Type 1 Diabetes Mellitus
George S. Eisenbarth
FORMS OF TYPE 1 DIABETES
During the past two decades, an accumulating body of information has led to the recognition that the predominant form of insulin-dependent diabetes mellitus (IDDM) is of immune etiology, that the disease can present at any age, and that individuals are not insulin-dependent early in the course of disease (1,2,3,4,5,6). Given this body of information, an expert committee of the American Diabetes Association has divided the “idiopathic” (e.g., not pancreatitis, mitochondrial DNA mutations) forms of diabetes resulting from β-cell loss and/or severe insulin secretory deficiency into type 1A and type 1B (1). Type 1A is the immune-mediated form and is best characterized by the presence of islet autoantibodies (see below) and insulitis (7,8), with selective destruction of islet β-cells (e.g., glucagon-producing cells are spared). In addition, type 1A diabetes is strongly associated with specific human leukocyte antigen (HLA) alleles (4,9,10) and almost always progresses to severe insulin deficiency (11). The term type 1B diabetes is reserved for forms of diabetes with severe insulin deficiency without evidence of β-cell autoimmunity (1). An interesting form of potential type 1B diabetes was reported in Japan (12). The patients had lymphocytic infiltration of the exocrine pancreas (with increased serum amylase levels) and an acute development of diabetes such that,
at the time of diabetes onset, levels of glycosylated hemoglobin (HbA1c) were normal despite marked hyperglycemia. Japan has one of the lowest incidences of type 1A diabetes in the world, and thus type 1B forms of diabetes are likely to represent a larger proportion of their cases of childhood type 1 diabetes than that seen in other countries. In Europe and the United States, fewer than 10% of non-Hispanic white children presenting with diabetes do not have type 1A diabetes. In contrast, among African-American children and Hispanic-American children, almost one half do not have type 1A diabetes, with the majority believed to have early onset type 2 diabetes and relatively few having type 1B diabetes. In particular, a subset of African-American children can present with ketoacidosis but do not express islet autoantibodies, lack HLA alleles associated with type 1A diabetes, and often can be treated with oral hypoglycemic medications after resolution of their acute illness (13,14,15).
at the time of diabetes onset, levels of glycosylated hemoglobin (HbA1c) were normal despite marked hyperglycemia. Japan has one of the lowest incidences of type 1A diabetes in the world, and thus type 1B forms of diabetes are likely to represent a larger proportion of their cases of childhood type 1 diabetes than that seen in other countries. In Europe and the United States, fewer than 10% of non-Hispanic white children presenting with diabetes do not have type 1A diabetes. In contrast, among African-American children and Hispanic-American children, almost one half do not have type 1A diabetes, with the majority believed to have early onset type 2 diabetes and relatively few having type 1B diabetes. In particular, a subset of African-American children can present with ketoacidosis but do not express islet autoantibodies, lack HLA alleles associated with type 1A diabetes, and often can be treated with oral hypoglycemic medications after resolution of their acute illness (13,14,15).
The present “etiologic” classification of diabetes is not completely satisfactory, since specific diagnostic markers are lacking for many forms of “idiopathic” diabetes (type 2 and type 1B), and some individuals with type 1A diabetes may even lack islet autoantibodies if they are tested only once at diagnosis. In addition, no combination of HLA alleles is completely diagnostic for type 1A diabetes. Only 20% to 50% of the patients with type 1A diabetes have the highest-risk HLA genotype (DR 3/4 DQ2/DQ8 heterozygous) compared with the 2.4% of individuals without diabetes who have this genotype (16). Approximately 1% of children with type 1A diabetes have highly “protective” HLA alleles (e.g., DQB1*0602) compared with 20% of the general public with these alleles. Given “biochemical” assays for islet autoantibodies with specificity set at greater than the 99th percentile, the terms autoantibody-positive and antibody-negative diabetes are perhaps more precise descriptors. Caveats for such designations include the loss of islet autoantibodies prior to the development of overt diabetes that occurs in a subset of children and the observation that individuals can have both type 1 and type 2 diabetes (17,18). Figure. 23.1 illustrates first-phase insulin secretion (1+3 minute insulin on intravenous glucose tolerance testing) for a markedly obese teenager who progressed to overt diabetes. At the time of diagnosis of diabetes, the fasting insulin level was approximately 25 μU/mL, and although the sum of insulin at 1+3 minutes (50 μU/mL) was above the first percentile (of 48 μU/mL), there was essentially no glucose-stimulated insulin secretion. This child with islet autoantibodies and progressive loss of first-phase insulin secretion has both type 1A diabetes and evidence of insulin resistance (marked by high fasting levels of insulin and obesity).
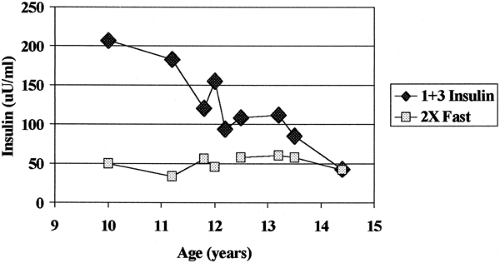 Figure 23.1. Progression to diabetes of an overweight teenager (BMI 30–35) with evidence of both type 1 and insulin resistance. Fasting insulin ranged between 17 μU/mL and 36 μU/mL during the prodrome to diabetes (normal children fasting insulin usually ≤5 μU/mL), suggesting insulin resistance. At time of diagnosis of diabetes, fasting insulin decreased to 21 μU/mL, and twice the fasting insulin was equivalent to the 1+3 minute insulin post-glucose, indicating no first-phase insulin response to glucose. |
The category of diabetes that will expand with increased knowledge is that of “other specific forms of diabetes,” which will include the subset of “immune-mediated diabetes.” At present the best example of a defined form of type 1A diabetes is that in patients with diabetes as part of the autoimmune polyendocrine syndrome type 1 caused by a mutation of the AIRE (autoimmune regulator) gene on chromosome 21 (19) (see below).
Does the classification of diabetes matter? Most children presenting with diabetes have type 1A diabetes, and most adults have type 2 diabetes. We believe it does matter when providing prognostic information (20), information concerning likely associated disorders [e.g., autoimmune disorders for type 1A diabetes (21) and manifestations of insulin resistance for type 2 diabetes], for assessing genetic risk for relatives (22), and in providing some guidance concerning therapy [insulin versus oral medications (e.g., insulin secretagogues and insulin sensitizers)] (23). Patients with islet autoantibodies and who have diabetes are probably best treated with insulin even if they are not currently “insulin dependent (24).” Oral agents rapidly lose their efficacy in adults with type 1A diabetes.
