4009, with permission.)
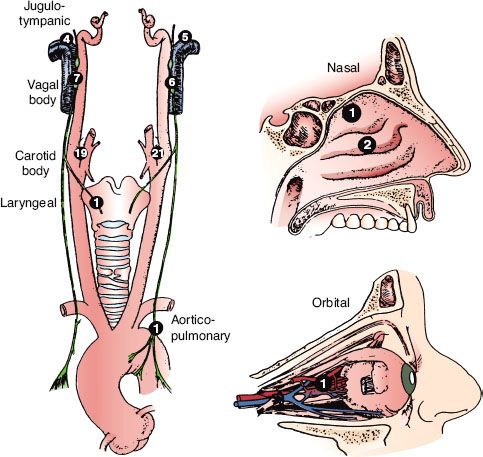
FIGURE 48.3. A: Late-phase arteriogram illustrating large glomus jugulare tumor with extension into the neck. B: Computed tomography scan with contrast enhancement showing intracranial component of lesion.
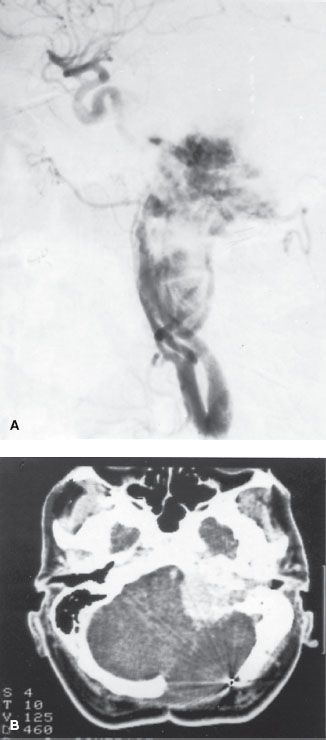
TABLE 48.1 DIAGNOSTIC WORKUP FOR GLOMUS TUMORS OF THE EAR AND BASE OF SKULL, HEMANGIOPERICYTOMA, ESTHESIONEUROBLASTOMA, EXTRAMEDULLARY PLASMACYTOMA, AND SARCOMA OF THE HEAD AND NECK

Epidemiology
The mean age at diagnosis has been reported to be 44.7 years for carotid body tumors and 52 years for glomus tympanicum.2 These tumors occur three or four times more frequently in women than in men, suggesting a possible estrogen influence.2,3,4 Glomus tumors may be familial; they occur in multiple sites in 10% to 20% of patients.5
Recent advances in genetics identified three loci associated with hereditary paragangliomas, and genetic screening may detect affected patients.6 Multiple paragangliomas of the head and neck are rare, with an incidence of 10% of all patients, but in familial cases it increases up to 35% to 50%.5 In the head and neck region, the most common association is bilateral carotid body tumors or carotid body tumor associated with tympanic–jugular glomus.7
Clinical Presentation
Glomus tumors may arise along the nerve roots. In the middle ear they may initially cause earache or discomfort.97 As they expand, eventually they produce pulsatile tinnitus, hearing loss, and, in later stages, cranial nerve paralysis resulting from invasion of the base of the skull in 10% to 15% of patients. If the tumor invades the middle cranial fossa, symptoms may include temporoparietal headache, retro-orbital pain, proptosis, and paresis of cranial nerves V and VI. If the posterior fossa is involved, symptoms may include occipital headache, ataxia, and paresis of cranial nerves V to VII, IX, and XII; invasion of the jugular foramen causes paralysis of nerves IX to XI. Chemodectoma of the carotid body usually presents as a painless, slowly growing mass in the upper neck. Occasionally the mass may be pulsatile and may have an associated thrill or bruit. As it enlarges, the mass may extend into the parapharyngeal space and be visible on examination of the oropharynx. Very rarely these tumors may be malignant.8 Metastases occur in 2% to 5% of cases.3
Diagnostic Workup
Diagnostic evaluation for glomus tumors of the ear and base of skull is outlined in Table 48.1. In the majority of glomus tympanicum, physical examination demonstrates a red, vascular middle ear mass, although occasionally it may be bluish or white (the latter resembling a cholesteatoma).2 Audiography may demonstrate conductive hearing loss in the ear involved by tumor as noted in 33 of 49 patients evaluated by Larson et al.;2 4 of 33 patients with conductive deficits also exhibited tympanic pulsations. Examination of the neck may occasionally demonstrate a mass in the neck that may be pulsatile or have a bruit or regional lymph node metastases.
Radiographic studies are invaluable in the diagnosis of these tumors. Plain mastoid radiographs never show the soft tissue mass in the middle ear, although they frequently demonstrate clouding of the mastoid air cells, suggesting mastoiditis.157 High-resolution computed tomography (CT) with contrast has a degree of sensitivity and specificity to diagnose this tumor when located in the middle ear or jugular bulb; masses as small as 3 mm have been demonstrated. Tumor enhancement is similar to that of the temporalis muscle (Fig. 48.3).2 In 46 patients with glomus tympanicum, there were no instances of local bony erosion; instead, the tumors engulfed the ossicular chain, bulged or protruded through the tympanic membrane, filled the middle ear, or extended into the eustachian tube orifice or aditus ad antrum. This pattern is in contrast to cholesteatomas, which typically destroy adjacent bony landmarks, including the ossicles, and progressively erode the petrous bones as they enlarge.2
Magnification angiography is a sensitive and specific means of detecting glomus tympanicum tumors. This procedure should be performed after high-resolution thin-section CT scan (with contrast material), only when there is a question regarding the nature of the lesion or the location of the carotid canal. Findings include a hypervascular middle ear mass that first appears in the middle to late arterial phase, persists through the capillary phase, and quickly disappears in the venous phase without demonstrably early draining veins. Biopsy of an aberrant internal carotid artery can result in major neurologic sequelae or death.
Vogl et al.9 reported on 40 patients with glomus tumors of the skull; diagnostic interpretations were correlated with histologic examination, digital subtraction angiography, CT, and clinical follow-up. Sixteen of 18 proven tumors were detected with spin-echo images alone. Although four high-flying jugular bulbs were misinterpreted as tumors because of similar signal intensity, combined evaluation allowed differentiation between tumor and sinusal blood flow in all cases.
Drape et al.10 described magnetic resonance imaging (MRI) findings in 31 patients with a clinical suspicion of glomus tumor; gadoterate meglumine was injected into 19 patients. Twenty-seven of 28 pathologically confirmed glomus tumors were detected with MRI; a peripheral capsule was present in most tumors. The investigators were able to differentiate three subtypes of glomus tumors (vascular, solid, and myxoid) on the basis of relaxation times and enhancement characteristics. Multidetector CT angiography was found to be more accurate in the diagnosis of six glomus tumors, with enhancement in the arterial phase, when compared with MRI.11
As GTs show high levels of somatostatin receptor (SSTR) subtypes 2 and 5, fluorine-(18F)-octreotate positron emission tomography (PET) may be useful for diagnostic purposes in a semiquantitative manner and for improving target volume delineation in radiation therapy planning. Astner et al.12 noted that preliminary findings with two different PET tracers for SSTR imaging have been reported: gallium-68 (68Ga)-DOTATOC (DOTA-d-Phe(1)-Tyr(3)-octreotide [somatostatin analog]) PET was shown to detect SSTR-expressing tumors with high sensitivity and specificity. A second PET tracer, Gluc-Lys18F-TOCA, allows fast, high-contrast imaging of SSTR-positive tumors with superior biokinetics and diagnostic performance as compared with indium-111 (111In)-DTPA (diethylene triamine pentaacetic acid)-octreotide and—as far as can be determined from the literature—comparable to 68Ga-DOTATOC.13,14,15
Cytochemical techniques demonstrate increased levels of serotonin, epinephrine, and norepinephrine in normal glomus tissue of the carotid body. Histologic staining techniques, including chromaffin and argentaffin reactions, identify patients with hormonally active tumors. This is important because the glomus tumor may coexist with a pheochromocytoma, which requires special preoperative preparation of the patient. Biopsy of glomus tumors may result in severe hemorrhage.
TABLE 48.2 GLASSCOCK-JACKSON CLASSIFICATION OF GLOMUS TUMORS

TABLE 48.3 MODIFICATION OF McCABE AND FLETCHER CLASSIFICATION OF CHEMODECTOMAS
Staging
The prognosis of these tumors is closely related to the anatomic location and the volume of the lesion, which is reflected in the Glasscock-Jackson classification16 shown in Table 48.2. An alternative classification proposed by McCabe and Fletcher17 is presented in Table 48.3.
