Chemical Carcinogenesis
4.1 INTRODUCTION AND HISTORICAL PERSPECTIVE
Although many factors contribute to cancer causation, this chapter focuses on providing an overview of the mechanisms by which exogenous chemicals may influence the risk of cancer initiation and tumor growth, and how knowledge of these mechanisms might be exploited to improve human health through prevention or intervention. The reader is directed to the bibliography for a list of critical reviews that summarize the past history, current status, and future prospects for the field of chemical carcinogenesis. The relative importance of environmental chemical exposures to the total burden of cancer risk remains highly contentious and it has been estimated that chemical pollution of the environment accounts for no more than 1% to 3% of all human cancers. However, such estimates do not consider chemical exposures in the workplace (5%) or cigarette smoke (30%) to be environmental pollutants, and they tend to have a primary focus on genotoxic chemicals as causative agents. Thus they may underestimate the importance of the interplay between the permanent tumor-initiating effects of low-level carcinogen exposure, the additional effects of nongenotoxic chemicals, and the potentially reversible modulating effects on tumor growth of diet, exercise, and other lifestyle factors.
Historically, epidemiological studies suggesting that chemicals can cause human cancer date to more than 230 years ago when Percival Pott observed that scrotal cancer was correlated with soot exposure in English chimney sweeps, and Butlin suggested subsequently that the better hygiene practices of European sweeps reduced their cancer risk. In 1895, Rehn reported a high rate of bladder cancer in German factory workers who were exposed to aniline-based azo dyes. The 20th century saw the identification of specific chemicals associated with increased risk of cancer, and provided methods for identifying cellular and molecular targets of the causative agents and for elucidating mechanisms involved in the conversion of normal cells to produce tumors. A seminal breakthrough was Yamagiwa’s production of skin tumors in rabbits by the direct application of coal tar in 1915, which led to the isolation, identification, synthesis, and biological testing of polycyclic aromatic hydrocarbons (PAHs) as chemical carcinogens, including dibenz[a,h]anthracene and benzo[a]pyrene (Fig. 4–1). In 1938, Hueper followed up Rehn’s observation by demonstrating that the aromatic amine β-naphthylamine (Fig. 4–1), a reagent used in manufacturing azo dyes, caused bladder tumors in dogs.
