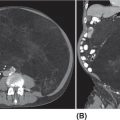
26520 Undifferentiated Pleomorphic Sarcoma/Myxofibrosarcoma/Other Fibrosarcomas Undifferentiated pleomorphic sarcoma (UPS) encompasses malignant neoplasms of mesenchymal origin, which demonstrate cellular pleomorphism and which lack evidence of cellular differentiation. The term UPS has been coined in replacement of the obsolete term malignant fibrous histiocytoma, as research evidence revealed the latter constituted a diverse group of neoplasms with various differentiation. Fibrosarcomas constitute a rare group of soft tissue sarcomas that usually affect deep tissues. For localized disease, the treatment of high-grade tumors includes a multimodality approach (perioperative chemotherapy, radiation therapy, and surgery), followed by close surveillance with scans and follow-up visits. For metastatic disease, patients will require systemic therapies as well as radiation therapy and surgery in selected patients. This chapter reviews clinical presentation, diagnostic approach, and general therapeutic approach for UPS, and discusses metastatic disease, atypical fibroxanthoma, and fibrosarcoma subtypes. Undifferentiated pleomorphic sarcoma, UPS, Myxofibrosarcoma, MFS, Fibrosarcomas, Atypical fibroxanthoma, Sclerosing epithelioid fibrosarcoma, SEF cellular differentiation, chemotherapy, clinical trials, fibrosarcomas, malignant fibrous histiocytoma, radiation therapy, soft tissue sarcomas, surgery, surveillance, undifferentiated pleomorphic sarcoma Clinical Trials as Topic, Drug Therapy, Fibrosarcoma, General Surgery, Radiotherapy, Undifferentiated Connective Tissue Diseases INTRODUCTION Malignant fibrous histiocytoma (MFH) is a term coined in 1964 to describe a group of soft tissue sarcomas (STSs) of mixed histiocytic and fibroblastic lineage.1 MFH was recognized for many years as the most common STS in adults and was associated with a metastatic rate of around 30% and a 5-year overall survival rate of approximately 70%.2 However, subsequent histopathologic and molecular studies demonstrated that MFH was quite diverse, and although tumors classified as MFH shared morphologic similarities, it was soon recognized that this diagnosis encompassed a diverse group of sarcomas with distinct clinical behavior. Underscoring this was evidence of patterns of incomplete mesenchymal differentiation in tumors classified as MFH, with some demonstrating features of smooth muscle, adipose tissue, and peripheral nerve sheath.3–6 In light of these findings, the term MFH is now considered obsolete, and the majority of cases previously classified as MFH have been reclassified as undifferentiated pleomorphic sarcoma (UPS).7 The term UPS encompasses malignant neoplasms of mesenchymal origin, which demonstrate cellular pleomorphism and which lack evidence of cellular differentiation. Most cases do not have an apparent predisposing factor, but prior radiation is a risk factor, accounting for about 2% to 3% of UPS cases, as are hereditary cancer predisposition syndromes, such as Li–Fraumeni syndrome.8,9 Overall, UPS constitutes 10% of adult STSs, with an incidence of around one new case per 100,000 per year and represents one of the most common STS subtypes diagnosed in older adults.10 Over the past decade, new light has been shed on the underpinnings of UPS, and this information will translate into new therapeutic horizons for patients suffering from this disease. CLINICAL PRESENTATION UPS typically presents as deep-seated lesions in the extremities or trunk, with lesions in the lower extremities being the most common. In contrast, superficial or subcutaneous lesions are rare in this sarcoma type. Clinical features of UPS are not specific, and the patients may present with a variable myriad of symptoms based on the location of the primary tumor. For example, UPS of the extremity tend to enlarge rapidly and painlessly.11,12 In contrast, patients with retroperitoneal tumors may develop constitutional symptoms including anorexia, weight loss, malaise, fatigue, and early satiety.11 The most common age of diagnosis for UPS is between the ages of 50 and 70 years,10 with cases of pediatric UPSs being infrequent.13 The majority of evidence suggests that males and females are approximately equally affected, although a slightly increased male prevalence