General Management
Li et al.18 published a historical perspective of various treatment modalities used to treat glomus tumors over the past 60 years.
Surgery
Surgery is generally selected for small tumors that can be completely excised. Glomus tympanicum tumors are particularly well managed with excision via tympanotomy or mastoidectomy. Percutaneous embolization of a low-viscosity silicone polymer has been used, frequently as preoperative preparation of the tumor embolization of feeding vessels allows meticulous microsurgery with virtually complete hemostasis.
Surgical treatment of a glomus tumor arising in the jugular bulb, however, often consists of piece-by-piece removal accompanied by significant intraoperative bleeding with damage to adjacent neurovascular structures and requires more complex surgical approaches involving the base of the skull. Preoperative embolization via a transarterial approach has proved beneficial but is often limited by vascular anatomy and unfavorable locations. Abud et al.19 reported experience with preoperative devascularization using direct puncture and an intralesional injection of cyanoacrylate (acrylic glue) under fluoroscopic guidance in nine patients with head and neck paragangliomas. Ozyer et al.20 performed devascularization with intralesional injection of N-butyl-cyanoacrylate (seven carotid and three jugular paragangliomas). The tumors were subsequently surgically removed.
The local tumor control rate with surgery alone is only about 60%, and there is significant morbidity, particularly cranial nerve injury and bleeding.
In a retrospective review of all skull-base surgery cases treated at Baylor University, 175 jugulotympanic glomus tumors and 9 malignant cases (5.1%) were identified.8 The 5-year survival rate was 72%.
Radiation Therapy
Irradiation is frequently used in the treatment of glomus tumors, particularly for those in the tympanicum and jugulare bulb locations. Tumors with destruction of the petrous bone, jugular fossa, or occipital bone or patients with jugular foramen syndrome are more reliably managed with irradiation.2,4,21,22 Some surgeons, such as Glasscock et al.16 have questioned the effectiveness of radiation therapy in the treatment of chemodectomas because on histologic sections, obtained even many years after irradiation, it is possible to find chromophilic cells remaining in the tumor. However, there is also evidence of fibrosis and decreased vascularity.23 Suit and Gallager24 demonstrated in a murine mammary carcinoma model that morphologically intact cells may have lost their reproductive ability after irradiation, which is the ultimate end point of cell killing. Furthermore, it is extremely unusual to observe clinical regrowth of a glomus tumor after irradiation, even if they do not regress completely.
Some reports describe successful combinations of surgery with preoperative irradiation, in an attempt to make an unresectable tumor operable, or postoperatively when obvious tumor could not be resected.
Radiation Therapy Techniques
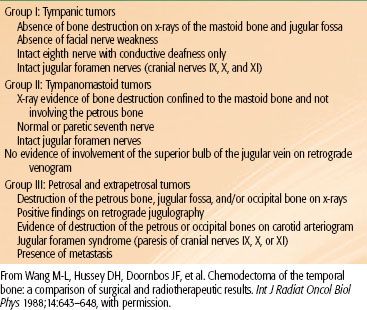
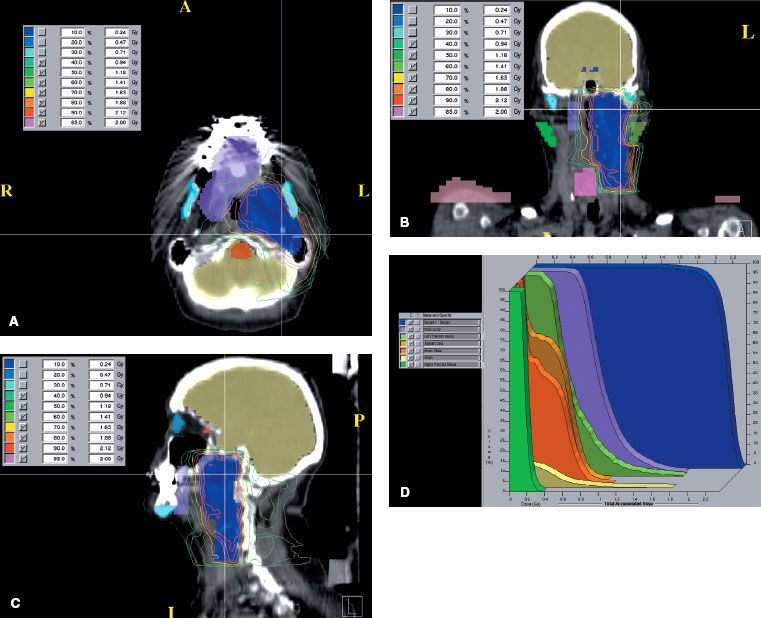
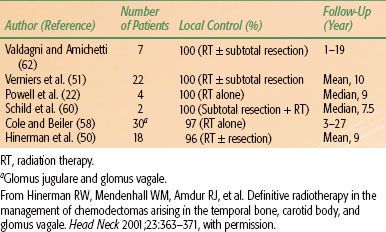
Radiation therapy techniques are determined by the location and extent of the tumor, which must be defined before treatment.21,25,26 Limited, usually bilateral, portals were used for relatively localized glomus tumors, whether or not the treatment is combined with surgery (Fig. 48.4). Dickens et al.27 used a three-field arrangement with a superior-inferior wedged and lateral open field, with a weighting of 1:1:0.33. A superior-inferior 60-degree and 45-degree wedged filtered field arrangement was also used. Electrons (15 to 18 MeV) with a lateral portal or combined with cobalt-60 (60Co) or 4- to 6-MV photons (20% to 25% of total tumor dose) render a good dose distribution (Fig. 48.5). Several prosthetic materials have been used to enhance irradiation dose homogeneity.28 In patients in whom tumor has spread into the posterior fossa, it may be necessary to use parallel opposed portals with 6- to 18-MV photons. Treatment is given at the rate of 1.8 to 2 Gy tumor dose per day with 5 treatments per week for a total tumor dose of 45 to 55 Gy in 5 weeks. Three-dimensional conformal radiation therapy (3D-CRT) or image-guided intensity-modulated radiation therapy (IMRT) are highly desirable techniques to treat these tumors, with excellent dose distributions (Fig. 48.5). Table 48.4 summarizes the doses of irradiation recommended by several investigators and the probability of tumor control for each.21,26,29
FIGURE 48.4. A: Portal used for relatively localized glomus tumor. B: Simulation film of patient with glomus tumor. C: Isodose distribution of a mixed-beam unilateral portal for a glomus tympanicum lesion (80% 16-MeV electrons, 20% 4-MV photons). (From Konefal JB, Pilepich MV, Spector GJ, et al. Radiation therapy in the treatment of chemodectomas. Laryngoscope 1987;97:1331–1335, with permission.)
FIGURE 48.5. Fifty-nine-year-old woman with an unusual malignant left glomus jugulare, who had a metastatic left upper cervical lymph node. She was treated definitively with intensity-modulated radiation therapy (66 Gy in 2-Gy fractions). Cross (A), coronal (B), and sagittal (C) sections showing dose distributions at primary site and left neck, sparing normal structures (D) dose–volume histogram:
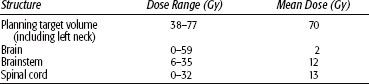
Leber et al.30 reported on 13 patients with glomus tumors treated with radiosurgery in 6, because of recurrences after surgical removal. Histology was not available in seven patients, diagnosis was made from neuroradiological features only. With mean follow-up of 42 months (range 14 to 72 months), there was no tumor progression and no clinical deterioration in any patient; 64% of the patients had improvement of symptoms, and in 36% the volume of the lesion decreased in size. There was no radiation-related morbidity. In recent years there has been an increasing number of reports in small series of patients with glomus tumors <2 or 3 cm treated with stereotactic radiation therapy, with tumor control over 80% and relatively minimal morbidity. The mean single dose is about 16 Gy (range 13 to 20 Gy).31,32,33,34,35,36,37 A report on fractionated stereotactic irradiation (6 MV x-rays) of 17 patients has been published, with a dose of 57 Gy.38 Pollock39 reported on 42 glomus tumors (19 primary treatment and 23 recurrences after initial surgery) treated with Gamma Knife (Elekta Corp, Stockholm) stereotactic radiation (12 to 24 Gy single dose at 50% isodose, depending on tumor size). Twelve lesions (31%) decreased in size and 26 were unchanged. The most common complication was hearing loss (19%). Wegner et al.40 treated 18 patients with carotid or jugular lesions with fractionated stereotactic CyberKnife (Accuray, Sunnyvale, CA) irradiation (21 Gy in 3 fractions or 25 Gy in 5 fractions) (Fig. 48.6). Tumor size was stable in 17 patients and decreased in 1 patient.
