Tumor Progression and Metastasis
10.1 TUMOR PROGRESSION
10.1.1 Cellular Aspects of Tumor Progression
Cancer is not a static disease. In many tumors (eg, colon, breast, cervical, pancreatic, melanoma), there appears to be an orderly progression from benign tissue to premalignant lesion to frank malignancy. In other tumors, premalignant lesions may not have been identified, but it is likely the tumor has passed through less-malignant stages before detection. The pathological and clinical criteria for tumor progression are often specific to a given type of tumor, but include local spread along tissue planes and into various tissue spaces and cavities. Tumors also have the capacity to invade and spread from their origins to other organs in the body; This process is referred to as metastasis. Increasing numbers and types of genetic abnormalities accompany tumor progression and metastasis.
More than 50 years ago, Foulds defined tumor progression as “the acquisition of permanent, irreversible qualitative changes in one or more characteristics of a neoplasm” that cause the tumor to become more autonomous and malignant. In 1986, Nowell proposed that such changes arise because cancer cells tend to be genetically unstable and described a conceptual model to explain the process of tumor progression (Fig. 10–1). The key features of this model are the generation of mutant cells within a tumor and the selection and outgrowth of more autonomous cells to become dominant subclones in the population, leading to progression of the tumor and increasing malignancy. Many studies have confirmed the genetic instability of malignant cells (see Chap. 5, Sec. 5.2 and Chap. 7, Sec. 7.4), and have identified somatic DNA copy number changes and mutations using genome-wide analyses (see Chap. 2, Sec. 2.7) that are becoming routine in cancer research and in the clinic. Consistent with this model, recent studies have identified different clonal populations within tumors, raising the possibility for a minor (resistant) subpopulation to cause tumor recurrence following therapy (Navin et al, 2011). The growth and development of various cells within a tumor is largely subject to constraints associated with interactions among the tumor cells, the stromal cells, and the extracellular environment. Thus the normal homeostatic mechanisms that control cell proliferation in the body (see Chap. 9, Sec. 9.2) are not completely lost in tumor cells, but rather the cells may become increasingly less responsive to them. In addition, tumor cells acquire autonomous means to grow, becoming less dependent on extraneous growth factors (Kopfstein and Christofori, 2006). These findings are consistent with the original concepts of Foulds that there are many different paths to malignancy. Tumors are thus evolving cell communities with properties that continue to change as they grow. The role of the stromal cell populations and the extracellular microenvironment is increasingly recognized as a critical element in tumor development and progression (Hanahan and Weinberg, 2011).
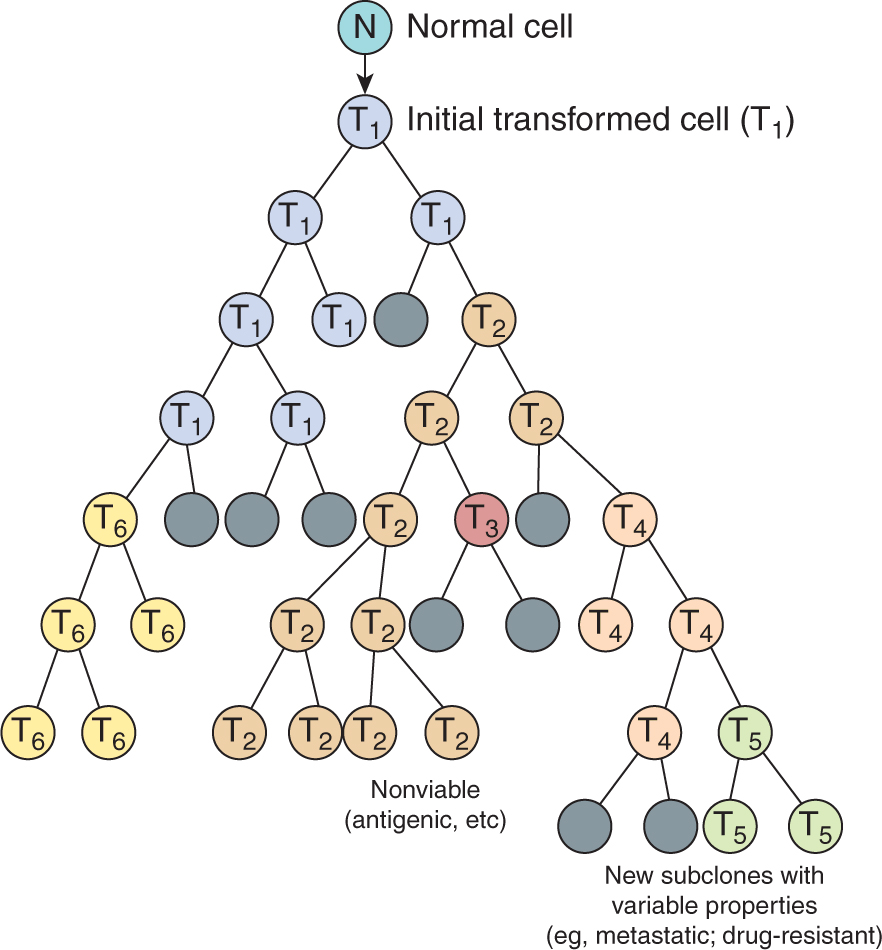
FIGURE 10–1 Schematic showing the clonal evolution of tumors. New subclones arise by mutation. Many of these may become extinct (indicated by dark shading) but others may have a growth advantage and become dominant. All of the subclones (indicated by T2 to T6) may share common clonal markers, but many of them have new properties leading to heterogeneity.
10.1.2 Molecular Genetics of Tumor Progression
Genetic instability of tumor cells may arise as a result of genetic and/or epigenetic changes. Epigenetic changes such as methylation of cytosine bases in DNA or modifications to chromatin structure (eg, by methylation, acetylation, sumoylation, or phosphorylation) can modify the expression of genes and are an important mechanism for “silencing” genes during normal differentiation (see Chap. 2, Sec. 2.3). Genetic changes may occur by point mutation, deletion, gene amplification, chromosomal translocation or other mechanisms (see Chap. 5, Sec. 5.2). A cell is continually exposed to both external and internal stresses, such as reactive oxygen species, which may cause DNA damage. Moreover, there are inherent errors made by DNA polymerases whenever DNA is being replicated. Normally such damage is either repaired by the various DNA repair mechanisms in the cell (see Chap. 5, Sec. 5.3) or damaged cells undergo apoptosis (see Chap. 9, Sec. 9.4). However, these mechanisms are not perfect, leading to a natural frequency of mutation in cells.
Many cancer cells appear to have an increased frequency of mutation because of deficiencies in their ability to repair lesions in DNA and/or decreased activation of apoptosis, so that mutated cells may survive and proliferate. For example, the breast cancer-related genes, BRCA1/2, are linked with DNA double-strand break repair. Oxidative lesions and deficiencies of mismatch repair have been demonstrated in tumor cells, particularly those from patients with certain types of colon cancer. A deficiency in mismatch repair can result in up to a 1000-fold increase in the mutation frequency. Failures in DNA damage repair may result in mutations or alterations in the expression of the many oncogenes and tumor-suppressor genes that are associated with different human cancers (see Chap. 7). Mutations in these genes are not necessarily more frequent than in other genes; rather the mutations are selected for in the process of cancer development, and thus are the ones that are most frequently detected. However, because of their genetic instability, cancer cells may also carry many “passenger” mutations that play little or no known role in their cancer phenotype.
The multiple changes that must occur in cells during tumor development and progression are illustrated by the model established by Vogelstein and colleagues describing the progression of colon cancer (Fearon and Vogelstein, 1990; Fig. 10–2). This model provides a paradigm for multistep carcinogenesis that has been applied to many other cancers (eg, breast, pancreatic, bladder, and lung), although recent studies have emphasized that the steps do not always occur in a specific order. These concepts have been reinforced by studies involving genetic manipulation of normal human primary cells that have demonstrated that sequential modifications involving activation of oncogenes or inactivation of tumor suppressors can result in their transformation (Hahn et al, 1999). Thus a molecular description of tumor progression envisages that cancers progress as a result of a series of (selected) genetic and epigenetic changes. Some of these changes are shared between different cancers, but changes unique to specific types of cancer also occur and the sequence of the multisteps may not always follow the same time line in different individual cancers of the same type.
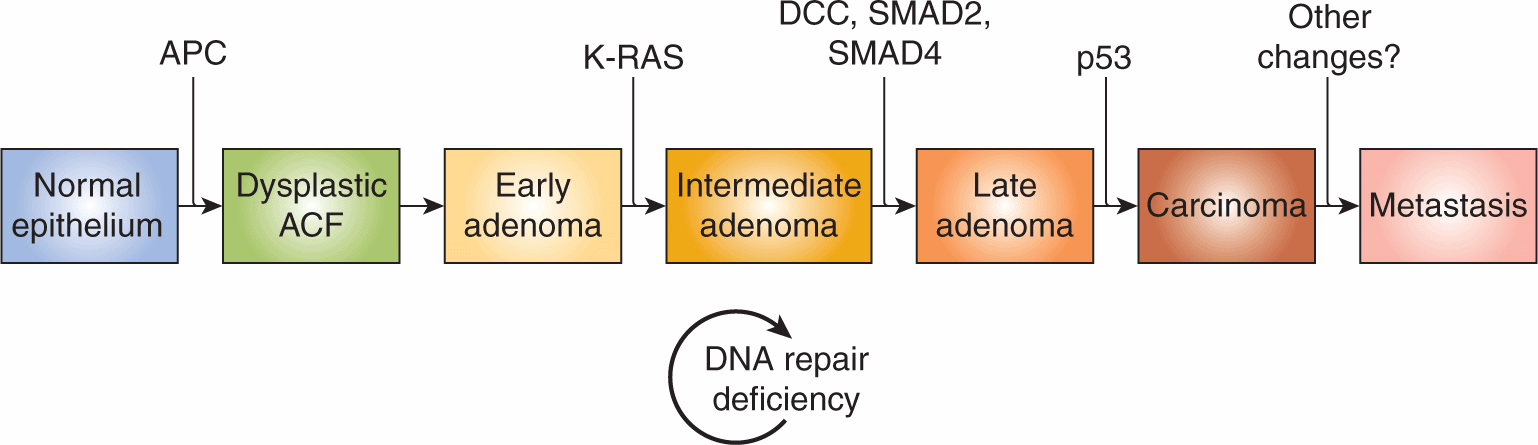
FIGURE 10–2 Genetic changes associated with colorectal tumorigenesis. Anaphase promoting complex (APC) mutations initiate the neoplastic process, and tumor progression results from mutations in the other genes indicated. Patients with familial adenomatous polyposis (FAP) inherit APC mutations and develop numerous dysplastic aberrant crypt foci (ACF), some of which progress as they acquire the other mutations indicated in the figure. The tumors from patients with hereditary nonpolyposis colorectal carcinoma (HNPCC) go through a similar although not identical series of mutations; DNA repair deficiency speeds up this process. K-ras is an oncogene that requires only 1 genetic event for its activation. The other specific genes indicated are tumor-suppressor genes that require 2 genetic events (1 in each allele) for their inactivation. Chromosome 18q21 may contain several different tumor-suppressor genes involved in colorectal neoplasia, with DCC (deleted in colon cancer), SMAD2 and SMAD4 genes proposed as candidates. A variety of other genetic alterations have each been described in a small fraction of advanced colorectal cancers. These may be responsible for the heterogeneity of biological and clinical properties observed among different cases.
As a result of their genetic and epigenetic changes, cells within animal and human tumors demonstrate considerable heterogeneity in their phenotypes. This heterogeneity includes morphology, karyotype, surface markers, biochemical pathways, cell proliferation, metastatic ability, and sensitivity to therapeutic agents. The ability of tumor cells to disseminate and form metastases represents the most malignant characteristic of a cancer. Therefore, it is important to understand how cancer cells metastasize and to determine the underlying genetic and molecular causes of metastasis. As described in Sections 10.4 and 10.5, metastases probably arise from a small subset of cells within a primary tumor that have undergone genetic or epigenetic changes that enhance their ability to metastasize. This feature of tumors has made it difficult to determine the cellular and molecular properties critical for metastatic spread, as the bulk of the tumor cell population may not reflect the specific properties of the individual cells responsible for the metastases.
10.2 TUMOR MICROENVIRONMENT
10.2.1 Extracellular Matrix
Most mammalian cells are in contact with an extracellular matrix (ECM). The composition and structure of the ECM is specific to location and developmental stage. For example, epithelial cells have specialized lateral, apical, and basal borders. The latter’s interactions with the basement membrane are instrumental for the formation, maintenance, and polarized differentiated state of the epithelial cell sheet. The basement membrane, a specialized form of ECM, is composed of laminin, type IV (and VII) collagen, entactin/nidogen, and heparan sulfate proteoglycan (HSPG), as well as smaller amounts of fibronectin, vitronectin, and chondroitin sulfate proteoglycans. To exert tissue-specific control, basement membranes vary in composition while maintaining a common set of structural and mechanical properties (Rowe and Weiss, 2009). There are at least 7 forms of laminin and 6 type IV collagen chains that interact with other ECM proteins to generate a 3-dimensional (3D) interlocking structural network. In contrast to epithelial cells, mesenchymal cells are not attached to each other or to a basement membrane, but are surrounded by an ECM that contains the interstitial collagens types I to III, elastin, proteoglycans, fibronectin, and vitronectin. Other specialized tissue-specific ECM molecules include tenascin, thrombospondin, and osteopontin. The highly organized 3D matrix provides an adhesive environment for cells and other molecules, such as growth factors. Interaction of cells with the ECM is essential for growth and survival, and the ECM can also regulate the differentiation of a variety of cell types. Depriving normal cells of such interactions results in the induction of apoptosis (anoikis) in epithelial and endothelial cells (Boudreau et al, 1995), or cell-cycle arrest in fibroblasts (Fang et al, 1996). Transformed cells are often defective in secreting fibronectin and in laying down an organized matrix and a common property of malignant cells is their ability to survive and proliferate with a lower dependence on interactions with an ECM than normal cells. Many studies report disruption of basement membrane continuity and collagen degradation at sites of tumor cell invasion, and breakdown of ECM integrity is considered a rate-limiting step in tumor progression and metastasis.