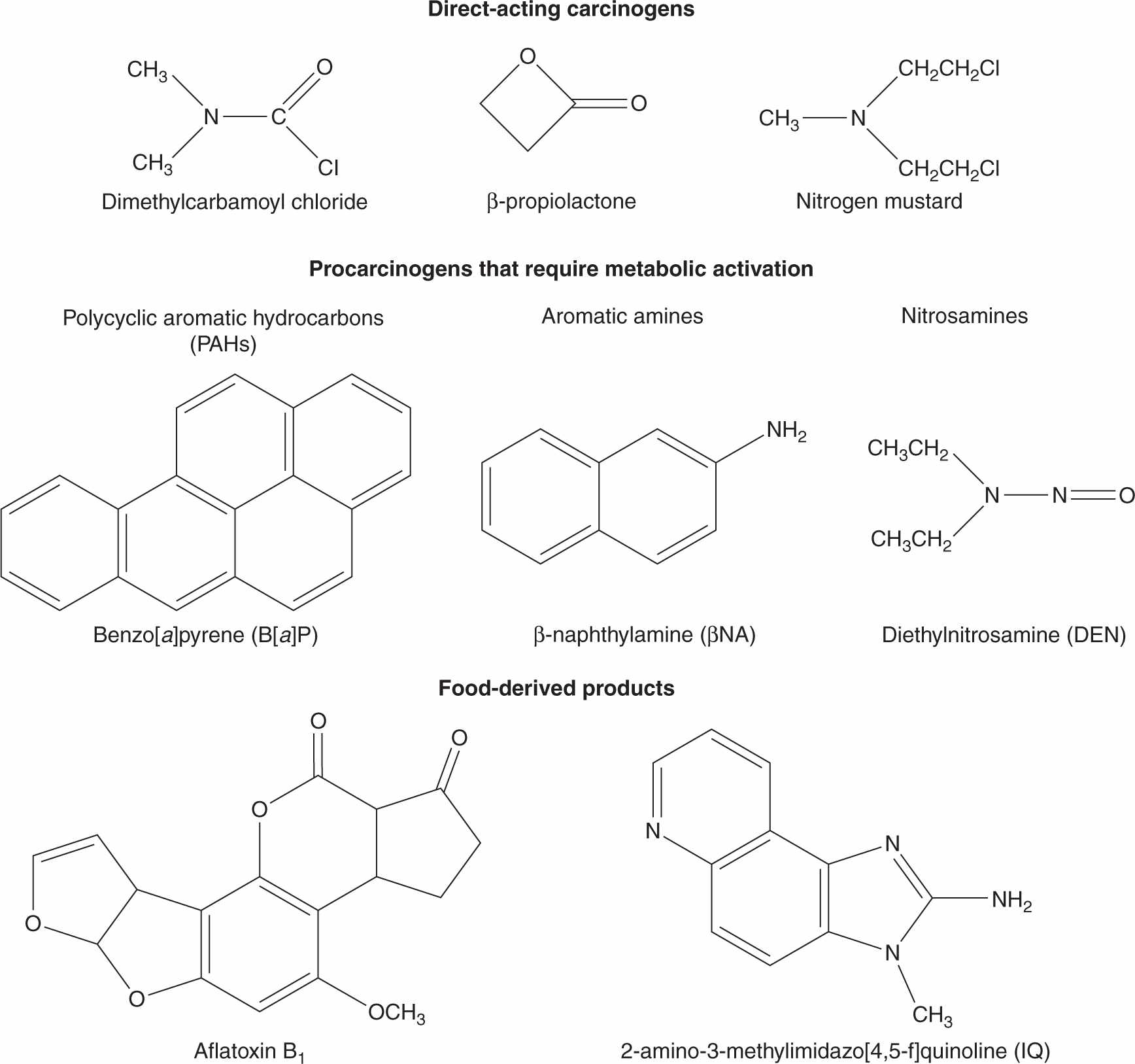
FIGURE 4–1 Structures of some direct-acting carcinogens and procarcinogens that require metabolic activation.
With the establishment of DNA as the mediator of genetic inheritance in 1953, it became clear that carcinogen-induced DNA damage leading to fixed mutations that could be transmitted to progeny cells was a key event in the process by which exposure to carcinogens led to uncontrolled cell division and tumor growth. The concept that many carcinogens require enzymatic bioactivation into electrophilic metabolites that bind covalently with DNA was established by the Millers in the 1960s. This was followed by investigations that identified specific DNA adducts of various chemicals including benzo[a]pyrene and aflatoxin B1, and demonstrated their binding to human tissues. Finally, in the 1990s, key target genes for carcinogens were identified (ie, oncogenes and tumor-suppressor genes—see Chap. 7) whose functionally altered products are key in the initiation, promotion, or progression of tumor growth.
4.2 BIOLOGY OF CHEMICAL CARCINOGENESIS
4.2.1 Tumor Initiation, Promotion, and Progression
Human epidemiological and experimental laboratory studies have long indicated that a latent period (often decades in humans) exists between the exposure to a chemical and the appearance of cancer. This led to the formulation of a sequential model that divided the carcinogenic process into 3 stages termed tumor initiation, tumor promotion, and tumor progression.
Tumor initiation was regarded as involving the interaction of a reactive chemical species (often a procarcinogen metabolite, see Sec. 4.2.3) with DNA to produce damage which, if not repaired before the next cell division, would lead to erroneous DNA replication resulting in fixation of mutations within the genome of individual cells. Thus 3 cellular functions are important in determining the likelihood of tumor initiation: the rate of procarcinogen activation, the efficiency and fidelity of DNA repair (see Chap. 5), and the capacity for cell proliferation. If a mutation disrupts the function of a gene whose product plays a role in maintaining the terminally differentiated function of the cell, the cell may acquire an altered (usually less differentiated) phenotype. Although initiation is irreversible, not all initiated cells will go on to establish a tumor, as many of these cells may die by apoptosis (see Chap. 9, Sec. 9.4), and further proliferation-enhancing signals are required for initiated cells to progress along the pathway to autonomous (cancerous) growth.