TABLE 48.4 LOCAL CONTROL WITH RADIATION THERAPY FOR CHEMODECTOMA OF THE TEMPORAL BONE (GLOMUS TYMPANICUM AND JUGULARE)

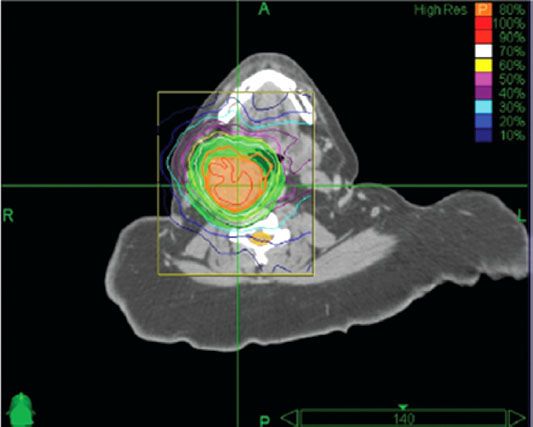
FIGURE 48.6. Dose distribution with stereotactic radiation therapy (radiosurgery) for glomus tumor. Patient was treated with 25 Gy in 5 fractions prescribed to the 80% isodose line; 98% of tumor received the prescribed dose. Thick orange line represents the 80% isodose. (From Wegner RE, Rodriguez KD, Heron DE, et al. Linac-based stereotactic body radiotherapy for treatment of glomus. Radiother Oncol 2010;97:395–398, with permission.)
Results of Therapy
The postirradiation change in tumor size is slow, with an increase in proliferative and perivascular fibrosis and minimal alterations in the chief epithelial cells. Histologic evaluation of tumor cell viability is not reliable.24 Despite the persistence of tumor both clinically and angiographically, amelioration of symptoms, absence of disease progression, and occasional return of cranial nerve function have been reported. Seventeen patients were treated for glomus tympanicum tumors at Washington University.3 In five patients, initial treatment consisted of irradiation alone, and all were tumor free at last follow-up (4.5 years in one patient) or at death. Seven of eight patients irradiated for surgical recurrence were free of disease 4.5 to 19 years after irradiation. The remaining four patients were treated preoperatively or postoperatively; only one had recurrence and was salvaged surgically and tumor free 10 years later. Of six patients with glomus jugulare lesions treated with irradiation, two with extensive lesions died of their disease, whereas the glomus tumor was controlled in four, including two patients with intracranial extension. Irradiation doses ranged from 46 to 52 Gy, with 86% to 100% tumor control with doses over 46 Gy and 50% (two of four) with doses below 46 Gy.
Wang et al.46 reported on 32 patients with tympanic chemodectomas; 13 treated with surgery alone, 15 with irradiation alone, and 4 with a combination of both modalities. The initial tumor control rate was 46% with surgery alone; ultimately 84% of patients were tumor free after salvage with additional surgery. Although 78% survived 10 years, 31% developed complications. Of the patients treated with irradiation, 84% had initial local tumor control; 77% survived 10 years, and only 11% developed complications. The doses of irradiation used were slightly higher than those reported by others (mean 58.32 Gy). However, no improvement in tumor control was noted with higher doses. Complications occurred in two patients receiving 66 Gy.
Zabel et al.47 described results in 22 patients with large chemodectomas of the skull base (8 after primary surgery and 4 after embolization), treated with fractionated stereotactic irradiation (median total dose 57 Gy with median fraction dose 1.8 Gy). With a median follow-up of 5.7 years, 5- and 10-year actuarial tumor control was 90%, with 7 patients (32%) having a partial response and 13 patients with (59%) stable tumors. No patient developed new neurological deficit.
In a compilation of several studies, Kim et al.48 noted a 25% local failure rate in 83 patients treated with <40 Gy and 1.4% local failure in 142 patients receiving >40 Gy.
Powell et al.22 reported on 84 patients with chemodectoma of the head and neck, 46 of which were in the glomus jugulare and tympanicum, treated with irradiation alone (45 to 50 Gy in 25 fractions). Local control of the lesion was 73% at 5 years. Thirty patients were treated with surgery after irradiation with no recurrences (median follow-up of 9 years). Four patients, treated with surgery alone, developed recurrences by 7 years. Four carotid body and glomus vagal tumors treated with irradiation were locally controlled at 1, 2, 8, and 11 years, respectively. In 13 patients treated with surgery alone, the 15-year local control rate was 54%.
Hinerman et al.49,50 updated a previous report with 104 patients who had 121 chemodectomas of the temporal bone, carotid bone, or glomus vagal treated with radiation therapy alone in 104 patients or subtotal resection with or without radiation therapy (17 tumors). Seventeen patients had undergone a previous treatment (surgery 14, irradiation 1, or both 2). Eighty-nine patients were treated with megavoltage radiation therapy, 15 with stereotactic fractionated, 6 with stereotactic single dose, and 11 with IMRT. Median dose with fractionated irradiation was 45 Gy with daily fractions of 1.5 to 2 Gy delivered with 60Co, 6-MV, or 8-MV x-rays, or a combination of different beam energies.50 There were six tumor recurrences, with a local tumor control of 95%. No severe treatment complications were noted. In the initial report,49 18 patients had 25 chemodectomas of carotid body and/or glomus vagal; 15 tumors originated in the carotid body and 10 in the glomus vagal. Pathologic confirmation of chemodectoma was obtained in 10 patients, and diagnosis was based on physical and radiographic findings in the remaining 8 patients. Twenty-two lesions were treated with radiation therapy alone and two received postoperative radiation therapy after surgical resection for gross residual tumor with malignant changes and lymph node involvement. Patients with benign glomus tumors received 45 Gy in 25 fractions, in most instances, whereas patients with malignant carotid body tumors received 64.8 to 70 Gy in 1.8-Gy fractions. Local tumor control was obtained in 14 of 15 carotid body and 10 of 10 glomus vagal (overall 96% tumor control).51
Ivan et al.52 published a meta-analysis based on 869 patients in 46 studies reported with glomus tumors, with follow-up ranging from 6 to 256 months. The tumor control rates were, for subtotal resection 69%, gross tumor resection (GTR) 86%, subtotal resection and radiosurgery 71%, and stereotactic radiosurgery (SRS) alone 95%. Posttreatment cranial nerve deficit was observed in 26% to 40% of patients treated with GTR and in about 10% of the SRS group. Guss et al.53 reported on a meta-analysis of 19 studies (335 patients) with glomus tumors treated with stereotactic radiation therapy (radiosurgery), eight publications with a median follow-up of 36 months; tumor control (unchanged or reduced tumor volume) was 95% to 96%, with various stereotactic radiation therapy (RT) techniques.
In 29 patients with post surgical recurrent glomus tumors (16 jugular, 7 carotid, 5 tympanic and one thyroid) Elshaikh et al.54 reported that the 5 year tumor conrol was 100% in 12 treated with radiation therapy and 62% in 17 treated surgically.
Cheesman and Kelly55 emphasized the importance of evaluating preoperatively the swallowing function of patients with glomus jugulare undergoing surgery, as it is common for these patients to develop postsurgical dysphagia.
The results of primary treatment for temporal bone chemodectoma are summarized in Table 48.5.
The initial results of treatment for carotid body or glomus vagal are summarized in Table 48.6. Complications were rare in patients treated with chemodectoma of the head and neck.