has also been reported.7 DIAGNOSTIC APPROACH Tumor biopsy remains the gold standard for morphologic diagnosis. Numerous ancillary techniques are useful in support of morphologic diagnosis, including immunohistochemistry (IHC), classical cytogenetics, fluorescence in situ hybridization (FISH), and genetic sequencing.31 Sarcoma diagnosis should be carried out by an experienced sarcoma pathologist. The diagnosis of UPS is based on exclusion, as the key feature of UPS is the absence of a specific line of differentiation. Several sarcoma types show a comparable degree of cellular pleomorphism, such 266as sarcomatoid carcinoma (SC) and pleomorphic forms of liposarcoma, leiomyosarcoma, and rhabdomyosarcoma, among others.11 These sarcoma types should be considered in the differential diagnosis and should be excluded to reach the diagnosis of UPS. RADIOGRAPHIC EVALUATION The radiologic evaluation of STS has changed over the past 30 years. Initially, the evaluation of these tumors was limited to radiographs. With the introduction of CT and MRI, the ability to accurately characterize STSs changed dramatically.14 There are no pathognomonic radiologic findings in UPS. The radiologic evaluation of UPS follows the general approach to STS and should include CT or MRI of the primary site and imaging of the lungs to evaluate for distant metastatic spread at presentation. Whole-body PET-CT is a useful adjunct to CT/MRI of the primary tumor31 and can be helpful for staging, prognostication, and subsequent evaluation of treatment response for patients receiving systemic therapy or radiotherapy.15 As the risk of central nervous system (CNS) metastasis is low—1% to 8%16—imaging of the brain is not required at presentation unless patients demonstrate clinical symptoms suggesting CNS metastasis. TUMOR CHARACTERISTICS Although there have been several studies to elucidate the molecular aberrations driving UPS, the critical pathways involved in oncogenesis of UPS remain incompletely understood.10,17–20 Early studies of UPS/MFH revealed that these tumors are characterized by complex genomic profiles with frequent gains and losses of whole chromosomes or chromosome regions as well as focal amplifications and deletions.21–23 Loss of tumor suppressor genes in UPS has also been reported, with 71% exhibiting at least one mutation and deletion in p53 in a cohort of 109 patients24 or inactivation of the RB1 gene.25 Gene expression profiling has not identified significant recurrent alterations in UPS.12 However, Wang et al. described the increased activation of Hedgehog and Notch signaling pathways in cells with stem-like tumor-initiating potential from UPS.26 The overexpression of DKK1, an inhibitor of the Wnt canonical pathway, has also been reported.27 Helias-Rodzewicz et al. and later the Cancer Genome Atlas Research Network (CGARN) reported CCNE1, VGLL3, and YAP1 genes were found to be overexpressed in a subset of UPS, indicating a proportion of them may be driven by the Hippo pathway.23,28 More recently, Delespaul et al. reported a small subset of UPS harbor fusions that involve the TRIO gene.29 Further studies are needed to understand the impact of various genomic and epigenetic events on the prognosis and optimal management of UPS.12 GENERAL THERAPEUTIC APPROACH Patients should be discussed in multidisciplinary sarcoma tumor boards, as it has been shown they will have adequate radiologic tumor assessments, tumor biopsies, and better adherence with clinical recommendations compared with patients who were not discussed at the sarcoma tumor board.30 Wide excision plus radiotherapy remains the cornerstone of treatment of nonmetastatic tumors, and it is considered the standard of care.31 Randomized trials have confirmed the role of these treatment modalities.32–34 Neoadjuvant or adjuvant anthracycline-based systemic chemotherapy is a consideration for patients with large (>5 cm), high-grade UPS per current consensus guidelines.31 A 2008 meta-analysis of 18 randomized trials evaluating adjuvant chemotherapy in patients with STS suggested benefits in both local control and risk of distant metastasis in patients receiving systemic chemotherapy. Combination regimens using doxorubicin plus ifosfamide were associated with a 46% relative risk reduction in death (odds ratio [OR] for