CLINICAL RAMIFICATIONS OF AN AUTOIMMUNE DIATHESIS
In that type 1A diabetes is of immune etiology, it marks an inherited failure in the maintenance of self-tolerance. It is thus not surprising that individuals with diabetes and their relatives are at increased risk for a series of autoimmune disorders (25,26,27,28). The presence of type 1A diabetes is also a “marker” for the presence of specific HLA alleles that are associated with many immunologic disorders. The associated disorders are so prevalent among patients with type 1A diabetes, and some are so devastating when not diagnosed early, both a high index of suspicion and screening for asymptomatic disease are warranted. Four important disorders are Addison’s disease (28), celiac disease (29,30,31,32,33,34,35,36), thyroid autoimmunity (37,38,39,40), and pernicious anemia (41,42,43). In addition, two major autoimmune polyendocrine syndromes are recognized, termed autoimmune polyendocrine syndrome type-I (APS-I) and APS-II (44).
Addison’s Disease
When Addison’s disease develops in a patient with type 1 diabetes, insulin requirements often decrease, with accompanying hypoglycemia. Such a decrease in insulin requirements can occur in the absence of the characteristic hyperpigmentation associated with Addison’s disease. Excellent radioassays for 21-hydroxylase autoantibodies associated with Addison’s disease now exist that have a sensitivity greater than 95% and a specificity exceeding 99% (28,45). Figure. 23.2 illustrates the expression of 21-hydroxylase autoantibodies in patients with type 1A diabetes. We have screened more than 2,000 patients for 21-hydroxylase autoantibodies, and approximately 1 (1.5%) of 60 are positive for autoantibody. Among 30 with 21-hydroxylase autoantibodies, five had Addison’s disease at initial testing. For three of the patients, the diagnosis of Addison’s disease could have been made on the basis of symptoms and signs, including a severe hypotensive crisis in one (46). For one patient with apparent Addison’s disease, the diagnosis was made only at age 17, but the child had missed a year of school at age 10 because of fatigue. Although the diagnosis for this child presumably
could have been made before detection of 21-hydroxylase autoantibodies on screening, it was not. The typical clinical history for patients with Addison’s disease includes years of symptoms before diagnosis.
could have been made before detection of 21-hydroxylase autoantibodies on screening, it was not. The typical clinical history for patients with Addison’s disease includes years of symptoms before diagnosis.
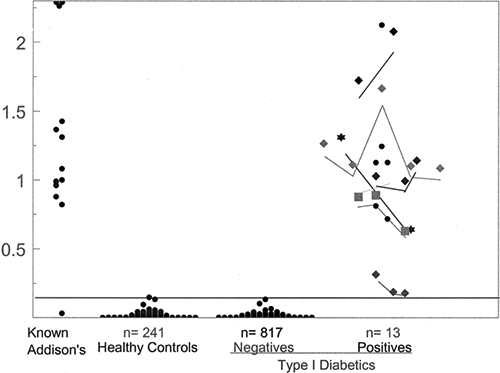 Figure 23.2. 21-Hydroxylase autoantibodies among patients with type 1A diabetes. Connected lines represent sequential measurements in the same patient. Positivity set at an index greater than all 241 healthy controls. A total of 957 patients with type 1 diabetes were included in the analysis, 15 of whom were positive. (Reprinted from Yu L, Brewer KW, Gates S, et al. DRB1*04 and DQ alleles: expression of 21-hydroxylase autoantibodies and risk of progression to Addison’s disease. J Clin Endocrinol Metab 1999;84:328–335, with permission. Copyright 1999, The Endocrine Society.) |
For the remaining 25 non-Addison’s patients with 21-hydroxylase autoantibodies, we annually measure adrenocorticotropic hormone and cortisol after Cortrosyn. Determined in part by their HLA alleles (highest risk with HLA genotype DR3 and DR4 with DRB1*0404, lower risk with DRB1*0401), some patients with autoantibodies are unlikely to progress to overt disease (28). Studies using immunofluorescent assays for adrenal autoantibodies (47) indicate that approximately one third to one half of those who are positive for this autoantibody are likely to progress to Addison’s disease and that the risk of Addison’s disease among patients with type 1 diabetes will exceed 1 per 200, in contrast to a risk in the general population of 1 per 10,000 (Table 23.1).
TABLE 23.1. Patients with Type 1 Diabetes Mellitus Screened for Autoantibodies: Examples of Associated Autoimmune Disorders in a Population of Patients with Type 1A Diabetes Mellitus | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Celiac Disease
Celiac disease, both in the general population and in patients with type 1A diabetes, is usually asymptomatic (29,31,48). In the United States, it is likely that celiac disease is not diagnosed in more than 90% of the patients with the disease. Similar to Addison’s disease and type 1 diabetes, celiac disease is associated with HLA alleles DR3 and DR4 (the DQ allele DQ2 in particular). As for Addison’s disease, there now exist excellent assays for autoantibodies associated with celiac disease. The endomysial autoantigen has now been cloned and found to be the molecule tissue transglutaminase (49). Approximately one third of patients with type 1 diabetes who are DQ2 homozygous are positive for transglutaminase autoantibodies (30). Twelve percent of patients with type 1A diabetes express transglutaminase autoantibodies, and one half of these have high levels of the autoantibody.
Approximately one-half of patients with type 1 diabetes who express transglutaminase autoantibodies have celiac disease, as determined by biopsy. A higher level of transglutaminase autoantibodies correlates with findings indicative of celiac disease on an intestinal biopsy. In the general population, 0.5% of the population have celiac disease (screening for transglutaminase autoantibodies followed by intestinal biopsy). Asymp-tomatic patients with transglutaminase autoantibodies show evidence of metabolic abnormalities, particularly decreased growth. Osteoporosis and gastrointestinal malignancy (50) are associated with untreated symptomatic disease.