TABLE 48.5 TEMPORAL BONE CHEMODECTOMAS: LOCAL CONTROL AFTER RADIATION THERAPY ALONE OR RADIATION THERAPY AND SURGERY
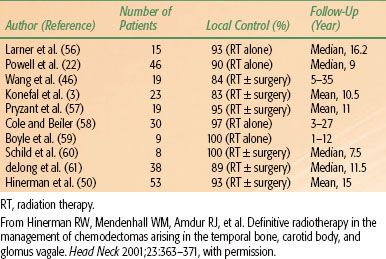
TABLE 48.6 CHEMODECTOMAS OF CAROTID BODY/GLOMUS VAGALE: RADIATION THERAPY ALONE OR RADIATION THERAPY AFTER SURGERY
 HEMANGIOPERICYTOMA
HEMANGIOPERICYTOMA
Hemangiopericytomas are rare soft tissue neoplasms that account for 3% to 5% of all soft tissue sarcomas and 1% of all vascular tumors. Some 15% to 30% of all hemangiopericytomas occur in the head and neck, and of these, approximately 5% occur in the sinonasal area. They may resemble meningiomas in the central nervous system (CNS), clinically and on imaging studies.63 Vagal paragangliomas originate within the first 2 cm of the extracranial stretch of the vagus nerve and are associated with the inferior ganglion.64 These tumors are believed to originate from the pericytes of Zimmerman extravascular cells, morphologically resembling smooth muscle, found around the capillaries or from primitive mesenchymal cells. The function of the pericyte is uncertain but is believed to provide mechanical support for the capillaries having contractile function.65
Epidemiology
Hemangiopericytomas is an unusual tumor; it represents approximately 1% of all vascular neoplasms; it occurs in both genders with equal frequency and is found primarily in adults. Only 45 cases of primary hemangiopericytomas of bone were described in the world literature in 1988.
In the head and neck, the most common sites are the nasal cavity and the paranasal sinuses, and, less frequently, the orbital region, the parotid gland, and the neck.66,67,68,69 Hemangiopericytomas represent 3% to 4% of all meningeal and <1% of CNS tumors.
Pathology
Hemangiopericytomas are composed of a proliferation of tightly packed pericytes around thin-walled endothelial-lined vascular channels, ranging from capillary-sized vessels to large, gaping sinusoidal spaces.66 The tumor has a tendency to grow slowly and invade locally into adjacent structures.68,70 Although they are always well circumscribed and partially or completely surrounded by a pseudocapsule, benign tumors may be difficult to differentiate from malignant tumors. However, prominent mitoses (>4 per high-power field), foci of necrosis, and increased cellularity are suggestive of malignancy; the definitive sign is local recurrence or development of metastases. In general, tumors of the CNS, lower extremity, and mediastinum tend to be more malignant, with local recurrence occurring in up to 50% of cases.66
Kowalski and Paulino71 reviewed 12 cases of hemangiopericytomas. Proliferation index was assessed using an immunoperoxidase stain for MIB-1 (Ki-67). The mitotic index per 10 high power fields varied from 0 or 1 to 15. Proliferation indices using MIB-1 ranged from 2.6% to 52.5%. Clinical follow-up revealed three cases with recurrence all possessing proliferation indices of approximately 10%, indicating a more aggressive subset of hemangiopericytomas. Vuorinen et al.72 found the proliferation index to be a poor predictor of prognosis. In a review of 23 cases, Sundaram et al.63 found that all hemangiopericytomas were negative for epithelial membrane antigen and S-100 and all were positive for vimentin.
Meningeal hemangiopericytomas almost always recur, despite seemingly complete removal, due to infiltrative properties of hemangiopericytoma cells and not just higher proliferation potential. They often metastasize.
Clinical Presentation
Soft tissue hemangiopericytoma is a firm, painless, slowly expanding mass that is often nodular and well localized. The skin overlying the mass does not have any discoloration or redness to indicate its vascular origin because the capillaries are emptied of the blood by compression of massive numbers of pericytes surrounding them.64,66
In the head and neck, the tumor may constitute a polypoid, soft gray or red mass that grows slowly and may cause nasal obstruction. Epistaxis and nasal obstruction are common symptoms. Orbital hemangiopericytomas account for 3% of orbital malignancies and most frequently occur with painless proptosis.73 Hemangiopericytoma rarely originates in the lacrimal sac; it occurs in a younger age group than that of hemangiopericytoma of other locations. Charles et al.74 reported on seven cases previously described and added one case.
Hemangiopericytoma may occur intracranially. When it arises in the brain, it is a solid mass attached to the meninges that grossly resembles a meningioma. These intracranial hemangiopericytomas carry a high risk of local failure (80%), as well as higher potential for dissemination. The mean time for local recurrence is 75 months.75
The incidence of metastasis, which depends on the site of origin, can be 50% to 80%. Late metastases occurring 10 years after diagnosis are not uncommon.
On plain radiographs, hemangiopericytoma appears as a soft tissue mass in the nasal cavity or other portions of the head and neck. A defect caused by pressure erosion of the surrounding bones may occur, and calcifications are rare. In the neck, the tumor appears as a well-circumscribed, homogeneously and intensely enhancing mass on CT. On MRI, the mass is iso- to slightly hyperintense to muscle on T1- and T2-weighted imaging. Multiple, branching flow voids are typically seen within the tumor on both T1- and T2-weighted images. On T2-weighted imaging, the punctate black flow voids in cross-section within the relatively bright tumor, creating a characteristic “salt and pepper” appearance in tumors >2 cm in diameter. Additionally, flow voids of large feeding arteries are seen at the periphery of the mass. Angiography demonstrates the characteristic appearance of a vascular tumor, with large feeding arteries, intense tumor stain, and early draining veins.64 On arteriography, according to Yaghmai,76 hemangiopericytoma is the only vascular tumor that has radially arranged or spiderlike branching vessels around and inside the tumor and a long-standing, well-demarcated tumor stain. Intracranial tumors typically have arterial blood supply from both meningeal and cerebral connections, with one to three main feeders supplying many small corkscrew-like vessels.75 The most distinctive and constant feature of this tumor is its hypervascularity, which may be demonstrated with contrast-enhanced CT.67 Intracranially, the diffusely enhancing tumor may closely resemble a meningioma on CT. However, some CT signs may suggest hemangiopericytoma rather than meningioma, such as a lack of calcification, scarce surrounding edema, and ringlike enhancement. Both CT and MRI scans are of special value in the delineation of the full extent of the tumor.
General Management
Complete surgical resection, if possible, combined with preoperative embolization of the tumor, is the treatment of choice. More extensive surgery is required in tumors that show features of malignancy. Many patients undergo surgical treatment after embolization of the feeding artery(ies).
For incompletely resected tumors, postoperative radiation therapy is used.77,78 The role of chemotherapy in this tumor is not well determined; a few reports have described partial tumor regression in some lesions treated with cytotoxic agents. Doxorubicin, alone or in combination-drug regimens, is the most effective agent for metastatic hemangiopericytoma, producing complete and partial remissions in 50% of cases. Other drugs prescribed when metastasis occurs are cyclophosphamide, dacarbazine, vincristine, and actinomycin-D.79 Park et al.80 reported on 14 patients with soft tissue hemangiopericytoma treated with temozolomide 150 mg/m2 orally on days 1 to 7 and days 15 to 21 and bevacizumab 5 mg/kg intravenously on days 8 and 22, repeated at 28-day intervals. Median follow-up period was 34 months. Eleven patients (79%) achieved a Choi partial response, with a median time to response of 2.5 months. The estimated median progression-free survival was 9.7 months, with a 6-month progression-free rate of 78.6%. The most frequently observed toxic effect was myelosuppression.
Radiation Therapy Techniques
The role of radiation therapy alone in the management of hemangiopericytoma is controversial. The main role of irradiation is as an adjuvant after complete excision of the lesion or postoperatively for minimal residual disease.69,81,82 The tumor has been considered relatively radioresistant. Tumor doses of 60 to 65 Gy in 6 to 7 weeks are required to produce local tumor control in postoperative cases.83 Orbital hemangiopericytoma has been cured by surgery and postoperative irradiation to 65 Gy.73
There appears to be a definite role for postoperative irradiation to the brain for primary hemangiopericytoma when radical surgery is performed because these tumors tend to recur after seemingly complete removal. Jha et al.82 reported local tumor control in all patients treated with adjuvant external-beam irradiation postoperatively. Radiation therapy also has been used as a salvage procedure after local recurrence following initial surgery or chemotherapy.
The fields of irradiation should be wide to encompass the tumor bed with a margin of at least 5 cm to safely avoid marginal recurrence. Portal arrangement and beam selection are similar to those used in treatment of malignant brain tumors or soft tissue sarcomas.