death 0.56, 95% confidence interval [CI] 0.36–0.85), which translated into an absolute risk reduction of 11%. Inclusion of ifosfamide appears to be important in reducing risk of death, as adjuvant chemotherapy with single-agent doxorubicin did not meet criteria for statistical significance.35 Tumor size and grade were subsequently demonstrated to be important factors to consider when evaluating patients for adjuvant doxorubicin plus ifosfamide chemotherapy. The European Organisation for Research and Treatment of Cancer (EORTC) 62931 study was a 351-patient randomized study of 267observation versus adjuvant doxorubicin plus ifosfamide chemotherapy in patients with grade 2 or 3 STS, >5 cm in size. This study only included 40 patients with UPS out of 281 cases reviewed.36 There was no difference in progression-free survival (PFS) or overall survival for all patients included in this study. However, subset analyses suggested a possible survival benefit from adjuvant chemotherapy, for patients with large (>10 cm), high-grade (grade 3) tumors.36 A subsequent patient-level reanalysis of the EORTC 62931 study, using the SARCULATOR nomogram to separate patients into low-, intermediate-, and high-risk cohorts, suggested that adjuvant chemotherapy may be beneficial to patients with the highest risk of recurrence following surgical resection alone.37 Given that doxorubicin plus ifosfamide is an intensive regimen that is not well tolerated, alternative chemotherapy regimens have been evaluated in the adjuvant setting for UPS. Recently, adjuvant chemotherapy with doxorubicin and ifosfamide was shown to be superior to gemcitabine plus docetaxel in patients with high-risk UPS.38 Several new adjuvant approaches are currently under investigation including the addition of oral tyrosine kinase inhibitors (e.g., pazopanib),39 other small molecule radiosensitizers (e.g., AMG-232),40 and immunomodulatory drugs (e.g., pembrolizumab)41 to radiation and wide excision. For patients with resectable, localized UPS who are not being treated on a clinical trial, wide surgical excision with adjuvant or neoadjuvant radiotherapy remains the standard of care; anthracycline-based chemotherapy with doxorubicin and ifosfamide should be considered for high-risk patients with good performance status and preserved organ function.31 SURVEILLANCE Essential goals of follow-up surveillance include early identification of recurrences, identification of treatment-related complications, and patient reassurance. After the definitive treatment for localized disease is completed, the patients will require periodic visits and scans (CT or MRI), because patient recurrence risk never returns to zero.42 The follow-up interval may vary depending on the institution. There is limited data regarding the best strategy for surveillance.43–46 The National Comprehensive Cancer Network (NCCN) guidelines outline the importance of a prudent follow-up schedule, tailored to the recurrence risk. Higher grade and larger tumors may require a more intensive follow-up. For patients with low-risk tumors (stage I), who completed treatment with curative intent, follow-up with annual scanning of the primary site for at least 5 years is recommended.31 Often these patients are seen every 3 to 4 months in the immediate postoperative period for the first 2 years, then every 4 to 6 months for the next 2 years, and yearly thereafter.47 Patients with high risk tumors (stage II–III) may have follow-up visits with imaging every 3 months for the first 1 to 2 years, then visits every 4 months for the next 1 to 2 years, followed by visits every 6 months for 1 to 2 years, and yearly visits thereafter.47 PROGNOSIS Sarcoma staging is essential not only because it provides an individual patient prognosis but also because it determines treatment. The staging system most often used for STS is the American Joint Committee on Cancer (AJCC) staging system. The AJCC recently published the eighth edition of its staging system, based on a retrospective analysis of 21,396 cohorts of adult patients with STS of the extremity or trunk.48 Prognostic factors include tumor size, grade, and distant metastases at initial presentation.49 The majority of UPSs are high-grade tumors, having a historical local recurrence rate ranging from 19% to 31%, a metastatic