The ECM provides structural elements that can interact with various molecules expressed on the cell surface (cell adhesion molecules; see Sec. 10.2.3). These latter molecules also interact with specific signal transduction pathways in the cell. Growth factors can be soluble, or embedded within the matrix, or anchored to the cell surface in close proximity to their cell surface receptors that are also directly linked to cell signaling (see Chap. 8). Within this complex milieu are also extracellular, plasma membrane-bound, and ECM-bound protein-ases. Normally, a balance exists in the biochemical activities of these various molecular entities, which is often disrupted within the tumor cell microenvironment. Aberrant proteolysis is an important means of offsetting the balance and can exert a “ripple” effect on multiple classes of molecules during tumor progression and metastasis. It is one hallmark of tumor cells that underlies their metastatic dissemination. Proteolysis is influenced by and localized via cell adhesion molecules, and it, in turn, acts on an array of substrates, including the ECM, cell adhesion molecules, growth factors, and cytokines, and their receptors and binding proteins. For example, increased matrix metalloproteinase (MMP)-mediated activity brings about remodeling of the ECM and basement membranes and influences cell adhesion by cadherins. As discussed in Section 10.5.6, loss of E-cadherin can promote epithelial-to-mesenchymal transition (EMT) in cells, potentially creating a phenotype leading to more aggressive cancers.
10.2.2 Cellular Proteinases and Their Inhibitors
As indicated above (and in Secs. 10.4 and 10.5), a series of tissue barriers (eg, basement membrane, interstitial connective tissue) are traversed by tumor cells during invasion and metastasis by processes involving proteolytic breakdown of the ECM. Mammalian proteinases fall into 4 major classes (serine-, cysteine-, aspartic-, and metalloproteinases), and many of these are associated with increased aggressiveness of tumor cells, and are functionally implicated during metastasis (Table 10–1). These diverse proteases have distinct structures and most have endogenous inhibitors that maintain a balance to keep proteolysis under strict control in normal tissues. Both the enzymes and their inhibitors may be aberrantly expressed by cancer cells and multiple other cell types within the tumor microenvironment.
TABLE 10–1 Proteinases in specific catalytic classes.
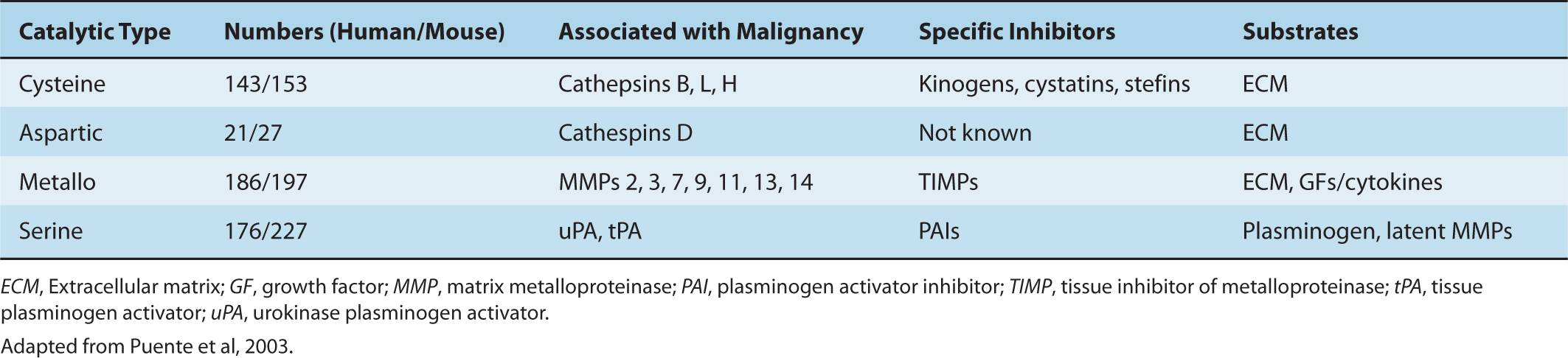
Metalloproteinases comprise the biggest family of proteases in the human genome with a total of 186 members. MMPs, including membrane type (MT)-MMPs, and are the major enzymes responsible for degradation of ECM proteins. Extracellular MMPs are often secreted in a latent form (pro-MMP), and subsequently activated. The MMPs chelate 2 zinc ions: 1 is present in the active site and the other associates with the pro-MMP to stabilize the inactive state (Egeblad and Werb, 2002). These enzymes can be autocatalyzed, activated by other MMPs, or activated by a serine proteinase. Cell-surface anchored proteases, such as MT-MMPs, are activated by proprotein convertases, some of which are also critical for the localization and cell-surface activation of soluble pro-MMPs. For example MT1-MMP is essential for the activation of pro-MMP2 at the cell surface. Interactions among different classes of proteases also generate complex proteolytic cascades which can amplify their activity. Within the metalloproteinase family, the ADAMs (a disintegrin and metalloproteinase) are unique in that they possess both adhesion and proteolytic domains (Werb, 1997). These transmembrane proteases act as “sheddases” to release cell surface anchored growth factors and cytokines in a process called ectodomain shedding; in addition, they are critical for the activation of receptor-mediated pathways such as NOTCH and epidermal growth factor receptor (EGFR) (Murphy, 2009).
Plasminogen activators (urokinase type [uPA] and tissue type [tPA]) are serine proteinases that act on circulating plasminogen to release plasmin and have long been associated with malignant cells. The activity of plasminogen and plasmin is localized to the cell surface by the uPA receptor (uPAR), which also associates with integrins and can bind to vitronectin in the ECM (Laufs et al, 2006). Increased expression of uPA has been correlated with metastasis in a number of cancers including human epidermal growth receptor 2 (HER2)-positive breast cancer (see Chap. 20, Sec. 20.3.3). The level of expression of plasminogen activator inhibitor (PAI)-1 also provides an independent unfavorable prognostic factor for the development of metastases for a population of hormone receptor- and lymph node-positive breast cancer patients (Schmitt et al, 2011). Among serine proteases, kallikreins are coded by a contiguous cluster of protease genes in the human genome and have been studied extensively for their utility as serum cancer biomarkers. For example, prostate-specific antigen (PSA) is the most commonly used kallikrein for routine evaluation of prostate cancer progression.
Cathepsins are cysteine proteases generally present on intracellular cell membranes localized to endosomal or lysosomal vesicles. Their proteolytic activity was presumed to be restricted to the intracellular compartment, but it was later discovered that cathepsins can also be found outside the cell during pathological conditions and their presence in body fluids is a prognostic indicator for several cancers. Slight structural differences between the eleven cathepsins are responsible for differences in substrate specificity and inhibition by their endogenous inhibitors. Cathepsins B, H, and L are particularly associated with tumor progression. An imbalance between the cathepsins and their inhibitors can occur during tumor progression and may be responsible for direct digestion of the ECM or activation of other proteolytic enzymes, such as uPA.
Proteinase inhibitors are produced by both malignant and normal cells. Examples of these inhibitors are PAI-1 and PAI-2, the cathepsin inhibitors, cystatins, kininogens and stefins, and tissue inhibitors of metalloproteinases (TIMPs). Under physiological conditions a balance between activated proteinases and their inhibitors keeps proteolysis under strict control; but when this balance is disrupted, (malignant) cells can invade tissues. Downregulation of TIMP-1 activity in immortalized murine fibroblasts, using transfected antisense RNA (see Chap. 2, Sec. 2.4.3), was found to confer invasive capacity and ability to form metastatic tumors in nude mice (Khokha et al, 1989). Increased levels of TIMP-1, TIMP-2, or TIMP-3 reduce the invasive and metastatic ability of malignant cells, and TIMP-1 provides increased resistance to metastatic colonization of organs. Gain- and loss-of-function studies have addressed the causal role of specific TIMPs and cystatins with various processes underlying tumorigenesis (Egeblad and Werb, 2002), and some studies suggest that TIMPs can function in a manner independent of their MMP-inhibitory function. However, the relationship between advanced malignancy and increased proteolytic activity (such as that arising from increased MMP or decreased TIMP expression) is not always clear. For example, increased MMP activity during cancer progression can be associated with a favorable prognosis, as is the case of MMP-12 in colon cancer, and increased TIMP expression has been identified to be a poor prognostic indicator in many studies (Egeblad and Werb, 2002). The recognition that proteolysis influences basic cellular processes, including cell division, differentiation, dissociation, and death (Hojilla et al, 2003) highlights the complexity of the proteolytic balance and the difficulty of predicting how changes in this balance may affect cancer progression.
10.2.3 Cell Adhesion Molecules
The cell–cell and cell–ECM interactions during invasion and metastasis depend upon several classes of molecules expressed on the cell surface, including integrins and cadherins, as well as the ligands that bind to these molecules. Cell adhesion molecules (CAMs) are transmembrane proteins with extracellular and intracellular domains, the intracellular domains are usually connected to the cytoskeleton or to signaling molecules. Although originally named for cell adhesion, CAMs have multiple functions, including a major role in signaling from outside to inside a cell and vice versa. The formation and breaking of adhesive bonds between tumor cells and their environment provides information to the cell about its environment and may lead to changes in the expression of specific genes that determine cell proliferation, invasion, or other processes.
Integrins are expressed in all cell types and are involved in the regulation of cellular functions during embryonic development, wound healing, inflammation, homeostasis, bone resorption, apoptosis, cell proliferation, tumor cell growth, and metastasis. They make up a family of widely expressed transmembrane receptors for proteins of the ECM, such as fibronectin, laminin, vitronectin, and collagens. These adhesion molecules are obligate heterodimers, comprising noncovalently associated α and β subunits, each of which spans the plasma membrane and typically possess a short cytoplasmic domain. Receptor diversity and versatility in ligand binding is determined by the extracellular domains through the specific pairing of 18 α and 8 β subunits, to form a family of 24 recognized heterodimers (Hynes, 2002). The cytoplasmic domain of the β subunit interacts directly with components of the actin cytoskeleton, such as α-actinin and talin, allowing its localization to focal adhesion plaques (FAPs) that form at points of contact between integrins and the ECM. FAPs represent the submembranous termini of actin stress fibers, indicating that integrins provide a structural bridge between the ECM and the actin cytoskeleton (Fig. 10–3). FAPs also contain a number of protein tyrosine kinases, such as the focal adhesion kinase (FAK), p125, and the integrin-linked kinase (ILK). It is well established that integrins act as part of signal-transduction complexes that allow cells to “sense” (and respond to) their extracellular environment (Desgrosellier and Cheresh, 2010). Integrin activation may act to prolong and intensify signaling from growth factor receptors and can be a positive mediator of angiogenesis (see Chap. 11, Sec. 11.4.7). In a recent study done with in vitro ECM gels, metastatic cells lost their proliferation capability when the α3β1 integrin was knocked down, resulting in defective FAK signaling. This suggests that the α3β1–FAK interaction might play a role in ECM signaling in primary tumor cells when they first migrate to other organs and thus promote metastasis (Shibue and Weinberg, 2009). The expression of integrin receptors is altered in malignant cells as compared with their normal counterparts, but the loss or gain of expression of a particular integrin has not been linked directly to malignant transformation.
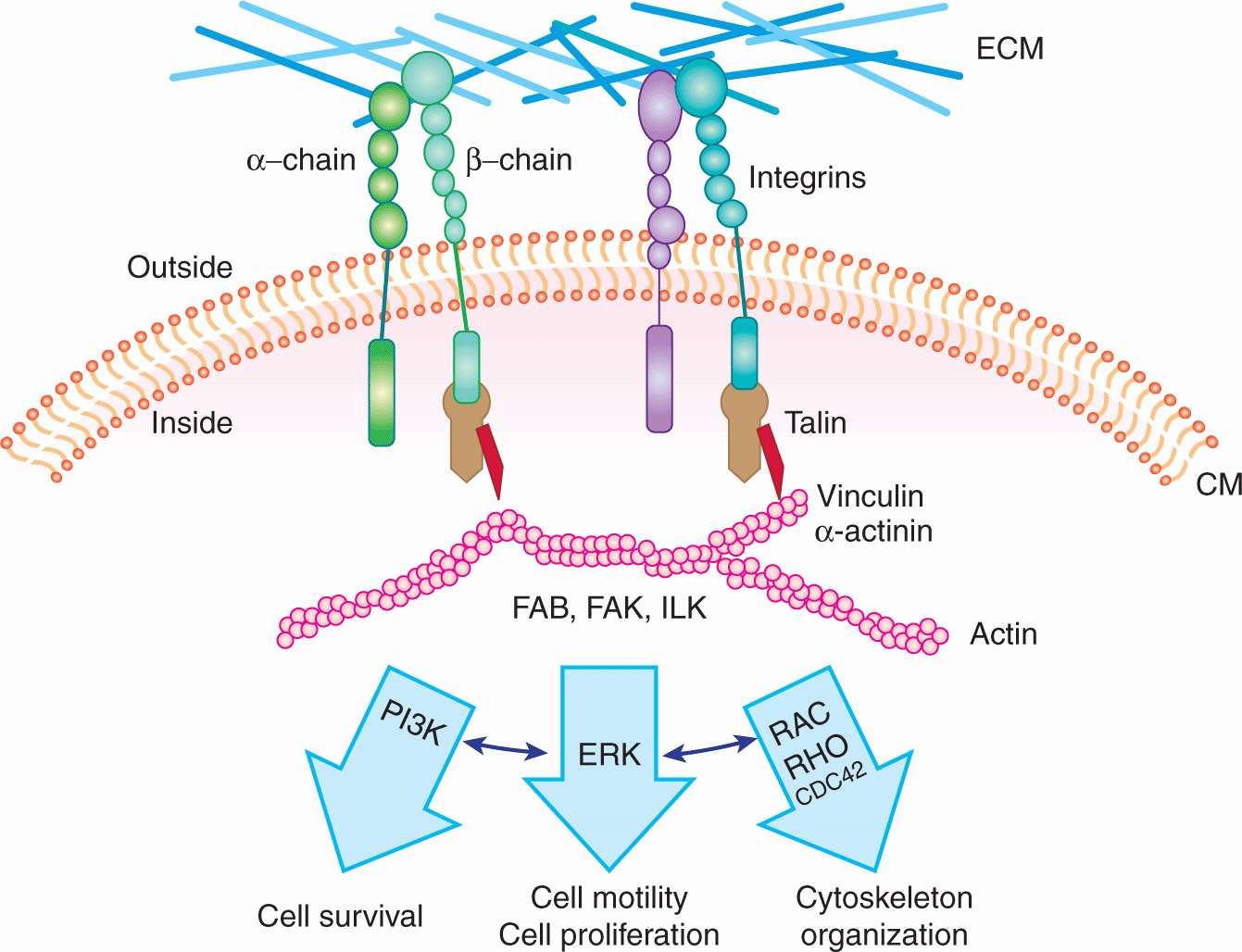
FIGURE 10–3 Schematic of integrin receptors with linkage to their major downstream signal transduction pathways. An integrin receptor contains 2 subunits (α and β); different combinations of α and β subunits lead to the structural and functional variety of the integrin receptors. Integrin receptors are important players in the “outside-in” signaling system, they can sense the changes in the environment, and transduce the signals into the cell through the signal transduction pathways shown.