Tumor promotion was viewed as the clonal expansion of an initiated cell as a consequence of events that alter gene expression, so as to provide the cell with a selective proliferative advantage. Although there is no single unifying mechanistic feature of tumor-promoting agents, they tend to be nongenotoxic and to cause, directly or indirectly, cells to divide but not to terminally differentiate or die, resulting in the survival and proliferation of preneoplastic cells and the formation of benign lesions such as papillomas, nodules, or polyps. Many of these lesions may regress spontaneously, but a few cells may acquire additional mutations that allow them to progress to a malignant neoplasm. Figure 4–2 shows the structures of 3 established tumor promoters: tetradecanoyl phorbol acetate (TPA), 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), and phenobarbital. Early studies of mouse skin carcinogenesis illustrated the tumor-promoting activity of croton oil, which contains TPA, when applied following low doses of the tumorinitiator benzo[a]pyrene. Administration of benzo[a]pyrene led to skin tumors only when followed by repeated dosing with croton oil, even though croton oil alone was not carcinogenic.
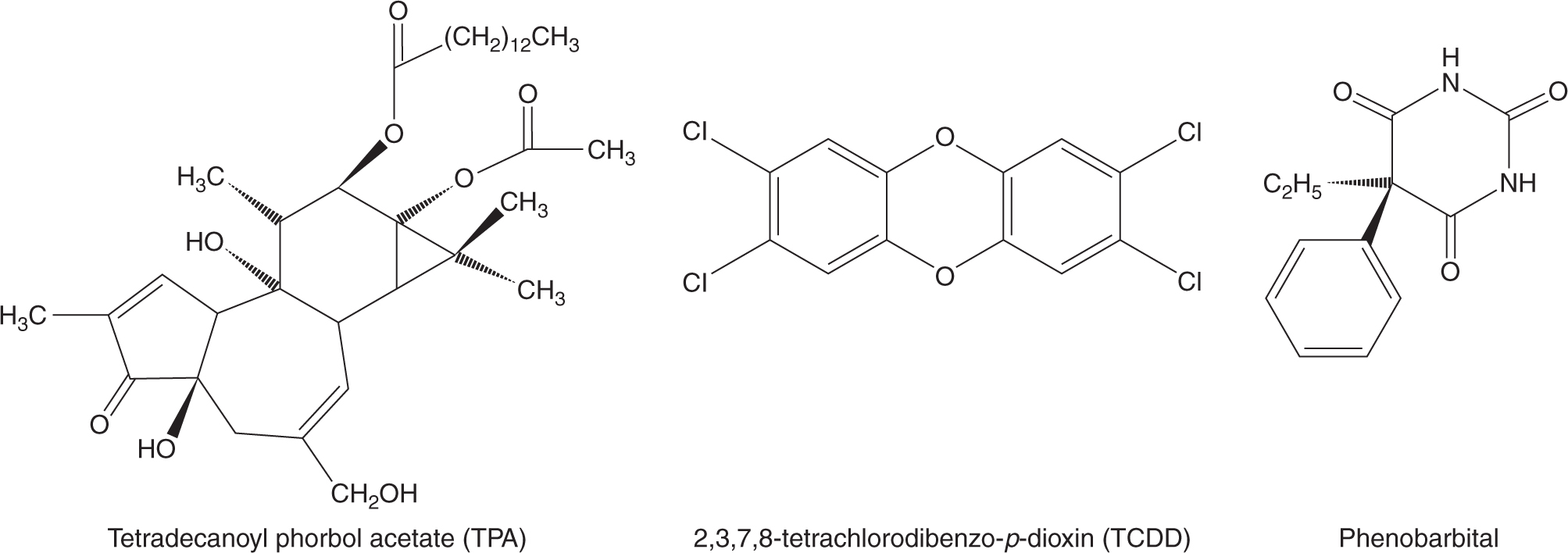
FIGURE 4–2 Structures of some established tumor promoters.
Tumor progression described the stage whereby benign lesions acquire the ability to further grow, to invade adjacent tissues, and to establish distant metastases.
Although this simple 3-stage model can be a useful conceptual framework for understanding carcinogenesis, we now recognize that the process does not so neatly compartmentalize within such stages: multiple sequential mutations in combination with epigenetic changes and prolonged alterations in the cellular microenvironment are required to convert a normal cell into a malignant tumor.