Results of Therapy
Billings et al.84 reported on 10 patients with hemangiopericytoma of the head and neck; seven tumors arose from soft tissue sites and three from the mucosa. All patients underwent wide excision of the primary lesion with a local recurrence rate of 40%. Three patients developed metastatic lung disease 0 to 8 years after initial diagnosis. Each patient who developed metastatic disease had abundant mitoses on pathological review compared with rare or absent mitoses in the lesions that took a more benign course.
Patrice et al.85 reported on 18 primary hemangioblastoma tumors (16 had no prior surgical resection and 2 were subtotally resected lesions) and 20 lesions treated after surgical failure with stereotactic irradiation (radiosurgery). Minimum tumor doses ranged from 12 to 20 Gy (median 15.5 Gy). With a median follow-up of 24.5 months (range 6 to 77 months), the 2-year actuarial survival was 88%, and the 3-year freedom from progression was 86%. Four of 22 patients died. Thirty-one of 36 evaluable tumors (86%) were controlled locally. None of the 18 primary tumors treated with definitive stereotactic irradiation failed. Of the 18 recurrent tumors, 13 (72%) were controlled. There were no significant permanent complications attributable to the stereotactic irradiation.
Spitz et al.69 published a report on 36 patients with hemangiopericytoma. The median follow-up was 57 months. Twenty-eight patients (78%) underwent complete and potentially curative resection. Of the nine patients (32%) who had local recurrences, four (44%) had epidural tumors and three (33%) had retroperitoneal tumors, but none had extremity tumors. Ten patients had recurrences at distant sites. Of the 13 patients who experienced any form of disease recurrence, four had recurrences after a disease-free interval of more than 5 years. The 5-year actuarial survival rate for the entire group of 36 patients was 71%.
Carew et al.86 reviewed the records of 12 patients with hemangiopericytomas of the head and neck: 5 had high or intermediate grade lesions and 7 had low-grade lesions. Nine patients were treated with curative intent; they underwent a variety of surgical resections dictated by tumor location and size. Four patients received postoperative radiation therapy, to a median dose of 60 Gy, for positive surgical margins (two patients), high-grade histology (one patient), or a recurrent lesion (one patient). The 5-year overall survival rate for patients treated surgically was 87.5%. A single mortality occurred in a patient with a recurrent high-grade lesion who failed at local, regional, and distant sites.
Staples et al.87 reported on 12 patients with localized hemangiopericytoma, 7 treated with surgery alone (only 1 had long-term tumor control and 2 were salvaged with radiation therapy), 4 with resection and postoperative irradiation (all with long-term tumor control), and 1 with surgery and chemotherapy. Local tumor control was achieved at all sites treated with doses >55 Gy. Mitotic activity was not a reliable predictor of biologic behavior.
Kim et al.48 evaluated 17 hemangiopericytomas in nine patients treated with Gamma Knife stereotactic radiation therapy. Mean and median marginal doses were 18.1 and 20 Gy (range 11 to 22 Gy), respectively, at the 50% isodose line. Mean clinical and radiological follow-up periods were 49 and 34 months, respectively. Successful tumor control was achieved in 14 of 17 lesions (82.4%). Actuarial local tumor control rates at 5 years was 67%. No adverse effects, such as radiation necrosis or marked peritumoral edema, were observed. Marginal dose (≥17 Gy) was the only statistically significant factor for local tumor control on univariate analysis.
Kano et al.88 in a retrospective review of 20 patients who had undergone stereotactic radiation therapy for 29 hemangiopericytomas. All patients had undergone previous surgical resection. In addition, 12 patients underwent fractionated radiotherapy before stereotactic radiation therapy. Of the 20 patients, 16 patients had low-grade hemangiopericytomas (20 tumors) and 4 had high-grade anaplastic hemangiopericytomas (9 tumors). The median target volume was 4.5 cm3 and the median marginal dose was 15 Gy (range 10 to 20 Gy). At an average of 48.2 months, the overall survival after radiosurgery was 85.9% and 13.8% at 5 and 10 years, respectively. Follow-up imaging studies demonstrated tumor control in 21 (72.4%) of 29 tumors. The progression-free survival rate after stereotactic radiation therapy at 3 and 5 years was 89.1% for low-grade hemangiopericytomas and 66.7% and 0%, respectively, for high-grade hemangiopericytomas. The factors associated with improved progression-free survival included lower grade and >14 Gy marginal radiation dose.
Olson et al.89 published a review of 21 patients with 28 recurrent or residual hemangiopericytomas on whom radiosurgery was performed. Prior treatments included embolization (6 cases), transcranial resection (39 cases), transsphenoidal resection (2 cases), and fractionated radiotherapy (8 cases). The mean prescription and maximum radiosurgical doses to the tumors were 17.0 and 40.3 Gy, respectively. Repeat radiosurgery was used to treat 13 tumors. With median follow-up of 68 months (range 2 to 138 months), local tumor control was 47.6% (10 of 21 patients). Of the 28 tumors treated, 8 decreased in size on follow-up imaging (28.6%), 5 remained unchanged (17.9%), and 15 ultimately progressed. Progression-free survival at 5 years was 28.7%, and it improved to 71.5% after multiple radiosurgery treatments. Prior fractionated irradiation or radiosurgical prescription dose did not correlate with tumor control. In 4 of 21 (19%) patients, extracranial metastases developed. Redmond et al.90 reported on 118 patients with hemangiopericytoma of the CNS, 9% of whom had distant metastases at the time of initial presentation; 112 patients underwent surgical resection (23% had GTR, 31% subtotal resection, and 46% had surgery not otherwise specified). Adjuvant RT was received by 31% of patients following GTR, 44% after subtotal tumor resection, and 50% of patients following surgery not otherwise specified. The 5- and 10-year overall survival for all patients was 76.7% and 50.1%, respectively. Patients receiving adjuvant RT (n = 42) had a significantly better overall survival than patients who did not (n = 67; 10-year overall survival was 66.2% vs. 40.7%; P = .05). There was no difference in 5- or 10-year overall survival for patients treated with subtotal resection plus RT (n = 16) compared with those treated with GTR alone (n = 14; 10-year overall survival 75% vs. 57.1%; P = .53).
 CHORDOMAS
CHORDOMAS
Anatomy
Chordomas are rare neoplasms of the axial skeleton that arise from the remnant of the primitive notochord (chorda dorsalis). About 50% arise in the sacrococcygeal area; 35% arise intracranially, where they typically involve the clivus, and the remaining 15% occur in the midline along the path of the notochord, primarily involving the cervical vertebrae.91
Epidemiology
Chordomas are more common in patients in their 50s and 60s but can occur in all age groups. In children and young adults, the prognosis and long-term survival appear to be better than in older patients. No risk factors have been identified. Male predominance is reported at a 2:1 to 3:1 ratio.
Natural History
Although slowly growing, chordomas are locally invasive, destroying bone and infiltrating soft tissues. Basisphenoidal chordomas tend to cause symptoms earlier and may be difficult to differentiate histologically from chondromas and chondrosarcomas and radiographically from craniopharyngiomas, pineal tumors, and hypophyseal and pontine gliomas. The lethality of these tumors rests on their critical location, aggressive local behavior, and extremely high local recurrence rate. The incidence of metastasis, which has been reported to be as high as 25%, is higher than previously believed and may be related to the long clinical history. The most common site of distant metastasis is the lungs, followed by liver and bone. Lymphatic spread is uncommon.
Pathology
Chordoma is a soft, lobulated tumor that may have areas of hemorrhage, cystic changes, or calcification. It is frequently encapsulated but may be nonencapsulated or pseudoencapsulated. Histologically, it is composed of cords or masses of large cells (physaliferous cells) with typical vacuoles and granules of glycogen in the cytoplasm and abundant intercellular mucoid material. Usually there are few mitotic cells.92 A chondroid variant of chordoma may exist, being prevalent in the spheno-occipital area. Patients with this type of histologic variant have improved survival.
Aside from the previously mentioned histologic features, the prognostic factors that most influence the choice of treatment are location and local extent of tumor.