Cadherins are intercellular adhesion receptors that play important roles in assembling adherens junctions and desmosomes. Distinct members of the cadherin family are principal constituents of each type of junction, mediating calcium-dependent adhesion between similar cells. There are more than 20 recognized cadherins and protocadherins. E-cadherin (CDH1), the major epithelial cadherin, contains 4 conserved extracellular domains, a fifth extracellular domain possessing conserved cysteine residues, a transmembrane domain, and a cytoplasmic domain. Calcium binding sites lie between adjacent extracellular domains. The cytoplasmic domain associates with cellular proteins such as catenins, which act to link cadherins to the actin cytoskeleton and to signal-transduction components (Cavallaro et al, 2002; Fig. 10–4). An important connection between cadherins, catenins, and tumor progression was made with the observation that the APC (adenoma polyposis coli) tumor-suppressor protein (see Fig. 10–2) and β-catenin form physiological complexes with axin and glycogen synthase kinase (GSK)-3β, in which β-catenin can be phosphorylated by GSK-3β and targeted for ubiquitin-mediated degradation. This complex thereby acts to control the level of β-catenin in the cell. Besides binding APC and cadherins, β-catenin can also enter the nucleus and associate with LEF/TCP transcription factors, upregulating genes involved with cell growth such as c-Myc and cyclin D1. Mutations in APC, which are associated with the formation of adenomas, cluster within the β-catenin binding region, yielding truncated APC peptides that are unable to bind to β-catenin. This reduces its degradation and increases its availability to diffuse to the nucleus and activate cell growth. Degradation of β-catenin is also disrupted if the activity of GSK-3β is blocked by activation of the Wnt-signaling pathway (see Chap. 8, Sec. 8.4.1 and Fig. 8–12). Loss of E-cadherin and increased levels of N-cadherin (CDH2) are correlated with cellular invasiveness and, as discussed in Section 10.5.6, are associated with the so-called EMT. Disruption of cadherin-catenin complexes leads to disruption of the cytoskeleton, which, in turn, may affect signal transduction, as there is evidence that cytoskeletal proteins can act as a scaffold for the components of signal transduction pathways.
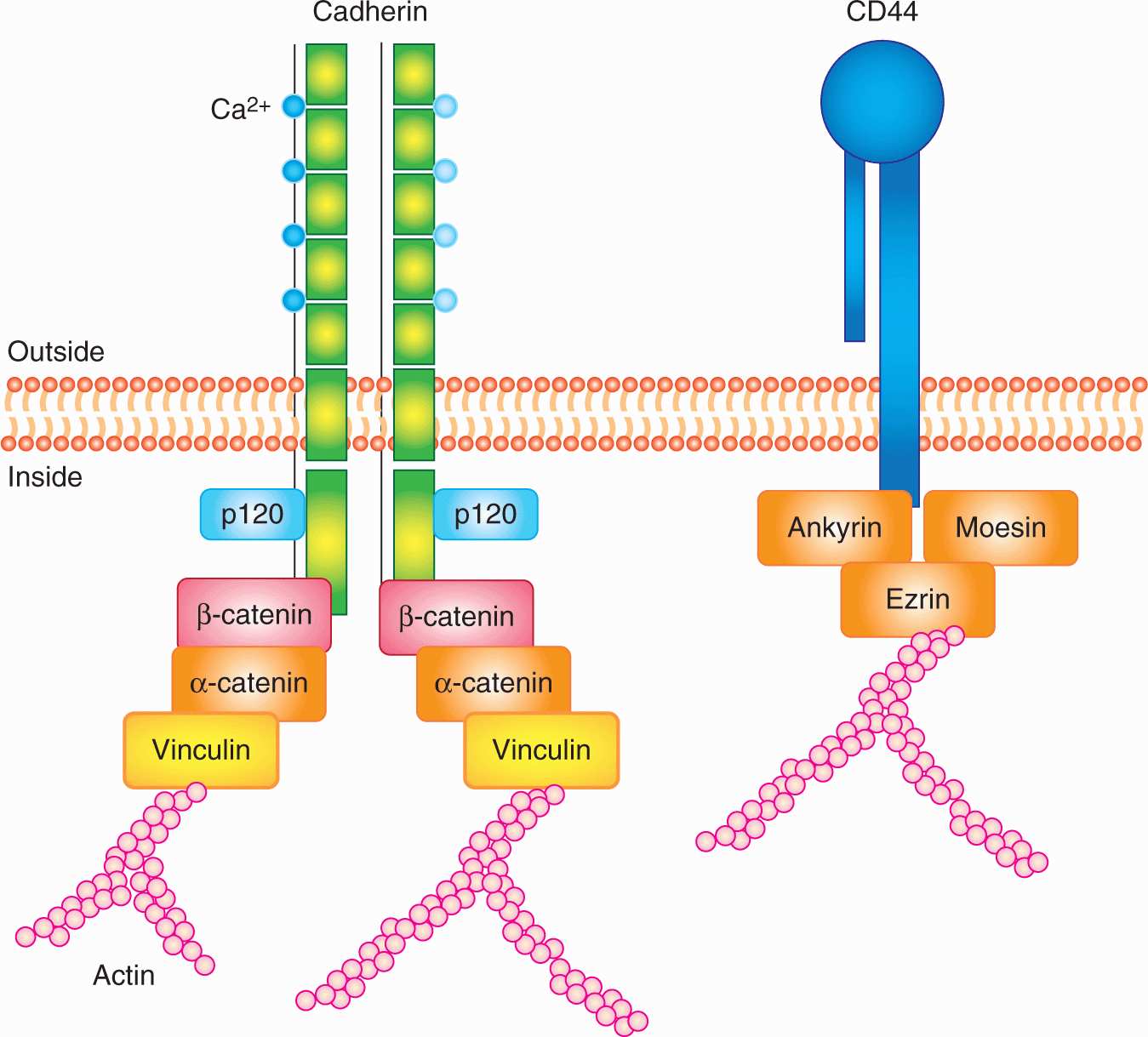
FIGURE 10–4 Schematic of the structures of cadherins and CD44. Cadherins are defined by their signature calcium binding domains. Different cadherins can have various number of repeats of calcium binding domains. Cadherins can also activate important signaling pathways, such as Wnt signaling, through their intracellular association with (p120, α and β) catenins. CD44 has many isoforms; the standard isoform is designated CD44s, while the splice variants are designated CD44v. The intracellular domain of CD44 can associated with ezrin, which is also an important player in metastasis. The function of CD44 is largely controlled by its posttranslational modifications.
CD44 is a cell-surface glycoprotein identified as the major receptor mediating cellular interactions with hyaluronate, a glycosaminoglycan component of the ECM. Its principal physiological functions include aggregation, migration, and activation of cell functions. CD44 is widely expressed and exists in multiple forms with variable glycosylation. All CD44 isoforms contain a cytoplasmic domain, which may link CD44 to actin filaments through interactions with ankyrin, ezrin, and moesin (see Fig. 10–4). Alternative splicing of the messenger RNA (mRNA) to produce these variable isoforms (CD44v) is regulated in a tissue-specific manner or by antigen activation in lymphocytes. CD44 isoforms are overexpressed by many tumors, and their expression has been correlated with clinical outcome in several human malignancies (Jothy, 2003). Recent studies suggest that CD44 is a surface protein that is expressed preferentially on subpopulations of cells that are able to induce high frequencies of tumor growth in xenograft models (tumor initiating or cancer stem cells; see Chap. 13, Sec. 13.4.5). The potential role of the different isoforms of CD44 has not been well defined in such cells. CD44 has been reported to be expressed at the invading edges of murine carcinoma cells and is required in melanoma cells for migration on type IV collagen and for invasion of the basement membrane. Osteopontin, a secreted glycoprotein widely implicated in invasion and metastasis, binds to CD44, inducing a variety of signaling cascades associated with adhesion, migration and invasion, suggesting its interactions with CD44 may play a role in its malignancy-promoting potential (Anborgh et al, 2010).
The ERM proteins (containing ezrin, radixin, and moesin motifs) serve an important role linking the actin network cytoskeleton and the cell membrane. They provide a means by which a cell can sense environmental changes and respond to growth factors. ERM proteins normally form aggregates with each other and assume an inactive conformation. Upon tyro-sine and threonine phosphorylation, ezrin becomes activated and switches into an active conformation, which allows for its translocation from the cytoplasm to the plasma membrane, bringing F-actin to the cell surface. The ERM proteins are especially abundant at cell protrusions such as membrane ruffles and microvilli. Ezrin is also reported to bind to CAMs such as CD43, CD44, and intercellular adhesion molecules (ICAMs). Many of these CAMs affect cell migration and invasiveness, and have been implicated in metastasis. Recently, ezrin was identified as a metastasis-associated gene that is related to prognosis of a number of cancers (Bruce et al, 2007). A high level of ezrin expression has been linked with poor prognosis in pediatric and adult osteosarcoma (Khanna et al, 2004). In a mouse model of osteosarcoma, it was shown that ezrin is required for metastasis, and possibly functions to provide a survival advantage to tumor cells when they reach a secondary site.
10.2.4 The Pathophysiological Microenvironment
As a solid tumor grows, the rate of cancer cell proliferation surpasses the ability of the existing vasculature to supply growth factors, nutrients, and oxygen, and to remove the catabolites produced by the cells. The result of this imbalance between supply and demand is a microenvironment containing regions of hypoxia (low oxygen concentration), low glucose levels, low pH, and elevated interstitial fluid pressure (IFP). There is spatial and temporal heterogeneity in the tumor microenvironment, both between different tumors and within an individual tumor. Furthermore, the tumor microenvironment has been linked to a more aggressive phenotype, playing multiple roles in tumor progression and metastasis (Finger and Giaccia, 2010; Lunt et al, 2009). Multiple clinical studies have demonstrated a connection between tumor hypoxia and disease progression in a variety of human tumors, including carcinomas of the cervix, prostate, and the head and neck, and soft-tissue sarcomas (see Chap. 12, Sec. 12.2.2). Furthermore, studies in animal models have shown that hypoxia in tumors increases metastatic dissemination to the lungs or lymph nodes. The mechanisms by which tumor hypoxia might increase meta-static potential are discussed in more detail in Section 10.5.7.
10.3 METASTASIS
10.3.1 The Spread of Cancer
The 2 major routes of metastatic spread are via lymphatic vessels and/or blood vessels (Fig. 10–5). Metastases are subdivided into 2 groups: those in regional lymph nodes, which are usually regarded as having disseminated via the lymphatic circulation, and those that arise at distant organs, which are spread via the blood circulation system. Metastasis is an ominous feature in cancer progression and most deaths caused by cancer are caused by metastases. The T (= Tumor) N (= Nodes) M (= Metastasis) system of cancer staging is now used widely in clinical management. In this system, cancers are divided into stages based on tumor size, presence of involved lymph nodes, and presence of distant metastases. Prognosis is closely correlated with the stage of the disease at diagnosis and treatment decisions depend on it. Local treatment is of limited effectiveness in the presence of metastatic disease and systemic therapies may be used to a greater extent.
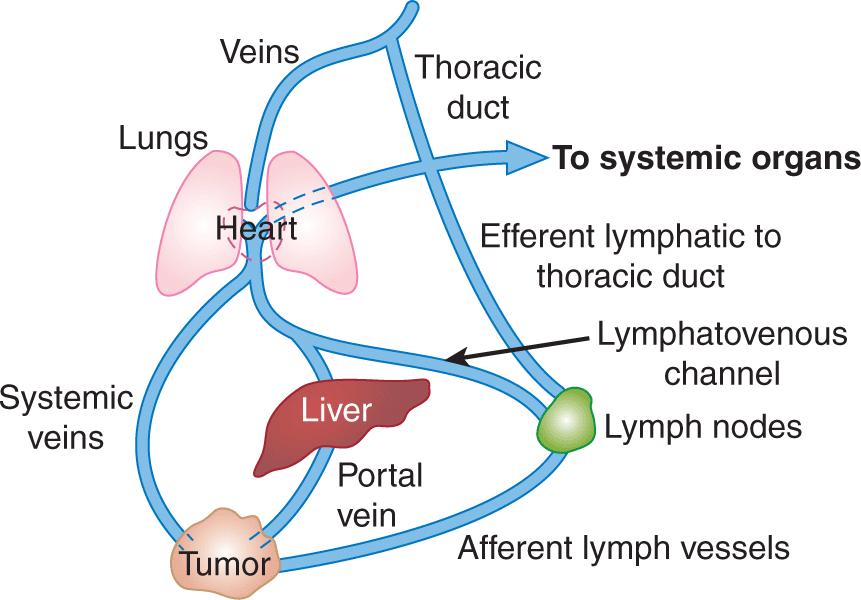
FIGURE 10–5 The major routes by which cancer cells can spread from a primary tumor are through the lymphatic or blood vessels. These 2 systems are interconnected as illustrated. The vascular drainage for tumors of the gastrointestinal tract is usually via the portal circulation, whereas for tumors at other sites in the body, drainage is via the systemic veins. (Adapted with permission from Sugarbaker,1980.)
The kinetics of tumor spread varies between individuals, and it does not always correlate with primary tumor size. This suggests that the ability to establish metastases is not present in all primary tumor cells. In addition to properties such as avoidance of cell-cycle arrest and apoptosis, cells that leave primary tumors and establish metastases require additional abilities (see Sec. 10.4). Tumor cells are generally very inefficient at forming metastases (see Sec. 10.3.3) and often are detected in the blood circulation of patients without evidence of metastatic disease. Oncogene-driven tumors in mouse models do not necessarily develop distant metastases (Klein, 2003). A recurring theme is that tumor cells often gain meta-static abilities by turning on endogenous gene programs that are involved in normal tissue development and homeostasis. An example is provided by epithelial tumor cells that undergo EMT, a process normally involved in embryonic development and wound healing, to gain motility and survival advantages for metastasis (see Sec. 10.5.6). Furthermore, the invasive and migratory functions that are responsible for normal epithelial cell branching (eg, in the mammary gland) can be hijacked by tumor cells to expand into surrounding tissue (Ewald et al, 2008). Tumor cells in hypoxic regions can express hypoxia-inducible factors (HIFs) to initiate the angiogenesis process to compensate for lower nutrient and oxygen supplies.