4.2.2 Genetic Instability and the Hallmarks of Cancer
Increased genetic instability and alterations in karyotype are often observed in tumor cells (see Chap. 5, Sec. 5.2). Inherited or acquired mutations in genes such as p53, retinoblastoma (Rb) or DNA mismatch repair genes can create a “mutator phenotype” of enhanced random mutation that accelerates the accumulation of further spontaneous or chemical-induced DNA damage that may be required for the development of cancer. This concept underlines the potential importance of DNA-damaging chemicals not only at the initiation stage but also at later stages of the carcinogenic process. The carcinogenic process requires that cells acquire Hanahan and Weinberg’s (2000) “6 hallmarks” of cancer: self-sufficiency in growth signals; insensitivity to growth inhibitory signals; evasion of apoptosis; limitless replicative potential; sustained angiogenesis; and potential for metastatic/tissue invasion. Acquisition of each of these properties may be driven by both genetic and epigenetic changes, and they relate to 5 overlapping models of carcinogenesis: mutational; genomic instability; nongenotoxic clonal expansion; cell selection; and microenvironment. Key to the focus of the current chapter is the recognition that exogenous chemicals may contribute to the genetic, epigenetic, and microenvironment alterations that are required for the acquisition of these features, and thus for tumor growth to proceed.
4.2.3 Genotoxic Carcinogens, Metabolic Activation, and DNA-Damaging Species
Chemicals can contribute to the initiation, promotion, and progression stages of the carcinogenic process either through their ability to damage DNA and produce somatic mutations in cells, or as a consequence of their ability to establish a cellular microenvironment that provides initiated cells with a growth advantage via metabolic changes, local vascular alterations, and/or the ability to evade apoptosis, terminal differentiation and contact inhibition. The distinction between genotoxic carcinogens as tumor initiators and nongenotoxic carcinogens as tumor promoters is now considered overly simplistic as DNA damage in key genes may occur—and for most cancers is likely required—also at later stages of tumor development.
Genotoxic carcinogens have a wide diversity of chemical structures (see Fig. 4–1) but they share the property of either being directly electrophilic (electron-seeking) or being capable of conversion to electrophiles. These reactive electrophiles interact with nucleophilic (electron-rich) groups on intracellular molecules such as DNA and proteins, forming either covalent adducts or oxidative damage. These types of damage to DNA, if not repaired before the next cycle of DNA replication, may lead to errors in replication and hence to fixation of the damage as nucleotide substitutions. If the reactive electrophile damages key cellular proteins the result may be cytotoxicity and necrotic cell death, which can eliminate cells that also have damaged DNA, but which may also trigger the development of an inflammatory microenvironment that promotes the proliferation of any surviving initiated cells (Sec. 4.2.7).
Some genotoxic carcinogens, including carbamic acids, nitrosamides, epoxides, lactones, imines, and mustards (see Fig. 4–1), are “direct-acting” because they are either already electrophilic or are spontaneously hydrolyzed into electro-philes. However, the majority of genotoxic carcinogens require enzymatic bioactivation to electrophilic or electrophile-generating metabolites in order to damage DNA. These reactions are catalyzed largely by drug-metabolizing enzymes whose normal physiological role is protective, converting lipophilic chemicals into water-soluble metabolites that can be more readily eliminated from the body via the urine or bile. Drug-metabolizing enzymes have evolved both multiplicity and catalytic promiscuity to ensure that most potentially harmful environmental chemicals will undergo biotransformation, inactivation, and elimination. Chemicals that are carcinogenic, however, are those whose structures lead to their inadvertent biotransformation into reactive electrophiles with the potential to damage DNA. Because biotransformation can produce many metabolites from a single chemical via multiple cooperating and competing pathways, the net effect of exposure to a carcinogen in a particular individual will depend on the balance of activating versus detoxifying pathways, which may, in turn, be influenced by both genetic variation and by environmental factors, such as chemically-mediated enzyme induction or inhibition.
Among the many classes of drug-metabolizing enzymes that have been implicated in metabolic activation of carcinogens, members of the cytochrome P450 (CYP) mixed-function monooxygenase superfamily have been studied most intensively. These Phase I enzymes catalyze the hydroxylation of carbon, nitrogen and sulfur atoms on chemical molecules to produce metabolites that are more polar and either stable and excreted, reactive (ie, epoxides, nitroso compounds), or possess structures that make them suitable substrates for further metabolism by Phase II conjugating enzymes (see below). Oxidative procarcinogen bioactivation may also be catalyzed by other non-CYP Phase I enzymes such as nicotinamide adenine dinucleotide phosphate (NAD(P)H) quinone oxidoreductase, aldo-keto reductase, and various peroxidases (Shimada, 2006).