Clinical Presentation
Chordomas tend to originate from the clivus and chondrosarcomas from the temporal bone.93 Clinical symptoms vary with the location and extent of the tumor. In the head, extension may be intracranial or extracranial, into the sphenoid sinus, nasopharynx, clivus, and sellar and parasellar areas, with a resultant mass effect. In chordomas of the spheno-occipital region, the most common presenting symptom is headache. Other presentations include symptoms of pituitary insufficiency, nasal stuffiness, bitemporal hemianopsia, diplopia, and other cranial nerve deficits. Volpe et al.94 reviewed the clinical features of 48 patients with chordoma and 49 patients with low-grade chondrosarcoma of the skull base. Twenty-five patients (52%) with chordoma and 24 patients (49%) with chondrosarcoma had ocular symptoms (diplopia or visual impairment) as the initial manifestation of the disease. Of the 59 patients (both groups) with diplopia, the diplopia was initially intermittent in 25 (42%). Headache and diplopia from abducens nerve palsy occurred in 22 patients (46%) with chordoma and 23 (47%) with chondrosarcoma.
TABLE 48.7 DIAGNOSTIC WORKUP FOR CHORDOMA

Diagnostic Workup
The diagnostic workup varies with the primary location of disease. Most patients have significant bony destruction, and some may have calcifications in the tumor; hence, plain films and, specifically, CT scans or MRI are very useful (Table 48.7).95 In most cases, the soft tissue component is much more extensive than initially appreciated, and a CT scan with contrast enhancement is required (Fig. 48.7A). CT and MRI are equivalent for demonstration of the presence and site of these tumors. MRI is inferior to CT in its ability to demonstrate bony destruction and intratumoral calcification (Fig. 48.7B).96 MRI is superior to CT regarding the delineation of the exact extent of the tumor, which allows for better treatment planning.95 Because of availability and lower cost, CT appears to be the technique of choice for routine follow-up of previously treated patients.96
Reliable signs of chordoma of the skull base are posterior extension to the pontine cistern; a lobulated, “honeycomb” appearance after gadolinium; the swollen appearance of the bone in the early stages; bone erosion on CT; and frequent extension to critical structures such as the circle of Willis, cavernous sinuses, and brainstem.96
General Management
Because of their surgical inaccessibility and relative resistance to radiation therapy, clivus chordomas represent a formidable therapeutic challenge. The general management of the patient is dictated by the anatomic location of the tumor and the direction and extent of spread. A surgical approach is recommended (when feasible), but complete surgical extirpation alone is unusual.97 Regression of preoperative symptoms without additional postoperative morbidity could be achieved by radical transoral tumor extirpation documented by MRI. Intracranial spread usually requires steroid coverage and therapy directed to correction of neurologic deficits that may be present. Because of the high incidence of local recurrence, combined surgical excision and irradiation is frequently used. No effective chemotherapeutic agent or combination of drugs has been identified.
Radiation Therapy Techniques
Irradiation techniques vary considerably, depending on the location of the tumor along the craniospinal axis. Basisphenoidal tumors usually have been treated by a combination of parallel opposed lateral fields, anterior wedges, and photon and electron beam combinations, depending on the extent of the neoplasm. Precision radiation therapy planning, using CT and MRI, is required because high doses of external-beam radiation therapy are needed. Three-dimensional-CRT or IMRT provide optimal dose distributions.
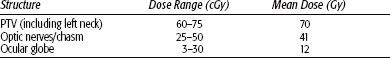
The tumor usually surrounds the spinal cord and infiltrates vertebral bones. A combined technique using protons or electrons to boost the initial photon fields is generally applied. In the treatment of chordomas surrounding the spinal cord, IMRT can provide high-dose homogeneity and planning target volume (PTV) coverage (Fig. 48.8). Frequent digital portal image-based setup control reduces random positioning errors for head and neck cancer patients immobilized with conventional thermoplastic masks. Gabriele et al.98 treated a patient with incomplete resection of a vertebral chordoma surrounding C2-3 with a total dose of 58 Gy in 2-Gy daily fractions. Beam arrangement consisted of seven 6 MV nonopposed coplanar IMRT fields using 120-leaf collimator in sliding window mode. To verify the daily setup, portal images at 0 degrees and 90 degrees were compared with the simulation images before treatment delivery (manual matching) and after treatment delivery (automatic anatomy matching). The mean dose to the PTV was 57.6 Gy covering 95% of the PTV with the 95% isodose. The minimum dose to the PTV (D99) was 53.6 Gy. The maximum dose to the spinal cord was 42.2 Gy and to the spinal cord planning risk volume (8 mm margin) 53.7 Gy. The mean dose to the parotids were 37.4 Gy (homolateral gland) and 19.5 Gy (contralateral gland). Because of the slow proliferative nature of chordomas, high linear energy transfer may prove useful in their management, as it will be discussed later. Brachytherapy can be used for recurrent tumors of the base of skull or adjacent to the spine when a more aggressive surgical exposure is offered.
Results of Therapy
Photons
Although survival in some patients with chordoma may be long term, the salient feature of this unusual neoplasm is local recurrence with eventual death. The course may be indolent, with multiple treatments for recurrences, but the overall 5-year disease-free survival rate is <10% to 20%. Catton et al.99 analyzed the long-term results of treatment for patients with chordoma of the sacrum, base of skull, and mobile spine treated predominantly with postoperative photon irradiation. In 20 base of skull chordomas, most of them irradiated with conventionally fractionated radiation to a median dose of 50 Gy in 25 fractions for 5 weeks (range 25 to 50 Gy), median survival was 62 months (range 4 to 240 months) from diagnosis with no difference between clival and nonclival presentations. There was no survival advantage to patients receiving radiation doses >50 Gy (median 60 Gy) compared with lower doses <50 Gy (median 40 Gy). Hyperfractionation regimens did not influence the degree or duration of symptomatic response or progression-free survival. Median survival after retreatment was 18 months.
Forsyth et al.100 reported on 51 patients with intracranial chordomas (19 classified as chondroid) treated surgically (biopsy in 11 patients and subtotal removal or greater in 40); 39 patients received postoperative irradiation. At the time of the analysis, 17 patients were alive. The 5- and 10-year survival rates were 51% and 35%, respectively; 5-year survival was 36% for biopsy patients and 55% for those who had resection. Patients who underwent postoperative irradiation tended to have longer disease-free survival times.
Gay et al.101 analyzed the outcome of 46 patients with cranial base chordomas and 14 with chondrosarcomas after extensive surgical resection, 50% of them treated previously; 20% received postoperative irradiation. Nine patients with chordomas and two with chondrosarcomas died during the postoperative follow-up period. The 5-year recurrence-free survival for all patients was 76%. Chondrosarcomas had a better prognosis than chordomas (5-year recurrence-free survival of 90% and 65%, respectively; P = .09). Patients who had undergone previous surgery had a greater risk of recurrence than did those who had not undergone previous surgery (5-year recurrence-free survival rates of 64% and 93%, respectively; P <.05). Those with total or near-total resection had a better 5-year recurrence-free survival rate (84%) than did patients with partial or subtotal resection (64%; P <.05). Postoperative leakage of cerebrospinal fluid was the most frequent complication (30% of patients) and was found to increase the risk of permanent disability. Patients who had undergone previous irradiation had a greater risk of death in the postoperative period (within 3 months of operation) and during follow-up.
Tai et al.91 reviewed the results of irradiation combined with surgery, irradiation alone, and surgery alone in 159 patients reported in the literature. An analysis of the optimal biologically equivalent dose was performed using the linear-quadratic formula on 47 patients. With conventional photon irradiation, no dose–response relationship was shown. Survival improved in patients undergoing surgery followed by irradiation.
Chetty et al.102 reported on 18 chordomas, 61% of them occurred in the sphenoid region. Follow-up for 12 patients ranged from 3 to 170 months. Various combinations of surgery and radiation therapy were used. Mean survival was 73.4 months, with a survival rate of 50% (6 of 12 patients).
Keisch et al.103 reported on 21 patients with chordoma treated at the authors’ medical center: 5 had clival tumors, 2 had nasopharyngeal tumors, and 1 had a lumbar spine tumor. Nine patients were treated with surgery alone, eight had subtotal resection and postoperative irradiation, and four received irradiation alone after biopsy. The 5- and 10-year actuarial survivals were significantly better in patients treated with surgery alone or surgery and irradiation than in those treated with radiation therapy alone (52%, 32%, and 0%, respectively; P = .02). Disease-free survival of patients with base of skull tumors was not significantly different among the treatment groups.