Celiac disease is a remarkable immune-mediated disorder in that an inducing environmental factor is known, namely the wheat protein gliadin (51,52,53). Removal of wheat from the diet results in the loss of transglutaminase autoantibodies and resolution of the intestinal lesions (54). A gluten-free diet also reduces the development of intestinal malignancies (55). There is evidence that oats need not be proscribed for patients with celiac disease (54). A wheat-restricted diet, combined with the
dietary restrictions associated with type 1 diabetes, is a considerable burden. Nevertheless, given the high prevalence of celiac disease among patients with type 1A diabetes, we recommend screening with measurement of transglutaminase autoantibodies. Following the diagnosis of celiac disease, lifetime avoidance of wheat and other gliadin-containing proteins is recommended.
dietary restrictions associated with type 1 diabetes, is a considerable burden. Nevertheless, given the high prevalence of celiac disease among patients with type 1A diabetes, we recommend screening with measurement of transglutaminase autoantibodies. Following the diagnosis of celiac disease, lifetime avoidance of wheat and other gliadin-containing proteins is recommended.
Thyroid Autoimmunity
Thyroid autoimmunity is very common among patients with type 1A diabetes. Approximately one fourth of patients with type 1A diabetes express thyroid autoantibodies. Only a subset of individuals with thyroid autoantibodies progress to overt thyroid disease (Hashimoto thyroiditis resulting in hypothyroidism or Graves disease). Thus, rather than assaying for thyroid autoantibodies, we routinely measure thyrotropin levels (approximately every 2 years with sensitive assays), allowing diagnosis of both early thyroid failure and Graves disease.
Other Autoimmune Disorders
In addition to their risk for Addison’s disease, thyroid autoimmunity, and celiac disease, patients or their relatives with type 1A diabetes are at risk for a series of other autoimmune disorders. With specific autoimmune diseases (e.g., myasthenia gravis, Addison’s disease, celiac disease, Graves disease), there is a reciprocal increased risk of type 1A diabetes. Anecdotal reports indicate that it is more frequent to observe type 1A diabetes in siblings of patients with multiple sclerosis than in the patients themselves. The high-risk haplotype for multiple sclerosis, DRB1*1501 and DQB1*0602, is one of the most important haplotypes protective against type 1A diabetes. Juvenile rheumatoid arthritis is a “non-organ”-specific disorder apparently also associated with type 1A diabetes. Disorders such as lupus erythematosus that are treated with glucocorticoids that induce insulin-resistant diabetes are difficult to assess for association with diabetes. Pernicious anemia occurs at an earlier average age in patients with endocrine autoimmunity, but even among patients with diabetes, pernicious anemia usually occurs in adults. A detailed study using the Belgian Diabetes Registry found pernicious anemia in approximately 10% of patients with parietal cell antibodies (38,42).
Autoimmune Polyendocrine Syndromes
Two important autoimmune syndromes are autoimmune polyendocrine syndrome type I (APS-I) (56) and autoimmune polyendocrine syndrome type II (APS-II) (27). Although APS-I is extremely rare, it is one autoimmune syndrome for which a mutation causing the disease has been defined (19,57)—a mutation of a gene termed autoimmune regulator, on the long arm of chromosome 21. APS-I characteristically develops in early infancy with mucocutaneous candidiasis, Addison’s disease, and hypoparathyroidism. An individual may have one or all three disorders, and his or her affected siblings may have different disorders of the syndrome. These patients are also at risk for autoimmune hepatitis. Approximately 18% of patients with APS-I develop type 1 diabetes (56). It appears that more patients have anti-glutamic acid decarboxylase (GAD) autoantibodies than progress to overt diabetes, but similar to patients with standard type 1A diabetes, those with multiple autoantibodies (insulin and islet cell antibody [ICA] 512 in addition to GAD) are at high risk of progression (20).
The APS-II syndrome is characterized by two or more organ-specific autoimmune disorders, including Addison’s disease, Graves disease, type 1A diabetes, and thyroiditis (27). This syndrome is strongly influenced by HLA alleles, and the majority of patients with Addison’s disease have high-risk alleles DR3 and DR4 (DQ2 and DQ8), similar to patients with type 1A diabetes (21). In comparison to patients with type 1A diabetes, patients with APS-II syndrome have an excess of DRB1*0404 alleles, while most patients with type 1A diabetes have DRB1*0401, 0402, and 0405 alleles. There is a tendency for patients with APS-II syndrome with Addison’s disease to develop type 1A diabetes later in life, and prediction of disease again relies on the presence of multiple islet autoantibodies. There is also a tendency for more patients with type 1A diabetes in the setting of APS-II to have “protective” or non-high-risk HLA alleles compared with the usual patient with only type 1A diabetes. At present, the genes outside of the HLA region that underlie risk of APS-II have not been identified, although some studies of type IA diabetes have reported an association of Addison’s disease and Graves disease with polymorphisms of the genes CTLA-4 (58,59,60,61) and MIC-A (62,63).
The XPID syndrome (X -linked syndrome of p olyendocrinopathy, i mmune dysfunction and d iarrhea) (64,65,66,67) was mapped to the X chromosome and results from mutations of the Foxp3 gene. This rare syndrome with overwhelming immunologic abnormalities results in type 1 diabetes, enteropathy with villous atrophy, chronic dermatitis, variable immunodeficiency, and death in infancy. The diabetes often presents in neonates. Mice with the scurfy mutation develop lymphoproliferative disease, increased cytokine release, and wasting potentially associated with high levels of tumor necrosis factor-α. Similar to the etiologic gene AIRE for the APS-I syndrome, the scurfy gene is thought to be a transcription factor. The specific genes whose abnormal regulation results in this spectrum of illnesses are unknown, but it is now known that the Foxp3 gene is essential for the generation of CD4+ CD25+ regulatory T cells.
STAGES IN THE DEVELOPMENT OF TYPE 1A DIABETES MELLITUS
Type 1A diabetes can develop at any age. For example, Cilio and colleagues have described a child with fulminant autoimmunity and diabetes presenting at birth and we have followed the mother of a child with diabetes who developed type 1A diabetes at age 69 (67). Given this wide spectrum of age at diagnosis, long-term prospective studies are necessary to define the course of the disease. Such studies have benefited from the discovery of islet autoantibodies (68) and from the realization that type 1A diabetes is a chronic autoimmune disorder (69). We divide the development of type 1A diabetes into a series of stages beginning with genetic susceptibility (Fig. 23.3) and ending with complete or nearly complete β-cell destruction associated with a lack of C-peptide secretion. Studies of monozygotic twins illustrate many of the stages associated with type 1A diabetes. In that monozygotic twins are identical for all germ-line-encoded polymorphisms and mutations, if diabetes develops in one twin of a pair, the other twin is at high risk of developing diabetes (70,71). However, because diabetes does not develop in all such twins or activate autoimmunity (as evidenced by a lack of islet autoantibodies) (72), a subset of genetically susceptible individuals do not progress beyond stage I.