Phase II conjugating enzymes such as the uridine diphosphate (UDP)-glucuronosyltransferases (UGTs), sulfotransferases (SULTs), arylamine N-acetyltransferases (NATs), and glutathione S-transferases (GSTs) can also produce either stable conjugated metabolites or unstable conjugates that spontaneously decompose to reactive electrophiles. The likelihood of either protection against, or enhancement of DNA-damaging potential by the activity of Phase I and Phase II enzymes depends on a combination of the structure of the particular chemical in question, the chemical reactivity of the metabolites produced, and the relative levels of expression of the various enzymes that may compete or collaborate in the activation and detoxification processes in a given tissue. For example, potential carcinogen-bioactivating oxidases such as CYP1A2 show liver-selective expression, while others, such as CYP1A1 and cyclooxygenase, are present at high levels in extrahepatic target tissues, such as the lung and bladder, respectively.
Three of the most extensively studied classes of chemical carcinogens that require metabolic activation are the PAHs, such as benzo[a]pyrene (B[a]P; Fig. 4–3), the aromatic amines, such as β-naphthylamine (βNF; Fig. 4–4), and the nitrosamines, such as diethylnitrosamine (DEN; Fig. 4–5). PAHs, aromatic amines, and nitrosamines together comprise a substantial fraction of the total number of chemicals that are either known or reasonably anticipated to be carcinogenic in humans according to the U.S. National Toxicology Program’s (NTP) 12th Report on Carcinogens (2011). Although many of the chemicals on the NTP list are reagents or by-products of industrial processes, others are present in foodstuffs or in the natural environment. Notable examples of these include aflatoxin B1, a potent liver carcinogen produced by Aspergillus fungi that contaminate improperly stored grains and nuts, and heterocyclic amines such as 2-amino-3-methylimidazo[4,5-f] quinoline (IQ), that form from the reaction of amino acids with creatine during high-temperature cooking of meat (Fig. 4–1). The single-step metabolic activation pathway of aflatoxin B1 is shown in Figure 4–6.
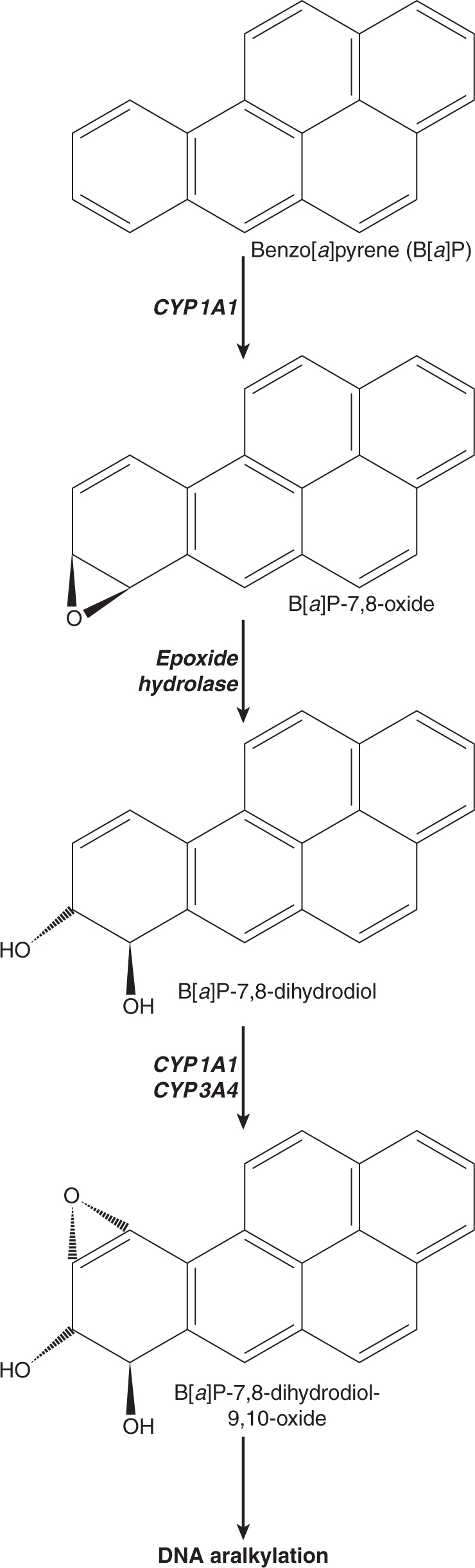
FIGURE 4–3 A proposed metabolic activation pathway for the polycyclic aromatic hydrocarbon Benzo[a]pyrene(B[a]P).