FIGURE 48.7. A: Contrast material-enhanced axial computed tomography scan demonstrates a large chordoma with extension into the posterior fossa and left parasellar region. B: Computed tomography scan photographed at bone windows shows the bony destruction and intratumoral calcifications. C: Treatment planning field arrangement for illustrated clivus chordoma using standard irradiation techniques with wedges on lateral ports.
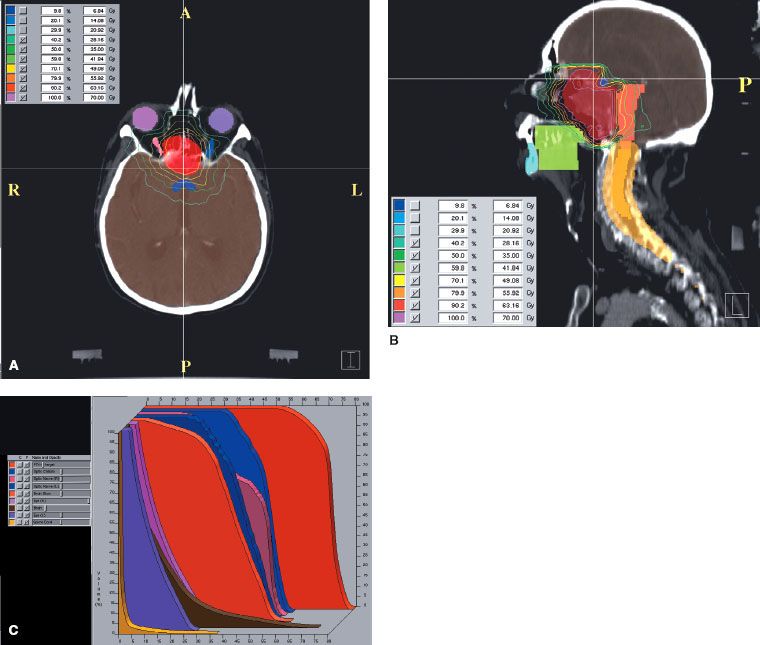
FIGURE 48.8. Chordoma of clivus in 81-year-old man treated with 70 Gy in 2-Gy fractions. Example of intensity-modulated radiation therapy plan: A: Cross-section in upper portion of planning target volume (PTV), demonstrating coverage of target volume with sparing of ocular structures. B: Sagittal plane dose distribution with excellent coverage of PTV. C: Dose–volume histogram:
Debus et al.104 reported on 45 patients treated for chordoma or chondrosarcoma with postoperative fractionated 3D stereotactic radiation therapy. Median dose at isocenter was 66.6 Gy for chordomas and 64.9 Gy for chondrosarcomas. All chondrosarcomas achieved and maintained local tumor and recurrence-free status at 5-years follow-up. Local control rate of chordomas at 5 years was 50% and survival was 82%. Clinically significant late toxicity developed in only one patient.
Bugoci et al.105 published results on 12 patients with skull-base chordoma treated with fractionated stereotactic RT and IMRT boost (total dose 74 Gy in 2-Gy fractions). With median follow-up of 34 months, local tumor control at 3 years was 65% and overall survival 92%.
Den et al.106 described a multi-institutional study of 31 patients, 28 with clivus chordoma or chondrosarcoma, treated with various photon techniques (single-dose stereotactic, fractionated 3D-CRT, IMRT). Median fractionated total dose was 65 Gy. Actuarial 3-year local tumor control was 68% and overall survival 63%. Patients receiving 65 Gy or a higher dose had a 5-year tumor control of 78% and overall survival of 90%. No grade 2 or greater toxicity was observed.
Muthukumar et al.107 published a report on 15 patients with skull-base chordoma and chondrosarcoma treated with stereotactic radiation therapy (13 had previous surgical resection). Median minimum marginal tumor single dose was 18 Gy (12 to 20 Gy) and the number of isocenters ranged from 1 to 10 (average, 4). Dose to optic nerve or chiasm was ≤9 Gy. With median follow-up of 40 months, eight patients had clinical improvement, three were stable, and four had died. No significant morbidity was noted.
Protons
The best results in the treatment of chordomas have been obtained with radical surgery followed by high-dose proton irradiation.108 Berson et al.109 described 45 patients with chordomas or chondrosarcomas at the base of the skull or cervical spine treated by subtotal resection and postoperative irradiation. Twenty-three patients were treated definitively by charged particles, 13 patients with photons and particles, and 9 were treated for recurrent disease. Doses ranged from 36 to 80 Gy equivalent (GyE). There appeared to be significant benefit for patients with smaller tumor volumes (80% vs. 33% actuarial survival rate at 5 years). Patients treated for primary disease had a 78% actuarial local tumor control at 2 years versus 33% for recurrent disease.
Austin et al.110 evaluated 141 patients with chordoma and chondrosarcoma of the base of skull and cervical spine treated with proton and photon irradiation. The local disease was controlled in 111 patients. They reviewed 26 patients who had recurrent disease (21 nonchondroid chordomas, two chondroid chordomas, and three chondrosarcomas). The prescribed doses ranged from 67 to 72 cobalt Gray equivalent (CGE). Approximately 25% (6 of 26) of the cases failed in the prescribed dose region. More than half (15 of 26) failed in regions where tumor dose was limited by normal tissue constraints. Approximately 10% of the patients recurred in the surgical pathway and 10% were judged to be marginal misses. Overall, 75% of the patients failed in regions receiving less than the prescribed dose. All tumors that failed in the high-dose region had recurrences (10 of 26) and larger tumors (average volume of 102 cc) than those with base of skull disease (16 of 115) with an average volume of 63 cc.
O’Connell et al.111 reported on 62 patients with base of skull chordomas treated with proton beam irradiation (65 to 73.5 GyE); 29 patients (19 women and 10 men) experienced local failure, and 14 women (48%) and 7 men (21%) died of disease. On histologic analysis, the presence of >10% necrosis, prominent nucleoli, and tumor >70 mm were significant predictors of short-term disease-specific survival. Chondroid chordoma and conventional chordomas had equivalent outcomes.
Fagundes et al.112 updated the Massachusetts General Hospital experience with 204 patients treated for chordoma of the base of the skull or cervical spine. Sixty-three patients (31%) had treatment failures, which were local in 60 patients (29%) and the only site of failure in 49 patients. Two patients had regional lymph node relapse, and three developed surgical pathway recurrence. Thirteen patients relapsed in distant sites (especially lungs and bones). The 5-year actuarial survival rate after any relapse was 7%. There was no significant difference in survival for patients who had a local or distant failure. Two patients (1.4%) with local tumor control developed distant metastases in contrast with 10 of 60 patients (16%) who failed locally and distantly.
Terahara et al.113 reported on 132 patients with skull-base chordoma treated with combined photon and proton irradiation; in 115 patients dose–volume data and follow-up were available. The prescribed doses ranged from 66.6 to 79.2 CGE (median 68.9 CGE). Dose to the optic structures (optic nerves and chiasm), the brainstem surface, and the brainstem center was limited to 60, 64, and 53 CGE, respectively. Local failure developed in 42 of 115 patients, with the actuarial local tumor control rates at 5 and 10 years being 59% and 44%, respectively. In a Cox multivariate analysis, the model’s equivalent uniform dose suggests that the probability of recurrence of skull-base chordomas depends on gender, target volume, and target dose inhomogeneity; equivalent uniform dose was shown to be a useful parameter to evaluate dose distribution for the target volume.
Hug et al.114 analyzed efficacy of fractionated proton radiation therapy for 33 skull-base chordomas and 25 chondrosarcomas. Following various surgical procedures, residual tumor was present in 91% of patients; 59% demonstrated brainstem involvement. Target doses ranged from 64.8 to 79.2 CGE (mean 70.7 CGE). The range of follow-up was 7 to 75 months (mean 33 months). In 10 patients (17%) the treatment failed locally, resulting in local control rates of 92% (23 of 25 patients) for chondrosarcomas and 76% (25 of 33 patients) for chordomas. All tumors with volumes of ≤25 mL remained locally controlled compared with 56% of tumors >25 mL (P = .02). Of patients without brainstem involvement, 94% did not experience recurrence; whereas with brainstem involvement (and dose reduction because of brainstem tolerance constraints), the tumor control rate was 53% (P = .04). Actuarial 5-year survival rates were 100% for patients with chondrosarcoma and 79% for patients with chordoma. Grade 3 and 4 late toxicities were observed in four patients (7%) and were symptomatic in three (5%).