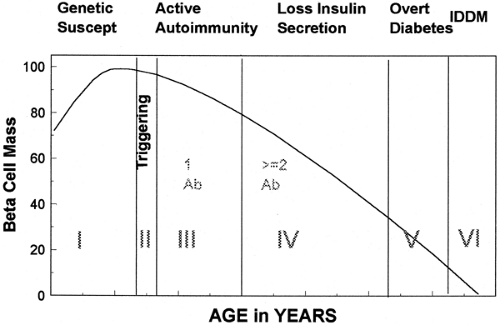 Figure 23.3. Stages in the development of type 1A diabetes. (Modified from Eisenbarth GS. Type I diabetes mellitus. A chronic autoimmune disease. N Engl J Med 1986;314:1360–1368, with permission. Copyright © 1986, Massachusetts Medical Society. All rights reserved.) |
Factors that induce the expression of islet autoantibodies are currently poorly defined. The great majority of individuals expressing islet autoantibodies that react with multiple islet antigens progress to loss of first-phase insulin secretion on intravenous glucose tolerance testing and eventually to overt diabetes (73,74). Islet autoantibodies can appear during the first year of life but also can first develop in adults. Figure. 23.4 illustrates
the course of an identical twin whose islet autoantibodies were first detected at age 42 at the time of diagnosis of diabetes, even though his twin mate developed diabetes at age 12. A subset of individuals who express islet autoantibodies are found not to progress to diabetes with long-term follow-up. These individuals usually express a single islet autoantibody, such as antibodies that react only with the molecule GAD (75,76). Progressive loss of first-phase insulin secretion characteristically precedes the development of hyperglycemia (77,78,79).
the course of an identical twin whose islet autoantibodies were first detected at age 42 at the time of diagnosis of diabetes, even though his twin mate developed diabetes at age 12. A subset of individuals who express islet autoantibodies are found not to progress to diabetes with long-term follow-up. These individuals usually express a single islet autoantibody, such as antibodies that react only with the molecule GAD (75,76). Progressive loss of first-phase insulin secretion characteristically precedes the development of hyperglycemia (77,78,79).
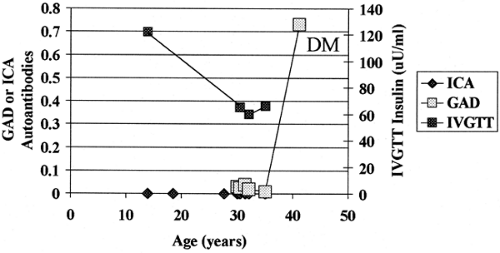 Figure 23.4. Late appearance of anti-islet autoantibodies in an initially discordant monozygotic twin of a patient with type 1A diabetes. IVGTT, intravenous glucose tolerance test; ICA, islet cytoplasmic antibodies; GAD, antibodies to glutamic acid decarboxylase. |
The detection of progressive loss of first-phase insulin secretion in the presence of islet autoantibodies led to the hypothesis that type 1A diabetes is a “chronic” autoimmune disorder (69). In that data concerning the prediabetic pancreas are limited, chronic progressive islet destruction remains a hypothesis. An alternative hypothesis is that chronic loss of first-phase insulin secretion results from functional inhibition of insulin secretion by the immune system and that the actual destruction of pancreatic β-cells is acute and follows a long latency period during which islet autoantibodies are expressed (80,81). We believe this is unlikely, given the available histologic data and the observation that loss of β-cell mass correlates with loss of first-phase insulin secretion (82). In particular, the pancreases of identical twins of patients with type 1A diabetes, when biopsied (on the occasion of living related donor transplantation), appear normal (83). Pancreases from individuals who have died at the onset of diabetes often have lost most β-cells but still have a significant mass of islet β-cells remaining (84). Several years after the onset of diabetes, as reflected by loss of C-peptide secretion, this remaining β-cell mass is lost. It thus appears that the chronic destruction that precedes type 1A diabetes continues after the development of hyperglycemia.
The histology of the pancreas in individuals at the onset of diabetes shows a diversity of islet lesions. Within islets, β-cells are selectively destroyed (84). This specificity of β-cell destruction is very dramatic, and for islets containing no β-cells, there is no insulitis. Thus, the same section of the pancreas may contain normal islets, islets with β-cells and insulitis, and islets lacking all β-cells and without insulitis (pseudoatrophic islets). This spotty process is conceptually similar to vitiligo, with destruction of patches of melanocytes. This destruction of β-cells of individual islets probably underlies the chronic progression to type 1A diabetes.
GENETICS OF TYPE 1A DIABETES MELLITUS
Familial Aggregation
The great majority of individuals in whom type 1A diabetes develops do not have a first-degree relative with the disorder (85% to 90%). If one has a first-degree relative with type 1A diabetes, the risk of diabetes is, however, much greater than that for the general population. In the United States, type 1A diabetes develops in approximately 1 in 300 individuals, compared with 1 in 20 first-degree relatives (85). The risk of diabetes in relatives can be stratified according to their relationship to the proband and by laboratory testing (e.g., HLA typing and determination of autoantibody expression).
Monozygotic twins of a patient with type 1A diabetes have an overall risk of developing type 1A diabetes of approximately 50% (72,86,87,88,89). This overall concordance for monozygotic twins is likely to be a composite of a number of different genetic syndromes resulting in type 1A diabetes, each syndrome potentially with a different concordance. In that only some of the genes contributing to the development of type 1A diabetes are currently known, concordance rates for specific genetic syndromes cannot be calculated accurately.
We have analyzed the development of diabetes in two large series of monozygotic twins, one from the United States (72) and the other from Great Britain (90). All twins in the study were selected to be discordant at the time of initial study. Both diabetes concordance and expression of islet autoantibodies were studied. Over time, diabetes develops in more twins. There is no duration of discordance at which the risk of diabetes disappears. Approximately 20% of those twins who became concordant did so after 15 years of discordance (91). With statistical methods to correct for pairs of twins who were both concordant at initial study and for discordant twins still being observed, the overall concordance is 50%. There is a marked difference in the hazard rate of progression to diabetes related to the age of the proband at diabetes onset (92). If the index twin (first diabetic twin) developed type 1A diabetes after age 20, concordance was less than 5%, compared with a concordance greater than 50% if the index twin developed diabetes before age 20. If the first twin developed type 1A diabetes before the age of 10, almost 5% of discordant twins per year developed diabetes in the first years of discordance, and the rate decreased to 1% per year only by 20 years of follow-up. In contrast, if diabetes developed in the first twin after age 20, the initial hazard rate was low and remained at approximately 1% per year for the duration of follow-up. These differences in concordance and hazard rates may reflect differences in genetic forms of type 1A
diabetes for groups of twins subdivided by the age of the proband twin at diagnosis of diabetes.
diabetes for groups of twins subdivided by the age of the proband twin at diagnosis of diabetes.