rate of 31% to 35%, and a 5-year survival rate of 65% to 70%. Only a minority of patients develop metastases after 5 years, with the common metastatic sites being lung (90%), bone (8%), and liver (1%). Regional lymph node metastases are uncommon.2,50–52 Several prediction tools have been developed in sarcomas. In 2002, Kattan et al. developed a nomogram, based on a prospectively followed cohort of 2,136 adult patients with primary STS who were treated at Memorial Sloan-Kettering Cancer Center (MSKCC; New York, NY). This nomogram can predict the likelihood of recurrence and overall survival for up to 12 years.53,54 The prognostic nomogram Sarculator (http://www.sarculator.com)55,56 calculates the probability of overall survival and distant metastasis at 5 and 10 years after surgery. It considers patient age, tumor 268size, tumor grade, and tumor.55 The Sarculator was previously retrospectively tested and validated in 1,452 and 2,300 patients, respectively55 and further validated in a prospective randomized trial investigating perioperative anthracycline-based chemotherapy for high-risk STS of extremities and trunk wall.57–59 Its potential use in the clinics has been recently demonstrated.37 METASTATIC DISEASE For patients with oligometastatic disease, metastasectomy or other definitive local therapy for metastases should be considered as part of a multimodality approach. There is conflicting data on the potential survival benefit of metastasectomy. In a retrospective study of 48 patients, there was no improvement in the overall survival for patients treated with metastasectomy, compared with those with unresectable disease.60 A recent retrospective study involving 112 patients with metastatic STS identified several statistically significant variables for improved overall survival including resection of metastatic disease, <4 pulmonary metastases, and the presence of lymph node metastases versus pulmonary metastases. The 5-year survival rate was 59% and 8%, respectively.61 A retrospective study of 66 patients with sarcoma reported that pulmonary metastasectomy led to a median survival of 25.5 months. However, recurrent metastasis was associated with poor prognosis.62 Despite the fact, recurrence is frequent after initial metastasectomy; a study based on a large prospective review of 539 patients suggested a potential survival benefit of repeat pulmonary metastasectomy in appropriately selected patients.63 Currently, no data support the optimal management of patients presenting with metastatic disease; the choice of local control modality may depend on factors such as performance status, patient preference, lesion location/accessibility, ability to preserve normal tissue function, and anticipated morbidity of treatment modality.31 In the metastatic setting, doxorubicin as a single agent or in combination with ifosfamide is likely to be the first choice of chemotherapy.31 The sensitivity of STS to single-agent doxorubicin was demonstrated since the early 1970s,64 and it has remained one of the most important systemic agents to the present time. Its dose-dependent effect has been well studied.65 Single-agent ifosfamide has a similar response rate compared to doxorubicin, ranging between 7% and 41% in patients who previously failed a doxorubicin-based regimen.66–70 A dose–response relationship has been shown for ifosfamide in metastatic STS with a threshold between 6 g/m2 and 10 g/m2.68,71,72 One of the most active chemotherapy regimens, in terms of response rate, is AIM (doxorubicin/Ifosfamide/mesna),73 but because of its potential toxicities, the use of this combination for palliation should be restricted to selected patients, for whom the specific goal is tumor shrinkage. Other drugs such as trabectedin, gemcitabine, docetaxel, and pazopanib also have shown activity in the advanced setting.31 These therapies represent the most commonly used regimens for patients with metastatic/locally advanced UPS. Patients with advanced UPS have the worst outcome compared with other histologic STS subtypes.74 Emerging data on the use of checkpoint inhibitors opens new potential treatment options for patients with advanced disease.75 Recently, Tawbi et al. published the results of the SARC028 Phase II study of single-agent pembrolizumab (anti-programmed death-1 