Ares et al.42 treated 42 patients with chordomas and 22 with chondrosarcomas of the skull base using spot-scanning protons (median doses 73.5 and 68.4 Gy, respectively, at 1.8 to 2.0 Gy relative biological effect). With median follow-up of 38 months, 5-year tumor control was 81% and 94% and overall survival 100% and 91%, respectively. Late toxicity consisted of one grade 3 and one grade 4 unilateral optic neuropathy and two patients with grade 3 CNS necrosis. No patient experienced brainstem toxicity.
Noel et al.115 reported on 49 chordomas and 18 chondrosarcomas treated with high-energy photons (two-thirds of dose) and 201 MeV protons (one-third of dose). Median total dose was 67 CGE (60 to 70 CGE). With median follow-up of 32 months, 3-year local tumor control was 71% for chordomas and 85% for chondrosarcomas, and 4-year overall survival 88% and 75%, respectively. Fourteen tumors (21%) failed locally.
Recently, heavy particles have been used to treat some of these patients.116, 210 Hasegawa et al.79 reported on 54 patients with skull base or paracervical tumors (31 with chordomas) treated with carbon ions (escalating doses from 48 to 60.8 GyE in 16 fractions over 4 weeks). In the 31 chordoma patients, 5-year local tumor control and overall survival were 78% and 85%, respectively. Patients were divided into two groups; a low-dose group (n = 10) irradiated with doses ranging from 48 to 57.8 GyE, and a high-dose group (n = 21) irradiated with 60.8 GyE. The 5-year local tumor control was 60% for the low-dose group and 93% for the high-dose group and the overall survival was 90% and 84%, respectively. One late grade 2 brain sequela was noted in a patient treated with 60.8 GyE.
Likewise, Schulz-Ertner et al.43 treated 24 chordomas and 13 chondrosarcomas with 3D planning carbon ions (median dose 60 GyE). With mean follow-up of 13 months, local tumor control at 2 years was 90%. Progression-free survival was 83% for chordomas and 100% for chondrosarcomas. No significant toxicity was observed.
Benk et al.117 described results in 18 children 4 to 18 years of age with base of skull or cervical spine chordomas who received fractionated high-dose postoperative irradiation using mixed-photon and 160-MeV proton beams. Median tumor dose was 69 CGE with a 1.8-CGE daily fraction. With a median follow-up of 72 months, the 5-year survival was 68%, and the 5-year disease-free survival rate was 63%. Patients with cervical spine chordomas had a worse survival rate than did those with base of skull lesions (P = .008). The incidence of treatment-related morbidity was acceptable: two cases of growth hormone deficit corrected by hormone replacement, one temporal lobe necrosis, and one fibrosis of the temporalis muscle, improved after surgery.
A report on proton therapy for base of skull chordoma published by the Royal College of Radiologists118 concluded that outcome after proton irradiation is superior to that reported for conventional photon irradiation. Radiation therapy schedules involving a mixed schedule of protons and photons have achieved an approximately 60% local tumor control rate at 5 years.
Sequelae of Treatment
In patients treated with high irradiation doses, as well as with charged particles, there is an increasing probability of sequelae, including brain damage, spinal cord injury, bone or soft tissue necrosis, and xerostomia. In a report by Berson et al.,109 three patients experienced unilateral visual loss, and four patients had radiation injury to the brainstem.
Santoni et al.119 reported on the temporal lobe damage rate in 96 patients (75 primary and 21 recurrent tumors) treated with postoperative high-dose proton and photon irradiation for chordomas and chondrosarcomas of the base of the skull. All the patients were randomized to receive 66.6 or 72 CGE with conventional fractionation (1.8 CGE per day, 5 fractions per week) using opposed lateral fields for the photon component and a noncoplanar isocentric technique for the proton component. Of the 96 patients, 10 developed temporal lobe damage (lateral in 2 and unilateral in 8). The cumulative temporal lobe damage incidence at 2 and 5 years was 7.6% and 13.2%, respectively. CT and MRI scans were evaluated for white matter changes; the MRI areas suggestive of temporal lobe damage in 10 patients were always separate from the tumor bed.
In patients receiving high-dose proton therapy for clivus tumors, Slater et al.120 observed a 26% incidence of endocrine abnormalities at 3 years and 37% at 5 years, with hypothyroidism being the most frequent sequela. The dose to the pituitary in patients with abnormalities ranged from 63.1 to 67.7 GyE.
 LETHAL MIDLINE GRANULOMA
LETHAL MIDLINE GRANULOMA
Natural History and Pathology
Lethal midline granuloma (LMG) or midline malignant polymorphic reticulosis is a clinical entity characterized by progressive, unrelenting ulceration and necrosis of the midline facial tissues.121,122 LMG is associated with Epstein-Barr virus, which has at least two subtypes with different biologic properties that can be identified by their genomic configuration. The occurrence of the rare subtype 2 in LMG may relate to a covert immune defect.123 Considerable controversy exists regarding various disorders characterized by a necrotizing and granulomatous inflammation of the tissues of the upper respiratory tract and oral cavity. It is now clear that if infections and other known agents such as cocaine use, sarcoidosis, environmental toxins, and various neoplasms can be excluded, three clinicopathologic entities remain: Wegener’s granulomatosis, LMG, and polymorphic reticulosis (PMR).15 A review of the literature suggests that cases described as idiopathic midline destructive disease and PMR are a large evolutionary spectrum from almost benign to fatal malignant lymphoma.124
Wegener’s granulomatosis is an epithelioid necrotizing granulomatosis with vasculitis of small vessels. Systemic involvement of the kidneys and lungs is common.
PMR is an unusual disorder with distinctive clinical and pathologic features. Histologically, PMR is characterized by an atypical mixed lymphoid infiltration of the submucosa with extensive areas of necrosis, sometimes extending to bone or cartilage. The lesion consists of variable zones of small lymphocytes with scattered immunoblastic forms, abundant plasma cells with occasional eosinophilia and histiocytosis.125 PMR has been considered a lymphoproliferative disorder; most, if not all, cases are peripheral T-cell lymphomas. Several authorities believe that PMR and systemic lymphomatoid granulomatosis are the same disease, with the latter predominantly involving the lungs.126
Idiopathic LMG describes a localized disorder not characterized by visceral lesions but by destruction of the midfacial area, which, if left untreated, is uniformly fatal. The histopathologic findings are nonspecific, with a relatively nondescript inflammatory reaction with acute and chronic inflammation and necrosis. Despite specific clinicopathologic features, the distinction between LMG and PMR is often difficult; although controversial, they may represent two phases of the same disease, with LMG remaining histologically benign or evolving into PMR. LMG occurs more frequently in men.98 Ages range from 21 to 64 years; almost half of the patients are in their 50s at presentation. Most patients have involvement of the nasal cavity (including destruction of the septum) and the paranasal sinuses (particularly maxillary antrum). The primary lesion may extend into the orbits, the oral cavity (palate, gingiva), and even the pharynx.
Characteristics of the three different diseases are outlined in Table 48.8.
Clinical Features and Diagnostic Workup
Clinical manifestations include progressive nasal discharge, obstruction, foul odor emanating from the nose, and, in later stages, pain in the nasal cavity, paranasal areas, and even in the orbits.
Examination discloses ulceration and necrosis in the nasal cavity, perforation or destruction of nasal septum and turbinates, and even ulceration of the nose. Edema of the face and eyelids may be noted, and the bridge of the nose may be sunken. Radiographic studies initially show soft tissue swelling, mucosal thickening, and findings consistent with chronic sinusitis.
CT is invaluable in demonstrating the full extent of the tumor, including bone or cartilage destruction. In 13 patients presenting with LMG, CT proved essential for determining the extent of the disease, guiding biopsy, and planning radiation therapy.127 MRI was also helpful for the latter because it could distinguish fluid retained within the paranasal sinuses from solid masses and tumor from granulation tissue; it was of little value for detecting bone lysis. Eight patients proved to have T-cell lymphoma, two had Crohn disease, in one the lesion was factitious, and two had granulomas without diagnostic histologic features.
TABLE 48.8 DIFFERENTIAL FEATURES OF THREE CLINICOPATHOLOGIC ENTITIES