Discordant dizygotic twins have a diabetes risk that is similar to that of non-twin siblings of patients with diabetes: approximately 6% (72). The difference between monozygotic and dizygotic twins most likely represents a difference in the inherited genes shared with the proband (100% for monozygotic twins; mean of 50% for dizygotic twins). There is one report that dizygotic twins have a very high prevalence of islet autoantibodies (87), but with reanalysis and exchange of sera, the high prevalence of islet autoantibodies was not confirmed. Dizygotic twins do not have increased expression of islet autoantibodies compared with that in their siblings (72), and when monozygotic twins express islet autoantibodies, they usually progress to overt diabetes (72,92).
Offspring of a father and siblings of patients with type 1A diabetes have a similar risk of type 1A diabetes, while offspring of a mother with type 1A diabetes have a somewhat decreased risk (93). The difference in risk between offspring of mothers versus fathers with type 1A diabetes has not yet been explained. Preliminary data from the Diabetes Autoimmunity Study in the Young (DAISY) (94) suggest that offspring of fathers with type 1 diabetes more often are DQ8/DQ2 heterozygous than are offspring of mothers with type 1 diabetes. This may contribute to a slightly higher diabetes risk (Table 23.2).
TABLE 23.2. Approximate Frequency of Islet Autoantibodies (Ab+) and Development of Diabetes Mellitus for Initially Nondiabetic Relatives | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||
As will be discussed, the ratio of the overall risk of diabetes of a sibling of a patient with type 1 diabetes is 6% and the general population risk is 0.4%, giving a sibling familial aggregation ratio, γ-s of 15 (6/0.4) (10). This number provides some estimate of the influence of being a sibling in terms of familial (shared genetic and sibling environmental background) risk of developing type 1A diabetes. For genetic disorders, a γ-s of 15 is a moderate association. The most important locus for type 1A diabetes is the HLA region of chromosome 6. Polymorphisms 5′ of the insulin gene also contribute to the familial aggregation. A major portion of the genetic risk of type 1 diabetes cannot currently be assigned to specific genes (10,95,96).
The Major Histocompatibility Complex
The most important determinants of type 1A diabetes are genes within the major histocompatibility complex (MHC) on chromosome 6p21 (33,97). The MHC is approximately 4 million base pairs long with more than 200 known or predicted transcripts (5). The region is divided into class I [containing the classical histocompatibility antigens HLA (human leukocyte antigens) A, B, and C], class II (containing immune response genes, HLA-DP, -DQ, and -DR), and the class III region with genes of the complement cascade, as well as other genes influencing immune function. There are also other genes within this complex not related to immune function, such as the gene for 21-hydroxylase, an enzyme of adrenal steroidogenesis, and the gene determining hemochromatosis telomeric to the MHC.
HLA-DQ, -DR, -A, -B, and -C molecules are extremely polymorphic, with hundreds of different alleles (e.g., as of January 2000, 241 alleles for DRB1 had been identified) (98). An individual inherits one allele of each gene from each parent. The series of alleles on a given chromosome inherited from a parent are termed a haplotype. By typing alleles of multiple family members, one can determine the four haplotypes of a nuclear family (two haplotypes for each parent). Alleles of different genes in a population are often not randomly associated with each other on the same chromosome; it is then stated that the alleles are in linkage dysequilibrium. For example, the class I HLA alleles A1 and B8 and the class II alleles DR3 and DQ2 (DQ2=DQA1*0501, DQB1*0201) are commonly associated with each other on chromosomes (99,100,101). The term linkage dysequilibrium implies that not enough evolutionary time has elapsed for these allelic associations to be randomized by crossing-over events between alleles. This “DR3” haplotype that encodes a high risk for diabetes has been termed an ancestral or extended haplotype (102, 103). Because all of these alleles, as well as alleles of other genes within this extended haplotype, are inherited as a unit, the specific combination of alleles determining diabetes risk is difficult to pinpoint. Other HLA haplotypes do not show as extensive linkage dysequilibrium, and the primary importance of DQ and DR alleles to diabetes risk is apparent.
The nomenclature for HLA alleles has changed almost with each modern edition of the Joslin text but is unlikely to change again because it is now based on variants of nucleotide sequence of the HLA genes rather than serologic typing (98). For DQ molecules, both the α and β chain of the molecule are polymorphic; thus to specify a DQ molecule, one must specify both chains (e.g., DQA1*0501, DQB1*0201). In contrast, for DR molecules, the DR α chain is not polymorphic and only the DR β chain is specified (DRB1*0301). If a chain has a “silent” nucleotide polymorphism (amino acid sequence not changed), a fifth digit is added (e.g., DRB1*03011). The nucleotide sequences are typically determined with direct sequencing and computer analysis. With automated sequencers and polymerase chain reaction amplification of DNA, direct sequencing is relatively simple. The direct sequencing of two alleles (one for each chromosome) of an individual can result in a small amount of ambiguity for certain combinations of alleles. This is usually not a problem, and the sequences provide
much more information concerning exact alleles compared with that obtained with older methods of typing for HLA alleles.
much more information concerning exact alleles compared with that obtained with older methods of typing for HLA alleles.
One can define HLA disease associations with individual alleles of single genes (e.g., alleles of DQB or DRB), with haplotypes (e.g., alleles of HLA-A, -B, -DQ, -DR on a single chromosome), and with genotypes (alleles of both chromosome 6 haplotypes possessed by an individual). In a physiologic sense, genotypes contribute to diabetes risk (104,105,106,107,108). With so many different alleles, and different haplotypes, there are millions of theoretical genotypes. In practice, the genotypes associated with type 1A diabetes risk or protection are relatively limited, and specifying genotypes for DQ (DQA and DQB) and DRB alleles provides the great bulk of relevant information (Table 23.3). The disease associations of genotypes can be complex. For instance, DQA chains coded for by one haplotype can combine with DQB chains of the other haplotype and influence diabetes risk. DQB1*0301 is in linkage dysequilibrium with DR4 but is not a high-risk allele (and is relatively protective). Thus, most white patients with type 1A diabetes have the DQB1*0302 allele on their DR4 haplotypes and not DQB1*0301 (109). The DQB1*0301 appears to be protective primarily in patients who also have a DR3 haplotype (with DQA1*0501, DQB1*0201) on their other chromosome. However, in combination with other haplotypes, such as the DR1(DQA1*0101, DQB1*0501), DQB1*0301 increases diabetes risk (110,111,112).