10.3.2 Organ Preference
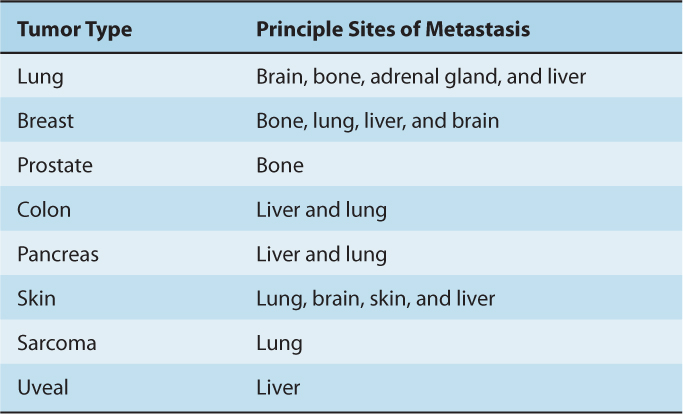
Distant metastases from different types of tumors tend to occur in specific target organs (Table 10–2). Paget’s soil-and-seed hypothesis postulated that tumor cell–host organ interactions can favor or hinder metastatic development. The structure and organization of capillaries and adjacent parenchyma and tissue function vary widely in different organs; therefore, organ-specific adhesive interaction between endothelial cells and tumor cells or between tumor cells and available growth factors in the organ can influence successful establishment of metastatic growth. This concept is supported by studies that show tumor cells expressing certain surface molecules have enhanced abilities to arrest, extravasate into, and grow in specific target organs (see Sec. 10.5). In addition, other factors, such as the dynamics of blood flow, are also likely to explain organ preference. In colon cancer, the mesenteric circulation delivers tumor cells from the bowels to the liver, a common site of metastasis, while blood flow carries osteosarcoma cells from the bone to lung capillaries, and osteosarcoma metastases to the lung are common.
TABLE 10–2 Typical sites of metastasis of the common tumors.
Organ specificity also occurs in animal models. Tumor cells that form large numbers of metastatic deposits in 1 organ (eg, the lung following intravenous injection) are not necessarily capable of doing so in another organ (eg, the liver following intraportal injection). Furthermore populations of tumor cells that have enhanced ability to form metastases in specific organs can be isolated by serially selecting cells from metastases in these organs. Cells forming metastases preferentially in the lung will “home” to a lung lobe even when it is transplanted ectopically into a subcutaneous site; such cells do not form metastases in other organs that are transplanted ectopically. The classic example of serial selection is the isolation of the B16F10 cell population from B16 mouse melanoma cells (Fidler, 1973). The procedure (Fig. 10–6) involved serial intravenous injection of the cells into isogenic animals, with selection at each stage for cells that had formed lung metastases. The cells forming lung metastases were grown in culture to expand their number before being reinjected into animals. After 10 such passages, a population of cells was obtained (termed B16F10 cells) that was approximately 10 times as efficient at forming experimental lung metastases after intravenous injection as the starting B16F1 cell population. Interestingly, these cells were not more capable of forming spontaneous metastasis when implanted at a local site, suggesting that selecting for increased ability to grow in lung does not affect the invasive properties necessary for initial escape from the primary tumor. However, selection for B16 melanoma cells with increased spontaneous metastatic ability (B16BL6) was achieved by selecting for cells that could invade through the wall of the mouse bladder in vitro, and then expanding these cells in culture and selecting again for invasion through the mouse bladder over multiple cycles. Other investigators have also been successful, using similar approaches, in selecting cell populations from a number of rodent tumors that have enhanced experimental metastatic ability in a variety of organs including lung, liver, ovary, and brain. However, such selection procedures do not always yield cells with increased metastatic ability (Ling et al, 1985; Stackpole et al, 1991), leading to the suggestion that some properties that contribute to metastatic ability may not be stably maintained within the tumor cell population during the selection procedures and may function only transiently to promote metastasis.
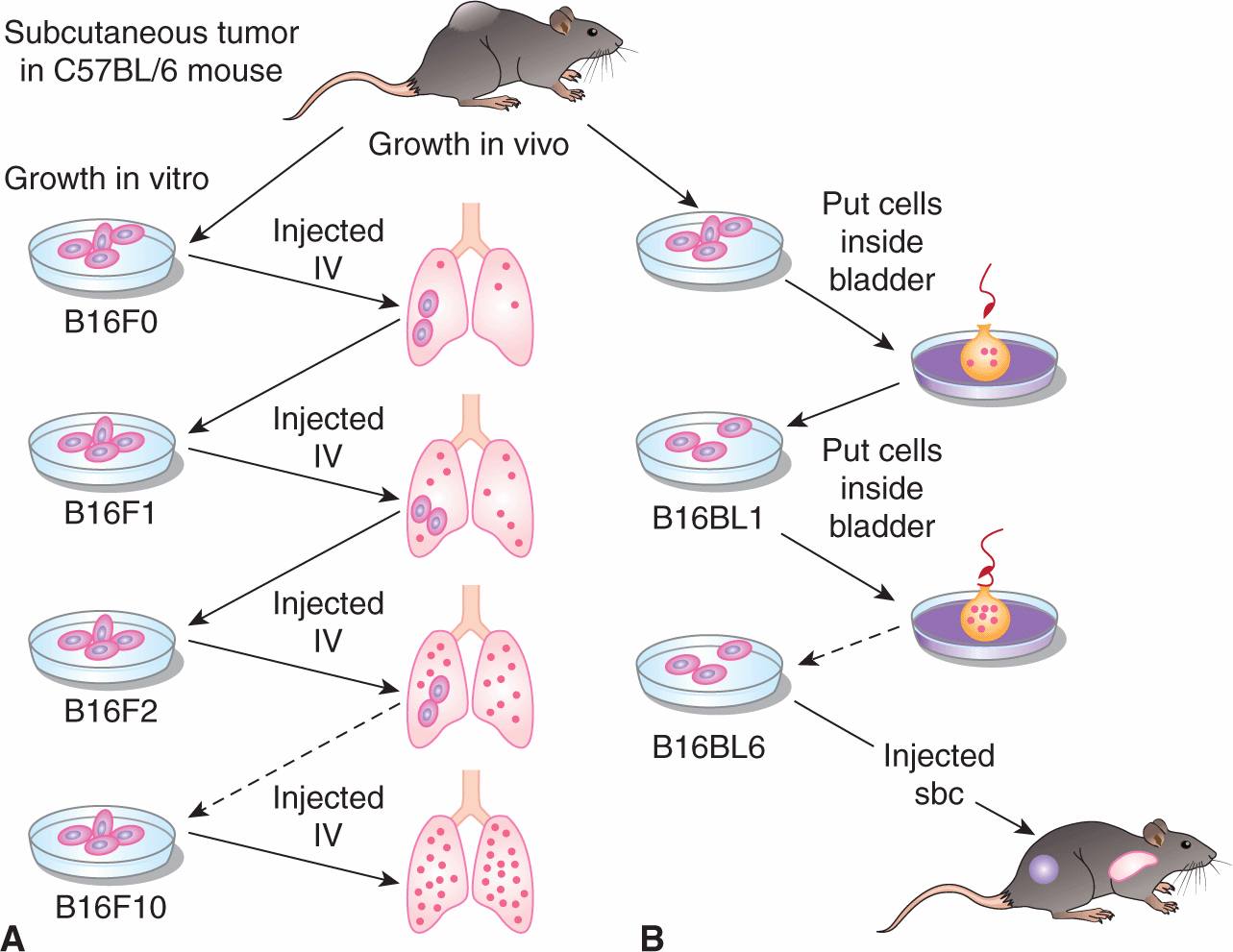
FIGURE 10–6 Procedures used for selecting highly metastatic cell populations from B16 melanoma cells. A) The B16F10 cells were selected by passing the cells 10 times through the lungs of mice, while (B) the B16BL6 cells were selected by requiring them to invade 6 times through the walls of mouse bladders. sbc, subcutaneous IV, intravenously.
The organ specificity of metastatic human tumor cells has been tested in immune-deficient hosts (eg, athymic nude mice or SCID [severe combined immune deficient] mice). These studies have demonstrated that the local site of growth of an implanted tumor may influence its capacity to seed spontaneous metastases and that tumors transplanted in orthotopic sites (tissue of the same pathological type as the tumor) are more likely to seed metastases (locally to lymph nodes or distantly to other organs) than tumors grown in ectopic sites (Fujihara et al, 1998). A refinement to these models is the transplantation of human tissue (eg, fetal bone) into SCID mice to generate (so-called) SCID-hu models. When human tumor cells are implanted locally or injected intravascularly into such mice, they show similar organ preference for metastasis to that seen in clinical practice and the metastases occur preferentially in the human tissue rather than the same murine tissue (Namikawa and Shtivelman, 1999).
Chemokines (molecules used by leukocytes to home to specific organs) may play an important role in organ specificity of metastasis. It has been reported that human breast cancer cells have high expression of the chemokine receptors CXCR4 and CCR7, while their respective ligands (CXCL12 and CCL21) are highly expressed in organs in which breast cancer cells have a high propensity to form metastases (lung, liver, regional lymph nodes, and bone marrow). Blocking of CXCL12/CXCR4 interaction significantly impaired formation of metastasis by a human breast cancer cell line in lung and lymph nodes in an experimental system, and this pathway is being targeted for cancer therapy. In a similar context, organ specificity may also occur as a result of specific molecular signatures that are expressed in the microvasculature in individual normal tissues (and tumors). The technique of phage display, which allows the identification and purification of small peptides that will specifically recognize and bind to the molecules involved is providing opportunities for imaging and directed therapy of cancer and other diseases (Mueller et al, 2009; Ruoslahti, 2002; Whitney et al, 2010).
10.3.3 Metastatic Inefficiency
The establishment of metastases by tumor cells appears to be an inefficient process. Blood samples taken from the renal vein in patients just prior to surgery for renal cell carcinoma allowed estimates that tumor cells were being released at rates of 107 to 109 cells per day (Glaves et al, 1988). Two of the 11 patients had no evidence of metastatic disease 30 and 55 months after the surgery. Similarly, patients with peritoneovenous shunts for malignant ascites have shown no evidence that release of large numbers of tumor cells into the blood increases the number of metastases observed. Studies in animals also support this concept, as only a few circulating cells are able to form metastases. In experimental metastasis (or colonization) assays, tumor cells are injected directly into the arterial or venous blood circulation and allowed to disseminate and arrest at various sites. It is rare that even 1% of such cells form tumor nodules and it is usually orders of magnitude lower.
The inefficiency of the metastatic process leads naturally to the question of whether metastasis is a random or a specific process. A small subpopulation of the cells in a tumor might express properties that confer a higher probability of being able to form metastases, but it is also possible that all tumor cells might have an equal (low) probability of forming metastases, but only a few manage to survive through the various stages of the process (see Sec. 10.4). Support for the random nature of the metastatic process derives from studies that have failed to demonstrate that cells obtained from metastases are consistently more metastatic than cells from the parent tumor (Weiss and Ward, 1990), which is not consistent with what would be expected if cells from metastases were expressing a stable phenotype that predisposed them to form metastases. Support for the specific nature of metastasis derives from experimental studies. When clones of B16 melanoma cells were isolated, expanded in culture and the cells tested for their ability to form experimental metastases, the variability in metastases for cells from a single clone was found to be much less than that observed when different clones were compared (Fidler and Kripke, 1977). These results indicated wide heterogeneity in metastatic ability between the different clones (Fig. 10–7) and were replicated in a number of cells lines. They suggest the presence of preexisting metastatic variants within a cell population. However, the finding that metastatic properties can be unstable cast doubt on this interpretation and gave rise to the clonal heterogeneity model of metastasis formation (Hill et al, 1984). This model proposes that, although some of the multiple properties necessary for a cell to metastasize may be relatively stably expressed, others are expressed only transiently, giving rise to unstable clonal heterogeneity. However, the wide variety of properties that have been demonstrated to promote metastasis makes it unlikely that any 1 model will adequate depict the whole picture. To better understand the process, metastasis is often divided into a few sequenced steps: invasion, intravasation and survival in the circulation, extravasation, and, finally, establishment of a new growth (Fig. 10–8). These steps provide a simplified view of the complex set of biological events involved in the process. In each step, expression of specific molecular factors may help the tumor cells to survive and proceed to the next step in the process (see Sec. 10.4).
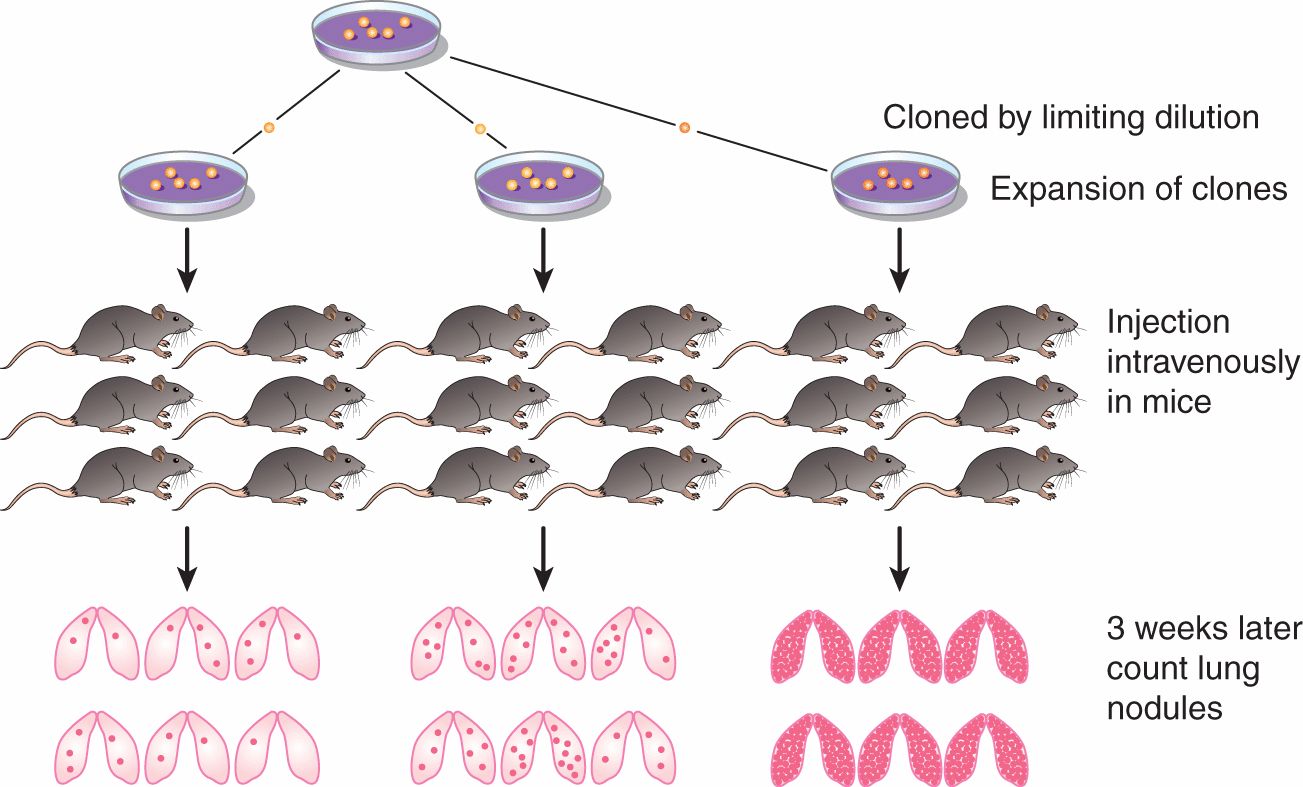
FIGURE 10–7 Clonal heterogeneity is demonstrated by establishing a series of clones from a tumor cell population and, after expansion, testing them for metastatic ability. Although there is some variability in the number of nodules observed in different animals injected with cells from the same clone, there is much greater variability between the clones.