antibody) in multiple sarcoma types, in which four out of 10 patients with UPS had an objective response rate (ORR) of 40%.20 A different trial evaluating the use of nivolumab, as a single agent and in combination with ipilimumab, also reported responses in patients with UPS.76 These promising results with checkpoint inhibitors suggest that other immunotherapeutic treatments may provide benefits to UPS patients; for example, MAGE-A3 has been described as a potential therapeutic target in patients with UPS.77 The dynamic evolution of the immunotherapy field forecasts the development of new clinical trials for UPS patients. ATYPICAL FIBROXANTHOMA (UNDIFFERENTIATED PLEOMORPHIC SARCOMA OF THE SKIN) This uncommon cutaneous neoplasm of mesenchymal origin was initially described in 1961.78 The relationship between atypical fibroxanthoma (AFX), pleomorphic dermal sarcoma (PDS), and UPS is not fully understood, and some authors consider AFX a less aggressive variant within the spectrum of UPS, and others consider it a distinct malignancy.23 269It occurs most frequently as a solitary nodule on sun-damaged skin of the elderly. Typically, it affects the superficial skin of the head and neck (particularly nose, cheek, and ear) and has been found to have low metastatic potential and excellent clinical outcomes.79–81 AFX most commonly arises in older adults (usually in the seventh or eighth decade of life) and has a predilection for men.81,82 Predisposing factors include solar exposure and radiotherapy.79,83,84 Ultraviolet-related mutations and photoproducts have been found in these lesions.85–87 Other risk factors reported include immunosuppressive agents88 and HIV.89 Children with xeroderma pigmentosum have developed ATX as well.90 Genetically, these tumors often harbors UV signature mutations, as well as COL11A1, ERBB4, CSMD3, and FAT1 alterations.91 Patients diagnosed with ATX should have a full skin examination, including palpation of lymph nodes. Radiologic imaging may be necessary for patients with signs or symptoms suggestive of distant disease. The mainstay of the treatment is surgical resection of the lesion with negative margins.84,92 Radiation therapy is reserved for unresectable lesions. In individual cases, combination chemotherapy with doxorubicin (Adriamycin) and ifosfamide has been successful.93 FIBROSARCOMAS Fibrosarcomas (FSs) have been traditionally defined as tumors with cells that resemble the aspect of normal fibroblasts. As the diagnostic criteria evolved over time, the reported incidence went from 12% of all adult STSs in 197494 to 1% in 2010.95 This STS group is composed by several different subtypes. Adult-Type Fibrosarcoma Adult-type FS is the most common in the third through fifth decades of life. Bahrami et al. found that patients with this sarcoma type ranged in age from 6 to 74 years, with a median age of 50 years.95 Like other sarcomas, adult-type FS causes no pathognomonic symptoms. Most patients present with a solitary, slow growing, painless mass in an extremity.11 These tumors may occur in any soft tissue site but are most common in the deep soft tissues of the lower extremities, particularly the thigh and knee, followed by the trunk and upper extremities, although some may arise in the head and neck.11 The diagnosis of FS is performed through an exclusion process after a tissue biopsy is obtained. FS does not exhibit any lineage-specific markers, such as cytokeratin or S-100 protein.11 The molecular genetic alterations of adult-type FS are yet to be fully characterized, but multiple rearrangements have been reported.96,97 In contrast to infantile FS,98 this tumor lacks a characteristic cytogenetical abnormality. As the diagnostic criteria of FS changed over time, the analysis of the results of old series became problematic as it included other sarcoma types. For that reason, more emphasis has been placed on the results of recent studies. A study published in 2010 showed that 12 of 24 (50%) patients with follow-up died of locally aggressive and/or metastatic disease; only six patients were alive without disease and six died of other causes.95 Hansen et al. reported the rate of local recurrences in seven of 21 (33%) patients with follow-up, and three of 21 (14.3%) patients developed metastases and died of a tumor.99 The lung is