TABLE 23.3. Risk of Type 1A Diabetes Mellitus Associated with DR and DQ Haplotypes | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
The prevalence of given genotypes (and haplotypes) varies among populations, but given the same DQ and DR genotypes, the influence of HLA on type 1A diabetes is similar for populations throughout the world (113). Thus, even in populations with a very low incidence of type 1A diabetes (e.g., Korea and Japan), when similar genotypes are present, they confer increased or decreased risk of diabetes. Investigators have attempted to encapsulate the HLA-DQ risk by reference to several specific amino acids. The most common rule is that aspartic acid at position 57 of the DQB is protective and that arginine at DQA 52 is associated with increased diabetes risk, but there are many exceptions (114). Figure. 23.5 illustrates the transmission of a series of DQB alleles from parents to children with type 1A diabetes (115). The transmission test corrects for differences in the prevalence of alleles in the population, as transmissions are counted only from parents heterozygous for the specific allele (115,116). If DQ alleles did not influence diabetes risk, all alleles would be transmitted 50% of the time to children with diabetes. As is apparent from the figure, there is a tremendous spectrum of transmission frequencies for different alleles, and a similar spectrum is documented by studies of large populations (117). The rules concerning aspartic acid at position 57 provide little predictive power. For instance DQB1*0402 is one of the alleles with the highest transmission frequency, DQB1*0301 is intermediate, and DQB1*0602 is one of the most protective alleles. All have aspartic acid at position 57 of their DQB chain. Since we currently can define all of the polymorphisms of these chains, knowledge of the complete sequence rather than of single amino acid polymorphism is preferred for assessing diabetes risk.
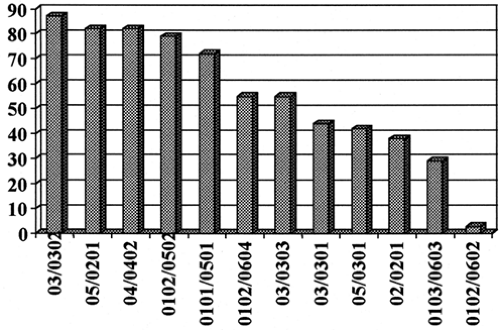 Figure 23.5. Percent transmission of DQA1/B1 alleles from parents heterozygous for the indicated allele to affected children. The first number refers to DQα and the second to DQβ. (Copyright © 1998 American Diabetes Association. From Kawasaki E, Noble J, Erlich H, et al. Transmission of DQ haplotypes to patients with type 1 diabetes. Diabetes 1998; 47:1971–1973. Reprinted with permission from the American Diabetes Association.) |
HLA-associated protection from type 1A diabetes is dominant relative to susceptibility (108,118). The three most protective molecules are DQA1*0102 with DQB1*0602, DQA1*0201 with DQB1*0303, and DRB1*1401. The latter two molecules are relatively rare, whereas in most populations, more than 20% of individuals have DQA1*0102, DQB1*0602 and fewer than 2% of children with type 1A diabetes have this allele. Note that both DQ and DR alleles can protect against the development of type 1A diabetes; this observation is similar to findings in animal models, in which alleles of the equivalent genes (I-A, DQ; and I-E, DR) both can provide protection (119,120). Transmission of haplotypes from parents to affected offspring was analyzed for the Norwegian NODIAB study (116) and the HBDI (Human Biologic Disease Interchange) repository (121). This allowed analysis of rare “recombinant” haplotypes such as DRB1*0101 or 1404 with DQA1*0101, DQB1*0503 in comparison to the usual DRB1*1401, DQA1*0101, DQB1*0503 haplotype (Table 23.4).
The DRB1*1401 allele was transmitted to none of 40 diabetic offspring, whereas DQB1*0503 without DRB1*1401 was transmitted to 2 of 4 diabetic offspring. In this case, DRB1*1401 apparently determines protection. DQB1*0602 was transmitted to 0 of 6 diabetic offspring for haplotypes without DRB1*1501; transmission of DQB1*0602 with DRB1*1501 was also rare (2 of 313). In contrast, DRB1*1501 without DQB1*0602 was transmitted to 5 of 11 diabetic offspring. For this haplotype, DQB1*0602 apparently provides protection (Table 23.4).
The DRB1*1401 allele was transmitted to none of 40 diabetic offspring, whereas DQB1*0503 without DRB1*1401 was transmitted to 2 of 4 diabetic offspring. In this case, DRB1*1401 apparently determines protection. DQB1*0602 was transmitted to 0 of 6 diabetic offspring for haplotypes without DRB1*1501; transmission of DQB1*0602 with DRB1*1501 was also rare (2 of 313). In contrast, DRB1*1501 without DQB1*0602 was transmitted to 5 of 11 diabetic offspring. For this haplotype, DQB1*0602 apparently provides protection (Table 23.4).
TABLE 23.4. Evidence that DQB1*0602 and DRB1*1401 Haplotypes Are Protective (Transmission of Haplotypes from Parent to Affected Child) | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
To date we have found 13 (1.5%) of 877 individuals with type 1A diabetes (as evidenced by positivity for islet autoantibody and the presence of diabetes) with DQB1*0602. Their DQB sequence did not differ from the standard sequence (97,118). One patient had the APS-I syndrome, although overall it is likely that, in patients with APS-1, DQB1*0602 is also protective for diabetes but not for Addison’s disease (26,56). Another child had high levels of 21-hydroxylase autoantibodies, often associated with polyendocrine autoimmunity. Thus, we suspect that some individuals with DQB1*0602 and type 1A diabetes are likely to have variant genetic forms of type 1A diabetes. Among the patients with DQB1*0602, the other allele is usually a high-risk HLA allele [10 (77%) of 13 DQ8 or DQ2]. Two of the three patients lacking DQ8 and DQ2 had DQB1*0402 (n = 2). Analysis of islet autoantibody-positive relatives of Diabetes Prevention Trial Type-1, for which the presence of DQB1*0602 is an exclusion criteria, indicates that 7% of cytoplasmic islet cell antibody (ICA)-positive relatives are positive for DQB1*0602 (122). There was an increased prevalence of DQB1*0602 among African-American ICA-positive relatives, again suggesting genetic heterogeneity (122).