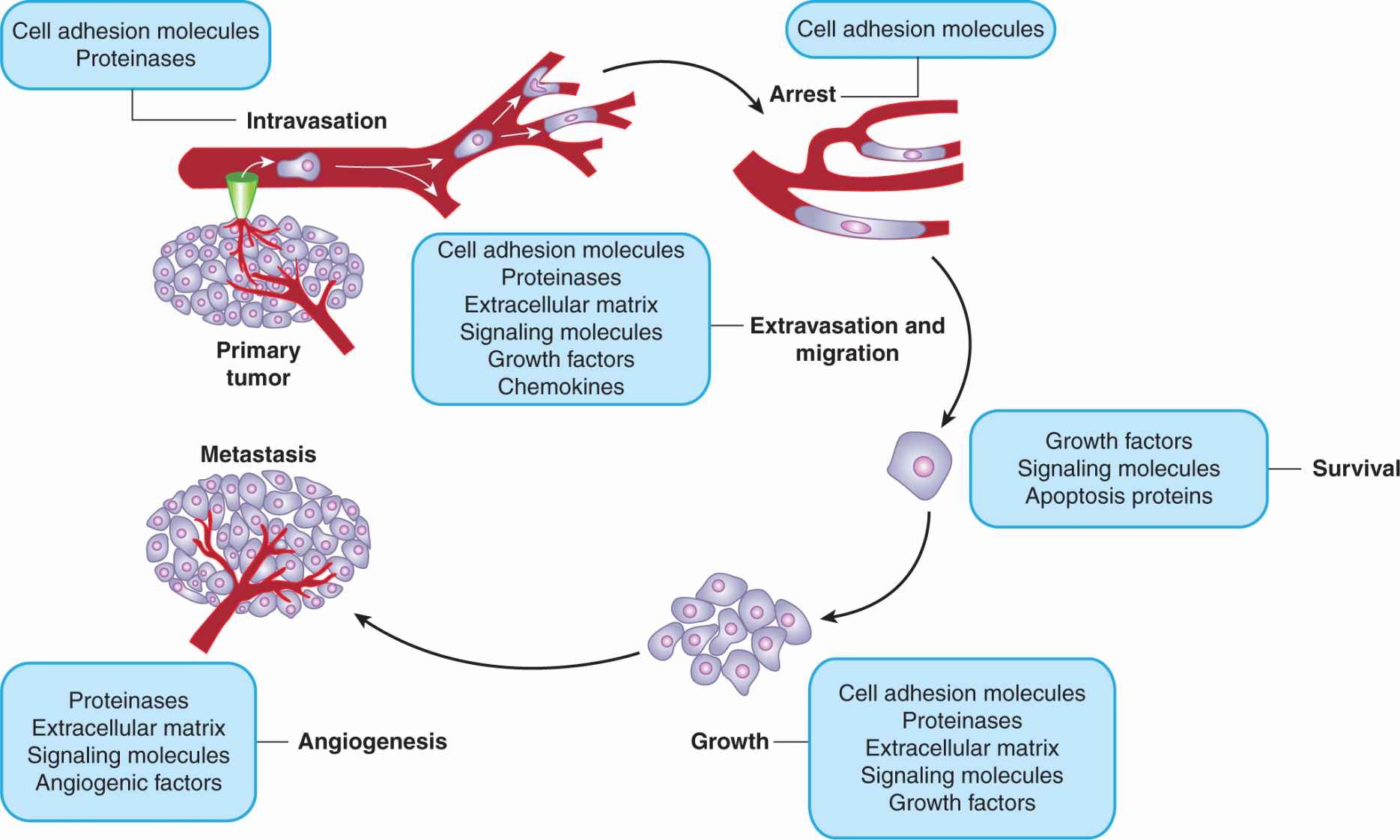
FIGURE 10–8 Schematic of the sequential steps in the metastatic process. Various types of molecules that are thought to be involved at each stage are listed.
10.3.4 Circulating Tumor Cells
Distant metastases usually arise through the distribution of tumor cells into the circulating blood system. Tumor cell dissemination can be an early event and may persist throughout the growth of the primary tumor, although it is likely that the survival time of such cells in the circulation is quite short (hours). It is now possible to detect (small numbers) of circulating tumor cells (CTCs) in the blood of many patients, and the numbers of such cells have been associated with overall survival in some patient groups. However, their isolation and characterization remains a significant challenge partly because of their low frequency (<1000 CTCs/mL of blood vs. millions of blood cells) (Pantel et al, 2008). The 2 main approaches for detecting these CTCs are immunological assays employing antibodies against cell-surface proteins specifically expressed on the cancer cells, and polymerase chain reaction (PCR)-based molecular assays detecting cancer cell-specific transcripts. The most common surface protein used in CTC detection is epithelial cell adhesion molecule (EpCAM), because many tumors are of epithelial origin, but other markers, such as various cytokeratins, have also been used. Identifying appropriate CTC markers is an ongoing research process and far from perfect (Wicha and Hayes, 2011). New sensitive methods such as in vivo multiphoton intravital flow cytometry may help to advance the detection of CTC in the blood of cancer patients.
Whole-genome copy number analysis and gene expression profiling have been used to analyze CTCs and have indicated that most CTC are dormant and in a nonproliferative state, possibly as a result of mechanisms of adaptive immunity (Koebel et al, 2007). However, CTCs can proliferate in cell culture systems if given appropriate growth factors, such as epidermal growth factor (EGF) and fibroblast growth factor 2 (FGF2). If the growth of CTCs is susceptible to reactivation in the patient this could clearly influence the overall outcome of treatment and the molecular nature of CTCs is being intensively investigated in this context. In breast cancer, ERBB2 expression on CTCs is associated with poor prognosis, and in gastric cancer, uPAR expression on CTCs is associated with metastatic relapse. This suggests ERBB2 and uPAR might play roles affecting the reactivation process of CTCs in their respective cancer types. CTCs that express angiogenesis and hypoxia-associated markers such as vascular endothelial growth factor (VEGF), VEGFR-2, FAK, and HIF-1, are also thought to be associated with aggressive disease and poor prognosis (Bednarz-Knoll et al, 2011). Some CTCs have been shown to exhibit EMT (see Sec. 10.5.6), which may enhance their ability to form metastases.
Because the detection of CTCs can have prognostic value in early stages of the disease, their presence is being included in international tumor staging systems. It is hoped that CTC detection might allow monitoring of the disease and potentially treatments could be customized based on the molecular profiling of these cells.
10.4 STEPS IN THE METASTATIC PROCESS
10.4.1 Detachment from the Primary Tumor, Local Invasion, and Intravasation
Detachment or shedding of cells into blood or lymphatic vessels (intravasation) may occur as a result of prior invasion of the tumor mass into vessels or because the abnormal vasculature of tumors permits passage of cells into the circulation. Angiogenic factors such as VEGFs and FGFs can induce vascular remodeling (see Chap. 11) and thereby facilitate intravasation. Tumor blood vessels are less well-organized and leaky in comparison to their normal counterparts, providing additional opportunities for cell penetration. The poor alignment of endothelial cells in tumor vessels has been reported to allow CTCs to home back to the original tumor, as has been observed in a mouse model of breast cancer (Kim et al. 2009).
This phenomenon of cross-seeding is enhanced by tumor-derived chemoattractive cytokines such as interleukin (IL)-6 and IL-8. Tumor cells may also fine-tune surface molecules to interact with endothelial cells so as to increase their survival. For example, tumor cells can downregulate KAI1/CD82, a surface molecule that interacts with the Duffy antigen receptor for chemokine (DARC) on the endothelial cell to induce senescence of tumor cells (Bandyopadhyay et al, 2006). Loss of CD82 is correlated with metastatic progression in patients, and the knockout of DARC abrogates the suppressive effect of CD82. Studies with certain human tumor (eg, melanoma and glioblastoma) cells have shown that these tumor cells can form vascular channels and endothelial cells, and may in this way gain ready access to the vascular space (Hendrix et al, 2002). Detachment of cancer cells from the primary tumor mass may involve decreased expression of adhesion molecules involved in the “homotypic” adhesion of cells to one another or may depend on the expression of motility factors (eg, hepatocyte growth factor [HGF], autotoxin, or autocrine motility factor), which are glycoproteins found to promote cell movement through interaction with cell-surface molecules linked to the Rho/Rac/Cdc42 guanosine triphosphatase (GTPase) intracellular signaling system. Disruption of E-cadherin expression in a mouse model of pancreatic cancer was found to enhance invasive properties and metastasis (Perl et al, 1998). Tumor cells may gain motility by overexpressing RhoC, a small calcium-dependent GTPase, that is normally responsible for sensing extracellular signals and controlling cytoskeletal actin organization. RhoC-deficient mice have normal primary tumor formation but reduced cancer cell mobility and drastically reduced metastases (Narumiya et al, 2009). The EMT process, in which a polarized epithelial cell assumes a mesenchymal cell phenotype, is also now considered to be coincident with the process of cancer cell migration (see Sec. 10.5.6). Local invasion and intravasation may also involve breakdown of the ECMs and basement membrane. Various proteases, including MMPs, play a major role in this process (see Secs. 10.2.2 and 10.5.1).
10.4.2 Survival in the Blood Stream
Once tumor cells break free of constraints at the primary site, they can gain access to the blood (or lymphatic) vessels (see Fig. 10–8). While in the blood vessels, tumor cells may travel with platelets to evade immune surveillance, ensuring their survival. Platelets are small, terminally differentiated anuclear cells shed from megakaryocytes. The basic biological function of platelets, such as blood clotting, is also exploited by tumor cells to their advantage. With the help of a number of other coagulation factors, activated platelets assemble and can form tumor cell–platelet aggregates. This aggregation, in combination with the adhesive properties of platelets, effectively arrests tumor cells in the capillaries of target organs. In addition, many platelet-surface molecules and platelet-derived factors enhance metastasis. For example, mice deficient in the platelet-specific receptor glycoprotein Ib-alpha (GPIb-α) have a 15-fold reduction of lung metastatic foci formation in an experimental metastasis model (Jain et al, 2007). In addition, α-granules released by platelets contain many growth-promoting factors, such as EGF, VEGF, HGF, and transforming growth factor (TGF)-β (Gay and Felding-Habermann, 2011).
Although CTCs can be detected in many patients, as discussed in Section 10.3.4, it appears that most tumor cells that reach the circulation are arrested in the first capillary bed that they encounter (first-pass capillary bed). Early experimental studies in which radiolabeled tumor cells were injected into the systemic or portal veins indicated that the majority of cells are arrested initially in the lung or liver capillaries, respectively. More recent work has used genes encoding markers, such as green fluorescent protein (GFP) or luciferase to provide direct visualization of tumor cell arrest following intravenous injection, or the growth of the tumor and its metastases in situ (Hoffman, 2002). Combined with such labels, the technique of intravital videomicroscopy (IVVM) has confirmed the early arrest of tumor cells in “first-pass” capillary beds and permitted dynamic study of events in the process of experimental metastasis as discussed below (MacDonald et al, 2002). It is this effect that probably accounts for the high level of metastases in lung and liver, but some tumor cells succeed in passing through the first-pass organ to become CTCs or to establish metastases in other (preferred) sites.
10.4.3 Extravasation
After tumor cells arrive at the distant organ, they may extravasate into the new tissue (see Fig. 10–8). As discussed in Sections 10.2.2 and 10.5.1, this process involves the activity of a variety of proteases, of which MMPs are believed to play a major role. Several groups have reported in vivo studies of extravasation using IVVM. Chambers and colleagues observed that the majority of arrested cells were able to extravasate, even cells with reduced proteolytic capability or nonmalignant fibroblasts, suggesting that this process may not be a major barrier to the metastatic process (MacDonald et al, 2002). Other IVVM studies, however, found that extravasation was a formidable barrier to metastasis, and tumor cells were observed to proliferate intravascularly. Yet others observed that tumor cell extravasation was dependent on CAMs and could occur in precapillary vessels upon stimulation of the endothelial cells by inflammatory growth factors. Because the structural organization of capillary walls varies in different organs, tumor cells may need specific capabilities for infiltration at different sites. Recent studies of gene expression in patients with advanced metastatic disease have indicated that tumor cells trapped in the lung capillaries produce factors such as epiregulin (EREG), cyclooxygenase-2 (COX-2), MMP-1, and cytokine angiopoietin-like 4 (ANGPTL4) to remodel the pulmonary vasculature. These factors modify the integrity of lung endothelia and provide entry points into the lung parenchyma. Genetic and pharmaceutical inhibition of these factors significantly reduced metastatic extravasation (Gupta et al, 2007). Some of these genes seem to be general metastasis-promoting factors; for example, COX-2 is also a potent mediator of metastasis to the brain, and RNA interference-mediated knockdown of COX-2 expression decreases brain metastatic activity of certain breast cancer cell lines (Bos et al, 2009). Genes such as α2,6-sialyltransferase can facilitate passage through the blood–brain barrier by enhancing adhesion to brain endothelial cells and can act as a specific promoter of brain metastasis. Various other cell-surface molecules, such as chemokine receptors (eg, CXCR4) and growth factor receptors (eg, c-MET), have also been reported to augment metastasis. Conceivably, they accomplish this by recruiting tumor cells to target organs and enhancing their survival and growth.
10.4.4 Initiation of a New Growth (Colonization)
Following infiltration, the majority of tumor cells die as a result of selective pressure from the foreign environment, but a few (rare) tumor cells may successfully initiate growth inside the target organ. This may be accomplished by their incorporation into sites in the organ that are receptive to tumor growth (metastatic niches; see Sec. 10.5.8). There is also evidence that tumor cells that initiate metastatic growth can modify the target organ to make the environment more favorable for their outgrowth. For example, tumor cells in the bone achieve this by disrupting normal bone homeostasis. Through the secretion of parathyroid hormone-related peptide (PTHrP), IL-6, tumor necrosis factor α (TNF-α), and other factors, tumor cells stimulate the release of receptor activator of nuclear factor-κB ligand (RANKL) by osteoblasts. RANKL, in turn, stimulates myeloid progenitor cells to differentiate into osteoclasts. Osteoclasts can digest bone structures and create space for establishment of metastasis. Moreover, the lytic action of osteoclasts releases TGF-β, insulin-like growth factor I (IGF-I), and bone morphogenetic proteins (BMPs). These factors then further enhance tumor cell survival, thus creating a vicious cycle. To further increase the efficiency of the cycle, tumor cells also suppress the production of the RANKL antagonist osteoprotegerin (OPG), increasing the efficacy of RANKL (Chen et al, 2010). Denosumab, a monoclonal antibody targeting RANKL that breaks this vicious cycle, was recently approved for treating patients with bone metastasis. In the lung, inhibitor of differentiation-1 (Id1) has been implicated in bypassing senescence and promoting tumor outgrowth by desensitizing the cells to p21-dependent cell-cycle arrest (Swarbrick et al, 2008). In a mouse cancer model driven by Ras and Id1, inactivation of Id1 caused widespread senescence in established tumors within 10 days.