the principal metastatic site, followed by the axial skeleton. As in other sarcoma types, most often metastases are diagnosed within the first 2 years after diagnosis.11 There are several additional factors associated with FS. Tumors arising from previously irradiated fields have been reported.95 It is not clear whether trauma is a contributing factor in the development of FS; however, multiple cases have been reported following trauma.100–103 As in other sarcomas, surgical resection is the most effective treatment when feasible. Myxofibrosarcoma The term myxofibrosarcoma (MFS) was proposed by Angervall et al. in 1997104 and its usage has developed through time. Currently, WHO definition includes tumors with different grades, a broad range of cellularity, nuclear atypia, and varying amounts of myxoid stroma unified by the multinodular growth pattern and characteristic curvilinear vasculature.7 More frequently, MFS presents as a slowly enlarging, painless mass in the lower extremities of patients in the fifth to seventh decades of life. It seems to be slightly more prevalent in men rather than 270in women.11,105 Like UPS, MFS does not have a specific line of differentiation; therefore, the diagnosis of MFS relies in a quantitative threshold of myxoid changes.105 MFS does not harbor pathognomonic genetic findings.106 MFS displays a longitudinal mode of spreading and can infiltrate microscopically far beyond what is grossly apparent, requiring careful surgical resection. MFSs are characterized by high local recurrence rates of 50% to 60%.107 Genetically, these tumors often have copy number alterations and mutations in TP53, RB1, CDKN2A, CDKN2B, and NF1, among others.106 As seen in other sarcomas, the metastatic potential is related to the histologic grade: intermediate-grade and high-grade neoplasms may develop metastases in approximately 20% to 35% of cases, whereas low-grade MFS rarely metastasizes.4,105 The overall 5-year survival rate is 70%.108 The presence of more than 5% of the myxoid component has been correlated with better outcome.109 Sclerosing Epithelioid Fibrosarcoma Sclerosing epithelioid fibrosarcoma (FES) is a very rare variant of FS, composed of epithelioid cells arranged in nests and cords and deposited in a densely hyalinized collagenous matrix. It was first described by Meis-Kindblom et al. in 1995.110 Genetically, these tumors harbor multiple fusion genes, including FUS-CREB3L2, EWSR1-CREB3L, FUS-CREM, and PAX5-CREB3L1.111 The age of presentation is wide, with a median age of 45 years, with no clear gender predilection.110,112 The greatest extent of patients develop a deep-seated mass affecting the lower extremity. The lesion may be painful in up to one-third of cases.11 SEF can also develop in the trunk, upper extremities, head and neck, and rarely bone.110,113–116 For localized disease, wide surgical excision remains the cornerstone of therapy. Radiation therapy has been used in the adjuvant setting for non-R0 resections. In the metastatic setting, doxorubicin-based therapies remain the first-line treatment. SUMMARY UPSs encompass malignant neoplasms of mesenchymal origin, which demonstrate cellular pleomorphism and which lack evidence of cellular differentiation. The term UPS has been coined in replacement of the obsolete term MFH as research evidence revealed the latter constituted a diverse group of neoplasms with various differentiation (Table 20.1). FSs constitute a rare group of STSs that usually affect deep tissues. For localized disease, the treatment of high-grade tumors includes a multimodality approach (perioperative chemotherapy, radiation therapy, and surgery), followed by close surveillance with scans and follow-up visits. For metastatic disease, these patients will require systemic therapies as well as radiation therapy and surgery in selected patients. The dynamic evolution of the immunotherapy and targeted therapy fields forecasts the development of new clinical trials for these patients. TABLE 20.1 Genetic Findings of Undifferentiated Pleomorphic Sarcoma, Adult Fibrosarcoma, Atypical Fibroxanthoma, Myxofibrosarcoma, and Sclerosing Epithelioid Fibrosarcoma
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Undifferentiated Pleomorphic Sarcoma/Myxofibrosarcoma/Other Fibrosarcomas
Gabriel Tinoco and David Liebner