The highest risk genotype for type 1A diabetes consists of DRB1*0301/DQA1*0501, DQB1*0201 (DQ2) with DRB1*0401 (or 0402, or 0405)/DQA1*0301, DQB1*0302 (DQ8) (9). This genotype occurs in approximately 2.4% of newborns in Denver, Colorado, and in approximately 40% of children with type 1A diabetes (16). A newborn with this genotype without a relative with type 1A diabetes is estimated to have a risk of diabetes of approximately 6%. In contrast, a sibling of a patient with type 1A diabetes with the same genotype has a risk of developing diabetes that is greater than 25%. We believe the greatly enhanced risk for a relative relates both to non-MHC genes inherited by such relatives and to other HLA alleles associated with increased diabetes risk. A number of other HLA genotypes are associated with high to moderate risk for type 1A diabetes, including DR 4/4, DQ 8/8 (DQ8 = DQA1*0301, DQB1*0302); DR 3/3, DQ 2/2 (DQ2 = DQA1*0501, DQB1*0201); DR 1/4, DQ 1/8; DR 1/4, DQ 1/7 (DQ7 = DQA1*0301, DQB1*0301). The DRB1 allele DRB1*0403 modifies the risk of diabetes associated with DQA1*0301, DQB1*0302 (DQ8) such that the haplotype DRB1*0403, DQA1*0301, DQB1*0302 is slightly protective (105, 123).
Genotypes consisting of both DR and DQ alleles appear to underlie the bulk of MHC-associated susceptibility. Nevertheless, there are likely to be alleles of other genes within the MHC contributing to susceptibility to diabetes. Class I alleles that present peptides to cytotoxic CD8 T lymphocytes are likely to influence disease risk or the age of development of type 1A diabetes. The allele A24 is reported to be associated with a younger age of onset of type 1A diabetes, and there is evidence that HLA-A2 influences diabetes risk (124). Alleles of the nontypical HLA allele, termed MIC-A, have been reported to be associated both with type 1A diabetes and with Addison’s disease (63). Alleles of several microsatellite markers (repeat elements) that simply mark chromosomal regions are reported to be associated with diabetes risk and subdivide haplotypes with fixed DR and DQ alleles. Such analyses are particularly difficult because of the extensive linkage dysequilibrium of alleles of genes within the MHC and the importance of class II alleles (DR and DQ, as well as influence of DP) (125,126).
The Insulin Gene
Another locus clearly associated with risk for type 1A diabetes is the insulin gene locus (127,128,129). Alleles of a variable nucleotide tandem repeat 5′ of the insulin gene influence the risk of type 1A diabetes. This tandem repeat has been divided into three major sizes, and the longest tandem repeats are associated with decreased risk of type 1A diabetes. The longer tandem-repeat group is also associated with increased expression of insulin messenger RNA within the thymus (130,131). In animal models, expression of minute amounts of insulin message within the thymus correlates with protection from diabetes and insulitis (132,133). An attractive hypothesis is that insulin is a key molecular target and that greater thymic expression of insulin leads to tolerance and thus decreased risk of diabetes.
Oligogenic versus Polygenic Inheritance
Genes within the MHC and the insulin locus account for approximately 50% of the familial aggregation of type 1A diabetes. The remaining familial aggregation is probably due to additional inherited polymorphisms/mutations, and the lack of 100% concordance of identical twins could be due to somatic mutation, random rearrangement of genes (e.g., rearrangements in the T-cell receptor), differences in environmental exposures, or stochastic events. We believe that non-MHC genetic loci are of major importance. For one of the best animal models of type 1A diabetes, the NOD mouse, the inheritance is polygenic, with more than 15 different genetic loci influencing susceptibility to diabetes (134,135,136).
Diabetes in the NOD mouse is a potential example of a single form of human type 1A diabetes. This is not strictly accurate, in that NOD mice are brother-sister mated for more than 20 generations (inbred) and thus have no genetic variability. NOD mice are not diallelic at any locus. Humans have different alleles at more than 100,000 loci, many within coding regions of
genes. The inbred nature of the NOD mouse may relate to the ease with which multiple small genetic changes, as well as therapeutic interventions, prevent diabetes (135). Such ease of prevention is less likely in humans. Other animal models do not show a polygenic form of inheritance but show an “oligogenic” inheritance, with a few major loci (including the histocompatibility complex) determining diabetes risk (137,138).
genes. The inbred nature of the NOD mouse may relate to the ease with which multiple small genetic changes, as well as therapeutic interventions, prevent diabetes (135). Such ease of prevention is less likely in humans. Other animal models do not show a polygenic form of inheritance but show an “oligogenic” inheritance, with a few major loci (including the histocompatibility complex) determining diabetes risk (137,138).
Whether human type 1A diabetes is polygenic or oligogenic is currently debated. Practical ramifications of the mode of inheritance are likely to relate to the ease of defining genes underlying susceptibility, identifying at-risk individuals, and discovering pathways for prevention of diabetes. We favor the hypothesis that type 1A diabetes is oligogenic but heterogeneous (139). Namely, in most families, one to three loci determine diabetes susceptibility but different families have different major “diabetogenes.” The bulk of the effort to define diabetogenic loci has involved the study of hundreds of families with two siblings (sib pairs) with type 1A diabetes. These studies have implicated more than 15 loci associated with diabetes risk but have been difficult to replicate (96). For example, several studies have suggested that a polymorphism of CTLA-4 may contribute to diabetes risk in certain populations but not in others (Table 23.5) (140,141,142,143,144,145). A missense mutation of the LYP gene (PTPN22; lymphocyte tyrosine phosphatase) is a major “autoimmune” allele (Arg620Trp) and has an odds ratio of approximately 1.7 for type 1 diabetes (145a).