10.4.5 Metastatic Latency and Dormancy
After entering the target organ a few cells may survive but enter dormancy (Goss and Chambers, 2010). The dormant cells can remain in target organs for years before resulting in a metastatic growth. This has been particularly observed in breast cancer. Immunosurveillance has been suggested as being responsible for limiting the metastatic growth of the dormant cells. Because dormant tumor cells are not actively dividing, they are often harder to treat, as most drug treatments preferentially target dividing cells (see Chap. 17, Sec. 17.4). Doxorubicin was shown to reduce macrometastases but was ineffective against dormant tumor cells in a mouse model of mammary cancer. A number of factors, such as VEGF and Id1, have been implicated in reactivating dormant tumor cells but more research is needed to understand this critical event, and target these cells therapeutically (Sleeman et al, 2011).
10.5 MOLECULAR MECHANISMS OF METASTASIS
10.5.1 Protease Activity at the Invasive Front
Proteolysis must function at the tumor cell surface to facilitate invasion and degradation of the basement membrane. Extracellular proteinases, transmembrane proteinases, cell-surface molecules, and intracellular factors all contribute to generating pericellular zones of proteolysis. Mechanisms known to underlie the proteolytic activation at the cell membrane include the activation of plasmin by uPA and its receptor uPAR, and the activation of pro-MMP-2 within a trimolecular complex generated by MT1-MMP, MMP-2, and TIMP-2 (Hernandez-Barrantes et al, 2000; Overall et al, 2000). MT1-MMP activity associated with tumor progression has been observed at the leading edge of migrating cells. For the cell surface activation of pro-MMP-2 in the trimolecular complex, MT1-MMP must itself be activated by a proprotein convertase called furin. Mature furin can cycle between the Golgi and the cell surface, and activate MT1-MMP at both locations. The association of pro-MMP-2 with α2β1 integrin-bound collagen was found to provide a reserve of the enzyme for subsequent activation of the trimolecular complex. By comprehensively comparing multiple MMPs in experimental systems that utilize 3D matrix composites, Weiss and colleagues found that MT1-, MT2-, or MT3-MMPs constitute the minimal requirement for basement membrane transmigration, and that MT1-MMP is the dominant protease mobilized for cancer cell trafficking through 3D interstitial ECM barriers (Rowe and Weiss, 2009). Several secreted MMPs, especially MMP-2 and MMP-9, have also been extensively studied in human cancer and experimental systems and have been shown to facilitate tumor cell invasion and motility, although their primary function in vivo may be to facilitate bulk ECM turnover during tissue remodeling (Kessenbrock et al, 2010).
The integrin αvβ3 may also localize active MMP-2 to the cell surface. Their colocalization was observed on newly developing blood vessels and on the tumor invasive front, and inhibition of their binding reduced tumor growth and angiogenesis (Silletti et al, 2001). Recently, it was shown that MT1-MMP catalyzes shedding of the α3 integrin ectodomain in ovarian carcinoma cells, and this associates with formation of multicellular aggregates, an important step in ovarian cancer metastasis (Moss et al, 2009). Shedding of MMP-9–dependent E-cadherin may also play a role in the dissemination of ovarian cancer cells. CD44 provides a means of anchoring active MMP-9 to the cell surface of invadopodia in breast cancer and melanoma cells, and was found to be critical for MMP-9-mediated cell migration (Dufour et al, 2010). The association of CD44 with hyaluronic acid also has been shown to increase MMP-2 secretion and CD44 has been found to recruit MMP-7 and direct localization of MT1-MMP to the cell membrane. These findings highlight the complex spatial coordination between adhesion molecules and enzymatic activity, which bring about controlled activation of metallo-proteinases and, ultimately, the digestion of ECM at the leading edge of invasive tumor cells.
10.5.2 Protease Activity in the Tumor Microenvironment
Beyond ECM degradation, studies using transgenic and knockout mouse models of MMPs and TIMPs have documented that proteolysis affects the early, as well as the late, stages of cancer progression. Protease activity impacts directly on cell growth, cell survival, angiogenesis, and inflammation (Fig. 10–9). Metalloproteinases and TIMPs alter the release of potent growth factors such as VEGF, TGF-β, and IGF-II, which are either sequestered in the ECM or exist in complexes with their binding proteins. Proteolytic cleavage of already synthesized factors results in altered bioavailability of growth signals to cancer cells and impact the process of cell proliferation. Activation of EGFR (see Chap. 8, Sec. 8.2), which is overexpressed in many human cancers, follows ADAM-mediated release of members of the EGF family of growth factors, including amphiregulin, TGF-a, and HB-EGF. Similarly, processing of Notch (see Chap. 8, Sec. 8.2.4), a master regulator of cell differentiation, requires a 3-step proteolytic activation with ADAMs performing the second cleavage of the receptor. MMPs and TIMPs also influence apoptosis signals, such as those feeding into Fas-mediated death receptor signaling (see Chap. 9, Sec. 9.4.4). In addition, proteolytic activity regulates capillary ingrowth, vascular stability and access of tumor cells to vascular and lymphatic networks. Overall, manipulation of the expression of TIMPs reveals that they universally inhibit angiogenesis, invasion, and metastasis, but their effects on cell proliferation and apoptosis are both tissue-specific and context-dependent (Cruz-Munoz and Khokha, 2008).
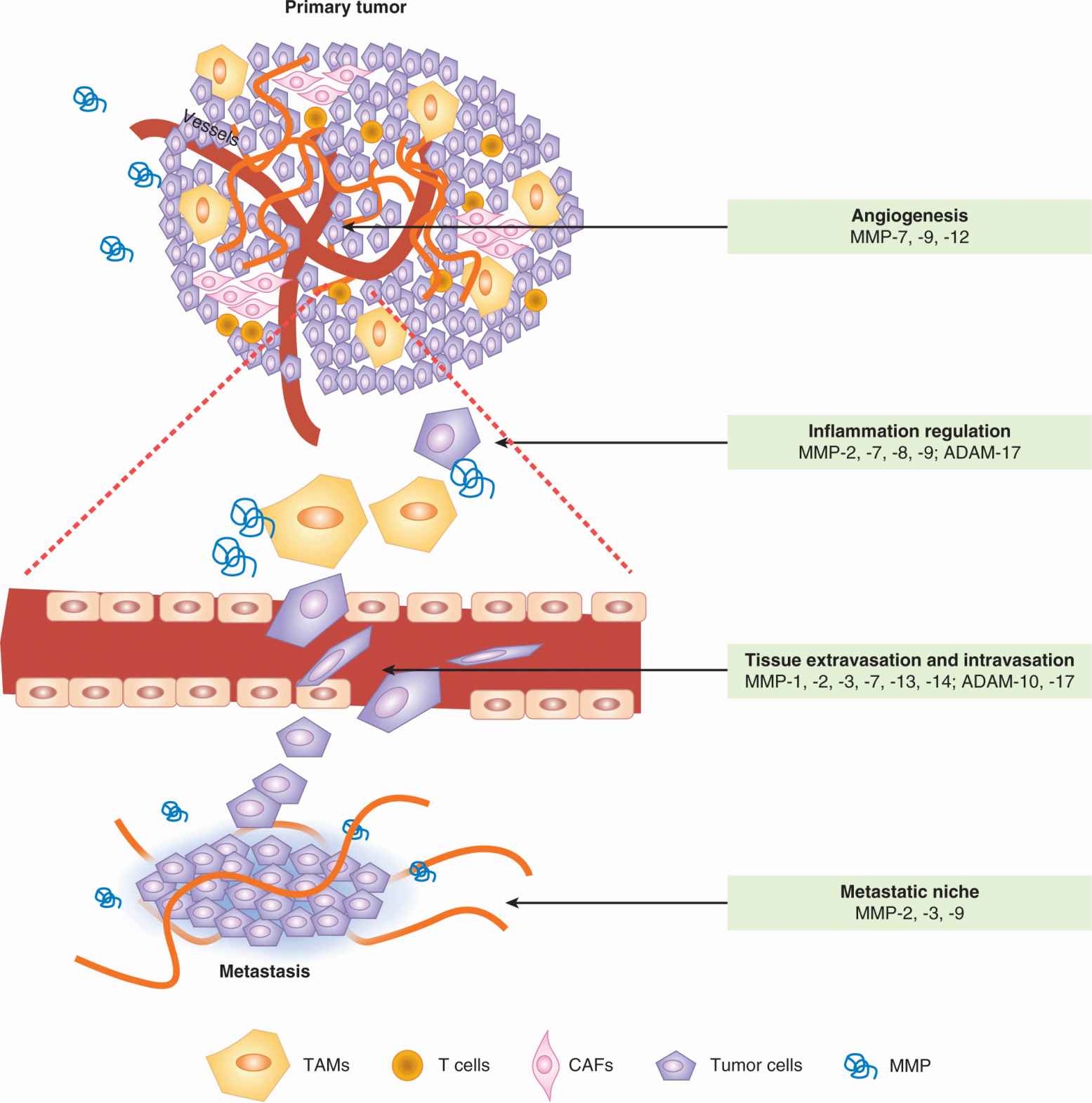
FIGURE 10–9 Metalloproteinases (MMPs) play multiple roles in tumor progression and metastasis. These enzymes are contributed by both stromal cells and tumor cells. Different MMPs are implicated in the various processes. Specific MMPs that promote each of the listed processes during metastatic dissemination are shown as examples. The figure shows vertically an expansion of intravasation and extravasation events in the vasculature of the tumor and metastatic organ, respectively. TAM, Tumor-associated macrophage. CAF, Cancer-associated fibroblast. (Adapted from Nguyen et al., 2009).
Several nonneoplastic host cells, including fibroblasts, endothelial cells, leukocytes, and bone marrow-derived cell populations, are recruited during tumor development, and the composite of these stromal and cancer cells creates a complex microenvironment (Fig. 10–10). Metalloproteinase activity contributes to generating this microenvironment and can further facilitate tumor progression. For example, metal-loproteinase activity, as regulated by the TIMP3-ADAM17 interactions, is important for the systemic release of molecules such as cell-surface-bound TNF-α. This pleiotropic cytokine is situated at the apex of cytokine cascades and NF-κB signaling that underlies immune cell crosstalk with cancer cells. Chemokines that influence immune cell motility are similarly processed by proteases. MMP cleavage of members of the monocyte chemoattractant protein (MCP) family of chemokines renders them receptor antagonists with inflammation-dampening effects (McQuibban et al, 2002). MMP-1 and MMP-3 process CCL8/MCP-2, which has antitumor activity in a melanoma model. MMP-8 is mainly produced by neutrophils, and was shown to exert a tumor-suppressive effect in a model of carcinogen-induced skin cancer (Balbin et al, 2003). Other chemokines, including CXCL1/KC (neutrophil-attracting) and CXCL11 (Th1-lymphocyte-attracting), are also substrates of MMPs and thus, proteolysis can affect neutrophil content or T-cell response (see Chap. 21, Sec. 21.4). Metalloproteinases can also cleave RANKL, which can subsequently promote metastasis to bone.
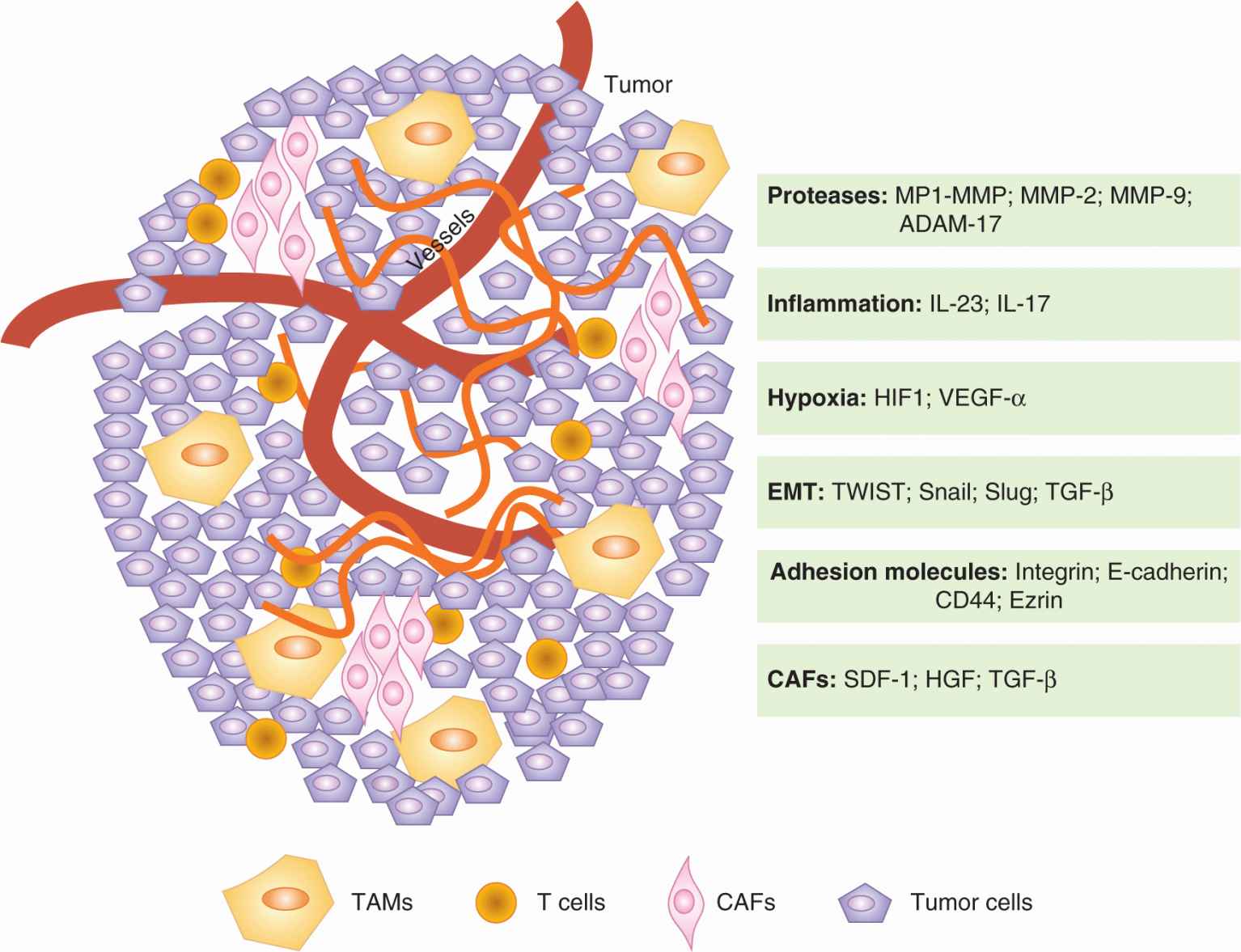
FIGURE 10–10 Mutual interactions between tumor cells and the microenvironment promote progression to metastasis. Tumor cells can express some of the factors listed above, such as Snail or Slug, to promote progression. Futhermore, tumor cells can influence their microenvionment and induce the production of tumor-promoting factors from neighboring or incorporated “normal” cells (CAFs and TAMs). Many of the critical factors for tumor survival and motility, such as TNF-a and TGF-β, are provided by the tumor microenvironment. CAF, Cancer-associatrd fibroblast; TAM, tumor-associated macrophage.