TABLE 23.5. Confirmed and Putative Loci Associated with Type 1A Diabetes Mellitus | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
If type 1A diabetes is heterogeneous and if no specific genetic syndrome accounts for more than 20% of cases of type 1A diabetes, one may need to define relevant genes by studying specific families before application to mixed populations. We have studied a Bedouin Arab family from Israel that has 21 members with type 1A diabetes. In this family, diabetes is associated with typical HLA alleles and with a locus on chromosome 10, currently termed IDDM17 (139). Individuals homozygous for related 10q25.1 haplotypes and with DR3 have a 40% risk of developing type 1A diabetes. In contrast, relatives lacking DR3 or who are not homozygous have a risk of type 1A diabetes lower than 2%. This suggests that a major diabetogene on chromosome 10 is segregating with diabetes and that diabetes develops from a combination of this locus and high-risk HLA alleles.
None of the putative non-MHC loci except for LYP and the insulin gene contribute to the assessment of risk for type 1A diabetes, and it is likely that genes, rather than loci, will need to be defined before such studies will be of major influence. The Genome Project should greatly facilitate studies to identify these genes, and it is now possible to create detailed maps of any region of the human genome and, thus, to analyze transmission and linkage dys-equilibrium for specific haplotypes.
ENVIRONMENTAL FACTORS
A number of potential environmental causes of type 1 diabetes have been reported, but few have been established beyond doubt. However, there are examples of autoimmune diseases that not only are caused by environmental factors but also can be prevented or treated through avoidance of these factors (e.g., celiac disease). These diseases occur only through a strong interaction between environmental factors and specific alleles of mainly HLA genes, suggesting the futility of defining “genetic” versus “environmental” etiology.
Celiac disease, discussed earlier in this chapter, is induced by ingestion of wheat protein gliadin (146,147,148) in a person with DQ2 or DQ8 haplotypes. Removal of wheat from the diet results in the loss of transglutaminase autoantibodies and resolution of the intestinal lesions (149). There is some evidence that the delayed introduction of wheat into infant feeding in recent years has prevented a substantial number of symptomatic cases, although some of such prevented cases may manifest with milder symptoms later in life. Rheumatic fever, another autoimmune disease, is induced by streptococcal infection of a host with an HLA-DR4 genetic background. It has been nearly eradicated in many countries with the introduction of antibiotics and guidelines for diagnosis and treatment of streptococcal infections.
Diabetes in patients with congenital rubella syndrome is perhaps the only form of type 1A diabetes proven to be caused by a viral infection (150,151,152). Non-congenital rubella infection does not appear to be associated with diabetes risk. At present it is not clear whether congenital rubella is associated with endocrine autoimmunity caused by direct infection of target tissues, the triggering of autoimmunity (153), or the induction of changes in the immune system that lead to autoimmunity (154). Although only a small proportion of cases of type 1A diabetes have been found to be caused by congenital rubella infection, this is the only type of autoimmune diabetes that we currently can prevent (using live attenuated rubella vaccine, a component of the MMR vaccine).
Figure. 23.6 contrasts prevalence of type 1A diabetes for a series of countries with dramatic differences in its incidence and compares such diabetes prevalence to diabetes risk, taking into account genetic information. The incidence of type 1A diabetes varies geographically by more than 45-fold (e.g., in Japan the approximate incidence is 1 in 100,000 compared with Finland, where it is 45 in 100,000) (155,156,157,158,159). Nevertheless, even the highest-risk Scandinavian countries have a prevalence of type 1A diabetes less than 3%, while an identical twin of a patient with type 1A diabetes from Japan has a diabetes concordance of approximately 30% (88).
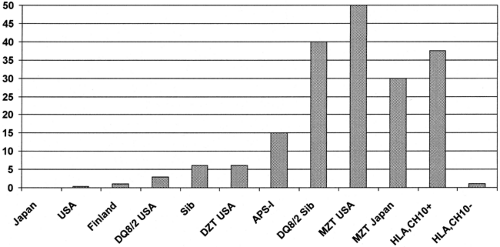 Figure 23.6. Prevalence of type 1A diabetes: geographic and genetic variation. DZT, dizygotic twin; MZT, monozygotic twin; HLA, Ch10+, patient from a Bedouin Arab family with HLA DR3 and homozygous for IDDM17 diabetes-associated haplotypes (Japan, 0.02%; USA, 0.3%; Finland, 1%) |
A sibling or offspring of a patient with type 1A diabetes with HLA DQ8/DQ2 has a diabetes risk of approximately 20% to 40% (17,160), and an individual with this genotype from the general U.S. population has a risk of approximately 6%. In addition, a dizygotic twin of a patient with type 1A diabetes has a risk of diabetes very similar to that of a sibling (72). This suggests that, if there are environmental factors triggering the development of type 1A diabetes, they account for a relatively small amount of the familial aggregation of the disease. This may explain the difficulty finding such environmental factors. One exception previously discussed is the rare congenital rubella syndrome that is strongly associated with the development of thyroiditis and type 1A diabetes (161,162,163) and recent reports from two studies that introduction of cereal before 3 months of age increases risk (160a,160b).
TRIGGERING OF AUTOIMMUNITY
The most prominent proposed environmental triggers for type 1A diabetes are infections (particularly viruses), vaccination, and dietary factors (163). There is lack of clear consensus concerning the etiologic role of any of these factors. For both the NOD mouse and the BB rat, the development of diabetes is enhanced when animals are raised in a germ-free environment (164). This suggests that, for both of these animal models, infections are not necessary for the development of diabetes. In fact, when either animal becomes infected with relatively common viruses, the development of diabetes decreases. The BB rat has a severe T-cell lymphopenia inherited in an autosomal recessive manner (165,166,167,168,169). For strains of BB rats termed DR (diabetes resistant) that lack the T-cell lymphopenia locus, diabetes can be induced with a number of immunostimulatory maneuvers, including infection with the rat Kilham virus or with induction of the expression of interferon-α with injection of poly-IC (poly-inosinic-polycytidylic acid, a synthetic double-stranded RNA) (170). Of note, administration of poly-IC induces diabetes and insulitis or just insulitis in a broad range of rat strains with “HLA” class II U alleles (diabetes and insulitis strains: lew1.WR1, PVG.RT1u, PVG.R8, WF, and WAG; only insulitis-LOU strain). In animal models, several forms of “vaccination” prevent diabetes (e.g., BCG vaccination for NOD mice), and dietary modification, usually with removal of complex proteins from the diet, decreases the development of diabetes (171,172,173). Thus, such manipulations are potential triggers of autoimmunity or may provide therapies (e.g., removal of triggering factors) of relevance to diabetes prevention.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