10.5.3 Ameboid Movement and Cell Motility
The idea that cell penetration of ECM-imposed structural barriers depends exclusively on proteolysis has been challenged by studies in collagen gels showing that tumor cells can acquire rounded, amoeboid-like shape and perform mechanical displacement of intact ECM fibrils by relying on increased cell deformation and reduced cell-ECM adhesion. This protease-independent amoeboid tumor cell migration is likened to that of leukocytes (Croft and Olson, 2008). The importance of this alternate mechanism of migration in the metastatic process is currently uncertain, partially because of concerns about whether these gels truly mimic the 3D ECM barriers found in vivo (Sabeh et al, 2009).
10.5.4 Cancer-Associated Fibroblasts
Mesenchymal cell types, including cancer-associated fibro-blasts (CAFs) and pericytes, coevolve with cancer cells during tumor progression and become an integral part of the paracrine communication (see Fig. 10–10). CAFs were thought initially to arise from local fibroblasts by acquiring a modified “activated” phenotype. However, later studies linked their origin to bone marrow-derived cells or transdifferentiation from epithelial or even endothelial cells. TGF-β is considered a critical activating factor for CAFs, which are typically identified through the expression of a set of markers (ie, α-smooth muscle actin [α-SMA]; fibroblast specific protein-1 [FSP-1]/S100A4; FAP; neuronglial antigen-2 [NG-2]; platelet-derived growth factor-β receptor [PDGFR-β]), whereas pericytes are more loosely defined but are thought to depend on PDGF and TGF-β signaling. These stromal cell types can contribute to cancer promotion by the delivery of key growth signals (HGF, FGF) and survival signals (insulin-like growth factors [IGFs]) that can counter death signals and activate downstream oncogenic signaling. They inherently provide ECM components for interaction with integrins resulting in the activation of specific signal transduction pathways. CAFs also overexpress metalloproteinases, chemokines (SDF-1 or CXCL12, IL-6, CXCL8) and angiogenic factors (VEGFs, FGFs) that can lead to the generation of a proangiogenic and proinflammatory microenvironment. Consistent with this, when they are coinjected with tumor cells CAFs promote xenograft growth. They have been observed at the sites of metastases, and SDF-1 is known to promote the recruitment of endothelial progenitors, whereas activated pericytes can affect vessel permeability, both of which are critical mechanisms in angiogenesis. The gene expression profiles of CAFs have demonstrated their heterogeneity in individual tumors and resulted in identification of CAF subsets, which may have prognostic value. The importance of understanding these cells, originally considered to be bystanders, is emphasized by the observation that mice deficient in specific CAF markers show decreased metastasis, and that CAFs may alter the drug-sensitivity of cancer cells (Ostman and Augsten, 2009).
10.5.5 Tumor-Associated Macrophages
Tumor-associated macrophages (TAMs) are important players in promoting cancer progression (see Fig. 10–10) and high TAM content in the tumor mass correlates with poor prognosis of patients (Qian and Pollard, 2010). In the local microenvironment, tumor cells can direct the differentiation of TAMs, which exhibit several protumorigenic and prometastatic functions, including induction of inflammation, secretion of growth factors and MMPs, promotion of angiogenesis, and suppression of cytotoxic effects (Sica et al, 2008). The inflammatory state of TAMs is controlled by the transcription factor NF-κB, which, in turn, is activated through toll-like receptors (TLRs; see Chap. 21, Sec. 21.2.2). TAMs exert most of their inflammatory effects through cytokines. Cytokines such as IL-6 cause the endothelial lining of the tumor vessels to become leaky, resulting in the recruitment of more inflammatory cells and providing escape routes into the bloodstream for tumor cells. TNF-α produced by TAMs can activate NF-κB and AP-1 family of transcription factors in the tumor cells, stimulating their cell proliferation and survival. Specific inhibition of NF-κB activity in myeloid cells through ablation of IkB kinase (IKK)-α resulted in a reduction of inflammation and inhibition of tumor progression. In contrast, inactivation of STAT3 (a transcription factor that functions to suppress inflammation—see Chap. 8, Sec. 8.3.1) in myeloid cells is associated with abundant expression of inflammatory cytokines such as TNF-α and IL-6 that have been shown to promote chronic colitis and invasive colorectal cancer in animal models (Grivennikov et al, 2009). Another crucial cytokine produced by TAMs is IL-23. IL-23 acts by enhancing the activity of Th17 cells and inhibiting the activity of T-regulatory cells. The Th17 cell is a T-helper cell subclass with strong inflammatory effects, and they are generally associated with tumor progression.
10.5.6 Epithelial-to-Mesenchymal Transition
Epithelium is a highly polarized structure composed of polygonal-shaped cells with abundant cell–cell tight junctions. It lines the outer surface, as well as the inner organ surface, of the body. Epithelial cells make contact with a basal membrane, and both cell–cell and cell–matrix adhesions are necessary for their survival. Different epithelial cell populations are heterogeneous, depending on the tissue/organ involved. In contrast, mesenchymal cells are spindle-shaped and highly migratory, which is essential for their role in supporting tissue/organ development. EMT (Fig. 10–11) is a process during which an epithelial cell loses its apical–basal polarity and becomes a mesenchymal-like cell with increased migratory ability, resistance to apoptosis, and increased production of ECM components (Kalluri and Neilson, 2003). During this process, epithelial cells often lose the expression of cell-surface and cytoskeletal proteins mediating adhesion, such as E-cadherin, cytokeratin, zona occludens 1 (ZO-1), and laminin. Instead, they gain proteins that are often seen on mesenchymal cell surface such as N-cadherin, vimentin, fibronectin, and α-SMA. The resulting mesenchymal-like cell eventually detaches from the basal membrane and migrates away from the epithelial layer. This process occurs extensively in the embryo at different stages of maturation and development of organs, and in wound healing (Hay, 1995). In tumors it is hypothesized that this embryonic program may be reactivated to drive an initial important step in metastasis, but may then be reversed mesenchymal to epithelial transition (MET) when the cell establishes a new growth (Kalluri and Weinberg, 2009). This latter (MET) step may help to explain why it has been difficult to observe evidence of the EMT process in human tumor specimens (Tarin et al, 2005).
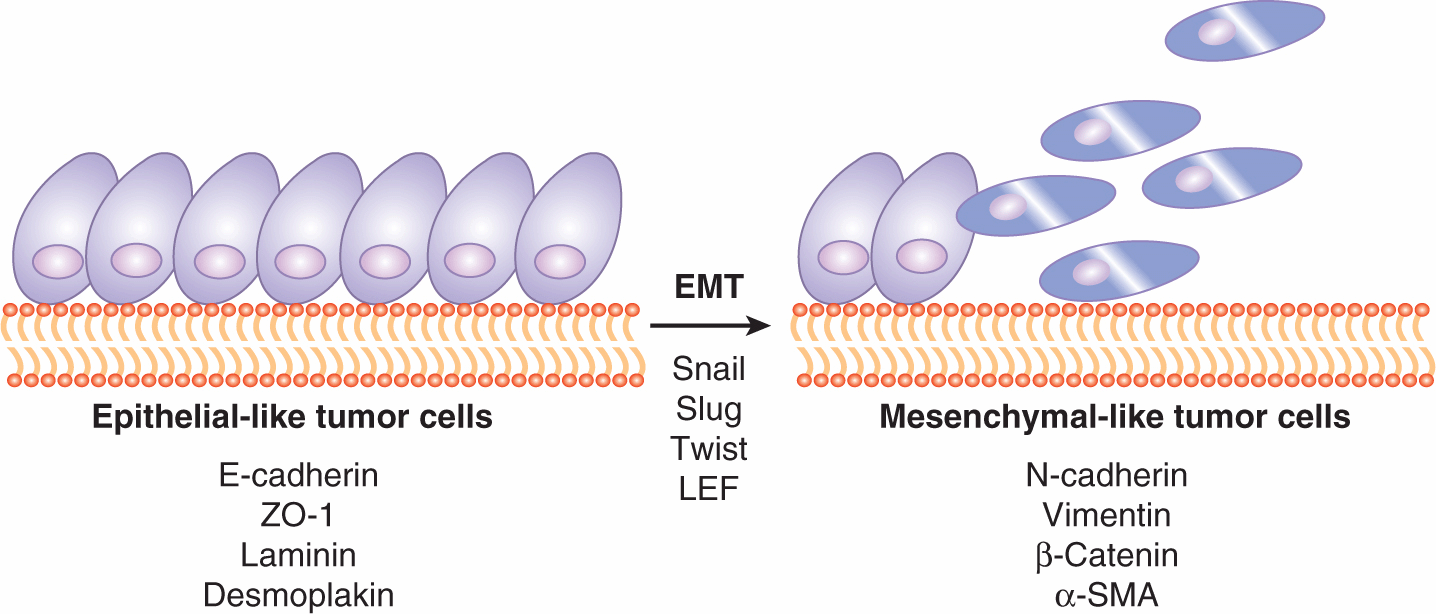
FIGURE 10–11 During tumor progression, epithelial tumor cells often undergo EMT to gain anchorage-independent survival and motility. Epithelial cells usually express markers such as E-cadherin, ZO-1, laminin, and desmoplakin. Through the aberrant expression of transcription factors such as Snail and Slug, these epithelial cells lose their epithelial markers and start to express mesenchymal markers, such as N-cadherin, vimentin, β-catenin, and α-SMA. Many of these markers are also functionally associated with the epithelial or mesenchymal characteristics. For example, E-cadherin and N-cadherin activate different intracellular programs that give rise to the epithelial-like or mesenchymal-like properties.
Various studies link loss of E-cadherin to EMT. Treatment with a monoclonal anti–E-cadherin antibody disrupted cell–cell junctions in MDCK cells, activation of a fusion protein that abolishes E-cadherin expression induced EMT in mouse mammary epithelial cells, while expression of E-cadherin induced a MET-like process with reestablishment of cell junctions and decreased proliferation in cells that had already undergone EMT. Cancer cell lines with loss of E-cadherin expression show higher tumorigenicity as xenografts in nude mice, and, in some cancers, E-cadherin levels relate inversely to prognosis. Mutations in the E-cadherin gene causing either loss or truncation of the protein have been identified in human breast and gastric cancers, possibly rendering these tumors more prone to EMT and metastasis. Similarly, epigenetic mechanisms such as transcriptional repression and promoter silencing by hypermethylation also contribute to E-cadherin downregulation in various carcinomas. Transcription factors that play roles in EMT such as TWIST, Snai1 (Snai1), and Slug (Snai2), also repress E-cadherin expression (Medici et al, 2008).
TGF-β, normally a negative regulator of epithelial cell growth, is another key player in EMT. In various in vitro studies, TGF-β has been shown to induce EMT-like changes in epithelial cell lines. Two pathways downstream of TGF-β are at least partially responsible for its transforming effect, Smad and p38/RhoA (see Chap. 8, Sec. 8.4.4). Studies using in vitro cell lines suggest that activation of p38 is indispensable. Other signals that induce EMT include HGF, which activates the receptor tyrosine kinase c-Met, as well as EGF and PDGF, activating their respective receptors. Multiple recent studies have linked noncoding microRNAs (see Chap. 2, Sec. 2.4.3) as regulators of EMT, both positively and negatively. MicroRNA family members, miR-200 and miR-205, prevent EMT by inhibiting Zeb-1 and Zeb-2, known repressors of E-cadherin expression. In contrast, miR-21 expression is elevated in many carcinomas and supports TGF-β-dependent EMT (Shi et al, 2010).
10.5.7 Role of Hypoxia
Hypoxia drives the expression of a large number of metastasis-related genes through the specific HIF1/2 transcription factors and has been found to relate to metastatic disease in many experimental models and in some clinical studies (Finger and Giaccia, 2010; Lunt et al, 2009). As discussed in Chapter 12, Section 12.2, cells in tumors may be exposed to hypoxia as a consequence of diffusion limitations (prolonged or chronic hypoxia) or of perfusion limitations (acute or cyclic hypoxia). Both types of exposure are reported to modify gene expression, and in experimental studies in vitro, both were found to affect metastatic properties of cells. In animal models or patients, it is currently not possible to determine directly whether cells which form metastases derive from areas of chronic or acute hypoxia, but studies that have deliberately induced increased levels of acute (cyclic) hypoxia in tumors in animal models have demonstrated increase development of metastases, suggesting that exposure to acute (cyclic) hypoxia can play an important role in the metastatic process (Lunt et al, 2009). Many specific mechanisms of metastasis are reported to be affected by hypoxia, as discussed below, but the specific genes involved may be different in different cell types.
EMT-promoting genes, such as TWIST and Snai1/2, can be upregulated by hypoxia in multiple cell lines, including ovarian, renal cell, pancreatic, and colon cancer lines, causing loss of E-cadherin. Other important adhesion molecules that can be mediated by hypoxia are the β1 integrins that have been found to correlate with invasive capacity in pancreatic cancer cell lines. Hypoxia also can induce the expression of uPA, uPAR, MMP-2, and MMP-9 in a variety of cell lines, leading to an increase in metastatic invasion both in vitro and in vivo. Blocking uPAR activity with a monoclonal antibody was reported to almost abolish metastatic disease in mice bearing human melanoma xenografts. Similarly, hypoxic exposure of a human breast carcinoma line in vitro resulted in the downregulation of TIMP and concomitant upregulation of MMP-9, causing increased invasive capacity, an effect that could be blocked by using an inhibitor of MMPs. Other studies demonstrated hypoxia-mediated upregulation of MMP-2 activity and a positive correlation with metastatic ability in lung and melanoma tumor models. Hypoxia has been shown to increase transcription of c-MET thereby sensitizing the cells to HGF, significantly increasing the invasive capacity of tumor cells.
The SDF-1/CXCR4 signaling complex plays a key role in tumor cell motility and homing. In vivo studies using a human breast cancer line showed that the formation of both spontaneous and experimental lung metastases could be significantly reduced using a monoclonal antibody against CXCR4, demonstrating its importance in the metastatic process. CXCR4 and SDF1 have been shown to be upregulated by tumor hypoxia, facilitating the development of metastatic disease. The presence of SDF-1 at secondary sites, such as lung, lymph nodes, and bone, concomitant with the expression of its receptor on CTCs may enable cell adhesion and extravasation at the secondary site. The ECM protein, lysyl oxidase (LOX), has also been identified as a hypoxia-regulated gene involved in meta-static disease (Erler et al, 2006). LOX was found to be positively correlated with tumor hypoxia in breast cancer patients, and there was a significant relationship between LOX expression and distant metastases. In vitro studies demonstrated a role for LOX in invasion and migration through regulation of FAK activity, suggesting multiple roles for hypoxia-induced LOX in the formation of metastatic disease. Another important ECM protein that is hypoxia-regulated is the secreted glycophosphoprotein osteopontin (OPN), which is expressed by multiple different cell types (osteoclasts, osteoblasts, epithelial cells, and endothelial cells). OPN has roles in cell adhesion, angiogenesis, prevention of apoptosis, and the anchorage-independent proliferation of tumor cells. Its expression has been found to correlate with increased metastatic potential in breast, prostate, colon, and head and neck cancers, and in soft-tissue sarcoma (Anborgh et al, 2010).
VEGF-A is a potent inducer of tumor angiogenesis (see Chap. 11, Sec. 11.4.1) that is upregulated in response to hypoxia by both HIF-1 dependent and independent mechanisms. Its receptors, VEGFR-1 and -2, are also induced under hypoxic conditions. The role of this protein in metastatic disease has been examined extensively, with some studies suggesting a link between VEGF-A expression and meta-static disease, and others not, consistent with the concept that angiogenesis is a result of multiple factors of varying importance in different tumor types. However, in driving the development of neovasculature, VEGF-A may provide a mechanism of transport for the tumor cells, as well as enhancing the intravasation and extravasation stages of the metastatic process because of increased vascular permeability. VEGF-A also plays a role in macrophage migration, and TAMs are reported to localize in hypoxic regions in tumors.
Using expression microarray, OPN was identified to be the most consistently upregulated gene in relation to tumor progression (Agrawal et al, 2002). High levels of OPN in the serum also have been reported to correlate with increased levels of hypoxia and poorer treatment outcome in head and neck and non–small cell lung tumors (Le et al, 2003; Mack et al, 2008).
10.5.8 Metastatic Niches and Microvesicles
A recent addition to the metastatic process is the concept that a “premetastatic niche” can be created in distant organs to which CTCs can “home” (Carlini et al, 2011). Although tumor cells are the driving force of metastasis, these new findings suggest that the host cells within the tumor microenvironment play a key role in influencing metastatic behavior. Specifically, bone marrow-derived hematopoietic progenitor cells expressing VEGFR-1 have been shown to precede the arrival of even single metastatic cells at distant sites. They are postulated to build a microenvironment suitable for tumor cell growth, although blockade of VEGFR-1 was insufficient to prevent metastasis in the widely used B16 melanoma and Lewis lung tumor metastasis models (Dawson et al, 2009). Molecular processes that have been identified in niche creation include the upregulation of specific integrins and their ECM ligands or increased expression of inflammatory chemoattractants. For example, α4β1 integrin expression on progenitor cells can allow their interaction with fibronectin-expressing metastatic cells. Secreted soluble factors are key players in bone marrow cell mobilization during metastasis, and S100A8 or S100A9 expressed in the niche can attract macrophages. Metalloproteinases are also likely candidates for promoting the formation of a metastatic niche, as this involves altered matrix proteins, VEGF, TGF-β, and TNF-α bioavailability, chemokine activity, and immune cell interaction. In addition hypoxia-induced LOX was recently associated with the development of meta-static niches (Erler et al, 2009). Mechanisms for how such niches are initiated, the extent of the role they play in the overall metastatic process, and whether factors secreted from the primary tumor are required for their initiation are currently unclear and require further investigation.
Membrane vesicles (exosomes) derived from both tumor and host cells have also been recognized to promote tumor growth and metastasis, possible through promotion of meta-static niches (Peinado et al, 2011). Microvesicles are generated by the outward budding and fission of membrane vesicles from the cell surface. They are released from cells upon activation, malignant transformation, stress, or death, and can be found in various biological fluids, including blood and urine. These structures contain “cargo” including proteins (receptors, antigens), lipids, and nucleic acids (DNA, mRNA, microRNA), and can be endocytosed by other cells or interact with their cell surface receptors through fusion. This process occurs frequently in platelets and tumor cells, and such exosomes can provide a means for horizontal transfer of bioactive molecules to stimulate tumor progression, immune response, invasion, angiogenesis, and metastasis. For example, microvesicles from platelets were found to induce angiogenesis and metastasis in lung and breast cancers, and those released from tumor cells contained tetraspanins that could recruit endothelial cells. Molecular information harbored in the circulating microvesicles is being explored for its prognostic and predictive significance (Pap, 2011).
10.5.9 Metastasis-Suppressor Genes
A small number of genes are reported as metastasis-suppressor genes. These genes are defined strictly as those whose products reduce the metastatic behavior of tumor cells without affecting their tumorigenic capacity. The detailed mechanisms by which these genes act are diverse and involve every step in the metastatic cascade. However, only a few genes have been investigated in enough detail to be certain about their mechanism(s) of action or even that they truly conform to the second part of the definition. The nm-23 (NME-1) gene was the first metastasis-suppressor gene isolated and it has several isoforms (Lee et al, 2009). Inverse correlations between expression levels and metastatic potential have been observed in a number of different cancers including breast cancer and melanoma. NME-1 has nucleoside diphosphate kinase (NDPK) activity as well as exonuclease and histidine kinase activity. It also appears to play a role in maintenance of genomic stability. Mutations in both NDPK and exonuclease activity still allow suppression of metastasis and the activities associated with them vary by cell type. It is unclear which of the various activities plays the critical role in suppressing cancer metastasis. The KAI-1/CD82 gene is a member of the Tetraspanin (TM4SP) family of adhesion molecules that play a role in lymphocyte differentiation and function. It has been reported to have a p53 binding site in its promotor, and the loss of KAI-1 correlated with loss of p53. Loss of expression is implicated in metastases in cancers of the prostate, breast and colon and in melanoma. KiSS-1 appears to be involved in cell signaling, as a posttranslationally-modified version of this protein, called metastatin, has been reported to bind to a G-protein–coupled receptor (Axor12) and preliminary evidence suggests that activation of this receptor can alter signaling through FAK. MKK4 is also a molecule associated with signaling, through the stress-activated protein kinase (SAPK) pathway (see Chap. 8, Sec. 8.2.4), and both these molecules may act to increase the likelihood that a tumor cell will be able to initiate growth at a new (metastatic) site. Breast metastasis suppressor-1 (BrMS-1), which is frequently altered in late-stage breast cancers, is a transcriptional repressor and causes downregulation of phosphatidylinositol (4,5) bisphosphate (PtdIns(4,5)P2). In vivo experiments have demonstrated that BrMS-1 inhibits several steps of metastasis, including the ultimate step, colonization at the secondary site. To date, however, whether gene regulation effects are direct versus indirect has not been clearly demonstrated. Hurst and Welch (2011) have reviewed clinical and experimental information about these and other possible metastasis-suppressor genes.
10.5.10 Genome-Wide Analyses and Metastatic Signatures
Recent advances in sequencing and microarray technologies (see Chap. 2) have enabled genome-wide analyses of primary tumors and researchers are increasingly using -omic approaches (eg, genome, transcriptome, proteome, or methylome) to study metastasis. One of the most common approaches is the gene expression profiling of tumors (transcriptome), and this approach has been applied to examining metastatic propensity of tumors. A seminal paper identified a group of 70 genes expressed in the primary tumors of breast cancer patients, which formed a profile that was capable of predicting survival and the likelihood of developing distant disease (van de Vijver et al, 2002). In a retrospective analysis, this profile was claimed to outperform the best predictions based on histological and clinical criteria, thus providing a capability to select early stage patients who needed adjuvant chemotherapy, and thereby avoiding exposing those who did not need such treatment to the toxicity involved (Knauer et al, 2010). Another study identified an expression profile involving 128 genes that distinguished between primary tumors and metastases of a range of different types of adenocarcinomas (Ramaswamy et al, 2003). This gene set was subsequently refined down to a group of 17 genes that retained a broad diagnostic ability to predict outcome in a range of tumor types (lung, breast and prostate adenocarcinomas and medulloblastoma but not lymphoma). Interestingly, predictive gene signatures from various studies often do not overlap with each other. This has been explained by the heterogeneity of the various patient pools from which the signatures are derived.
These technologies have been extensively applied to breast cancer, allowing specific subgroups with prognostic differences to be identified. Various tests using molecular profiling are currently commercially available to assist treatment decisions for patients with early stage breast cancer (Eroles et al, 2012). Clinical studies have demonstrated benefit in such tests for choice of drug treatment in patients with node-negative breast cancer, who may have undetected distant metastases, but there remain questions about which patient populations are most appropriate for these tests and whether they are applicable to other cancers (Oakman et al, 2010).
In addition to global RNA expression profiles, changes in genome-wide DNA copy number have been examined in cancers. Hu et al (2009) found consistent copy number gains on chromosome 8q22 in breast cancer patients with poor prognosis. Through computational analyses of this region, they were able to identify metadherin (MTDH) as a metastasis-promoter gene. Knocking down MTDH in several cell lines resulted in significant reduction of lung metastases in an experimental model of metastasis in mice. Hu et al (2009) also showed that gain of MTDH was associated with poor prognosis in an independent patient cohort. In another study on colorectal cancer, comparison of DNA copy number profiles from metastasis-free patients and patients harboring liver or peritoneal metastases identified copy number gains on chromosome 20q that preferentially associated with liver metastasis (Bruin et al, 2010).
Whole-genome sequencing is another powerful tool for understanding the metastatic process. In pancreatic cancer, comparison of mutations between primary malignancies and their corresponding metastases led to the identification of single-nucleotide changes that only existed in the metastases (Campbell et al, 2010). Furthermore, relying on extensive analyses of the sequencing data, an evolutionary path of the tumor cells was constructed and sequential mutations acquired by tumor cells that eventually led to metastases were identified (as illustrated by Fig. 10–2). Whole-genome methylation profiling is also being applied to metastasis research. Fang et al (2011) identified a methylation signature associated with low metastatic risk and high survival rate in breast cancer. The methylation status was able to account for many of the transcriptional differences between the poor- and good-prognosis patients, and was independent of other breast cancer markers. Its predictive potential is shared by other malignancies, such as glioma and colon cancer. As these new sequencing technologies become more affordable and more widely available, it is expected that they will lead to deeper insights in metastatic dissemination of each cancer type to specific distant organs.
10.6 TREATMENT OF METASTASES
Current cancer therapeutics largely focus on the primary tumor and it is generally assumed that the sensitivity of the cells in metastases is similar to that of the primary tumor. Specific treatment of identified solitary metastases may occur with focused radiotherapy or surgery, but there has been limited success in the development and clinical use of drugs specifically targeted against the metastatic process (Perret and Crepin, 2008). As described above, this process can be broken down into various stages and studies in preclinical models have demonstrated the potential to block metastasis formation by blocking essentially all the stages. However, clinical applications of some of these approaches have not shown much benefit to date. For example, an important step in the process is the arrest of CTCs, and many anticoagulant drugs have been assessed for their ability to inhibit metastases (Hejna et al, 1999). There have been consistent reports of reduced metastases in animal systems by treatment with heparin, warfarin, and inhibitors of platelet aggregation (prostacyclin and dipyridamole), but there is only limited information from clinical trials. One large trial of the use of warfarin in patients with lung, colon, head and neck, and prostate cancer led to little or no improvement in survival (Zacharski et al, 1984). Similarly, the use of peptides, blocking the integrin motif RGD, which is involved in binding of cells to fibronectin, were also largely ineffective in clinical studies, although they showed considerable promise in preclinical studies. New integrin antagonists not based on the RGD motif are currently in early clinical development.
Other stages of the metastatic process are intravasation and extravasation of the tumor cells into the circulation and from the circulation into a secondary site. Despite the important roles that MMPs appear to play in these processes, the development of synthetic inhibitors against metalloproteinases as cancer therapeutics has thus far not been very successful, although the clinical studies were largely in late stage patients and not specifically designed to study effects on formation of metastases (Zucker et al, 2000). The tremendous heterogeneity of metalloproteinase expression with respect to particular members and the cell type responsible for its expression and overlapping substrate profiles of metalloproteinases may be the reason for the lack of effect observed and more specific inhibitors are under development. The plasminogen activator system is also being investigated, particularly as uPAR and PAI-1 have been found to be strong predictors of treatment outcome in breast cancer in various tests and genetic screens. Clinical studies are in progress with inhibitors of this proteolytic system (Schmitt et al, 2011).
Agents targeted at the angiogenic process, which also is critical for the development of macrometastases, have also not proved very successful in clinical studies, although, again, many of these agents are tested in patients with advanced disease. Studies of the compound, razoxane, which “normalizes” the structure of tumor blood vessels and was found to be effective as an antimetastatic agent in animal models, did suggest improved outcome in patients with soft-tissue sarcoma when combined with vindesine and radiation and surgery (Rhomberg et al, 2008). Unfortunately, razoxane development was halted because of a possible link to leukemia.
One limitation to the successful development of clinical therapies to prevent metastasis formation is that microscopic metastases are often present prior to detection and treatment of the primary tumor. Prevention of secondary metastases (ie, metastases from metastases) might be useful in palliation, but the potential for increased cure through the use of antimetastatic agents is limited to patients in whom metastases are seeded after diagnosis and prior to eradication of the primary tumor or in cases where tumor cells have seeded to other sites in the body but are in a dormant state. Tumor cells are known to enter the circulation at the time of surgery, but seeding at the time of surgery is probably the sole source of metastases for only a small proportion of patients. The recent discovery of the development of “premetastatic niches” (see Sec. 10.5.8), may provide an opportunity for early (preventative) treatment for the development of metastases using inhibitors of components of this process such as LOX.
Related posts:
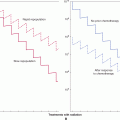
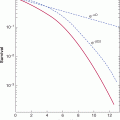
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