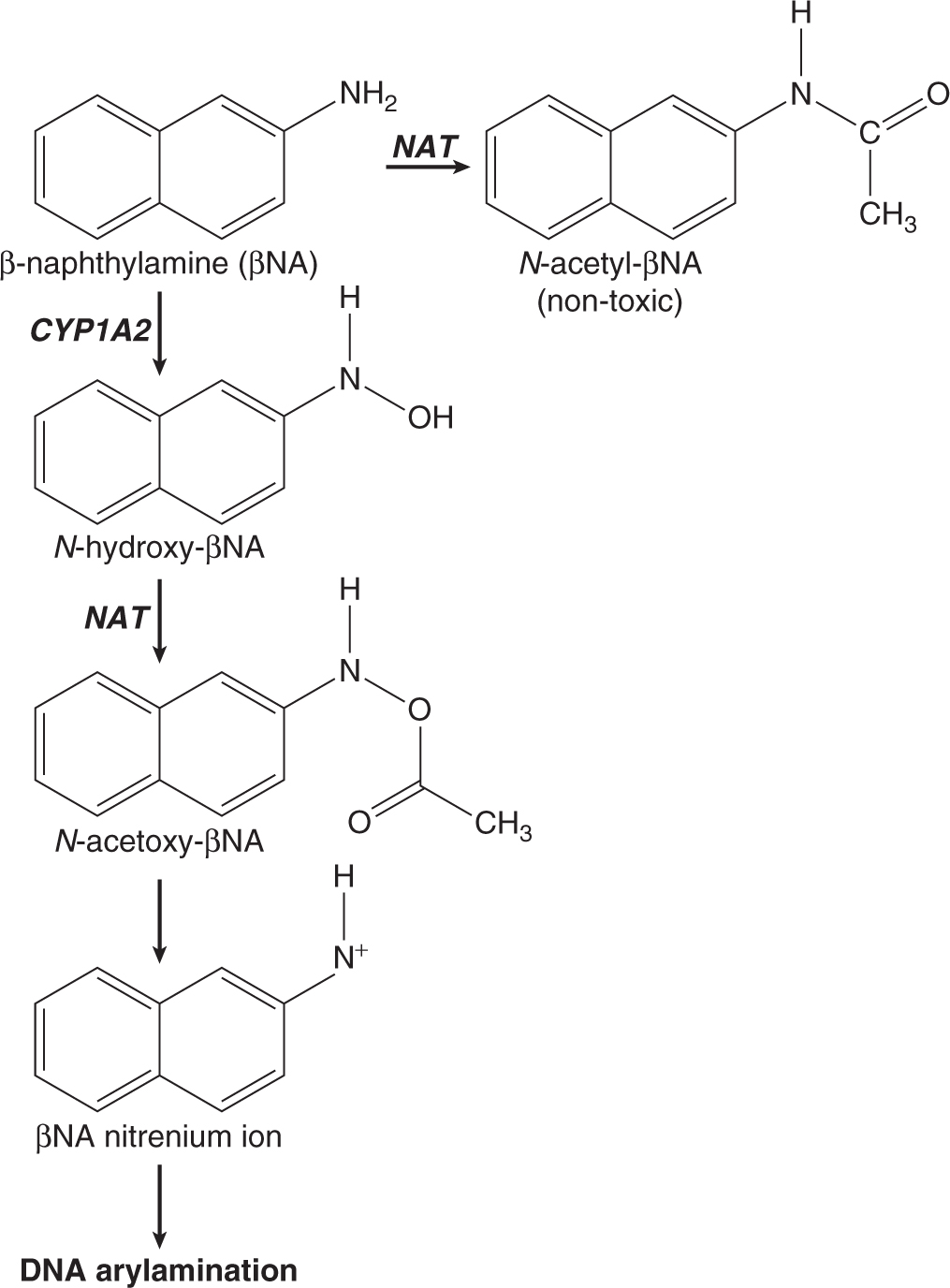
FIGURE 4–4 Proposed metabolic activation and detoxification pathways for the aromatic amine β-naphthylamine.
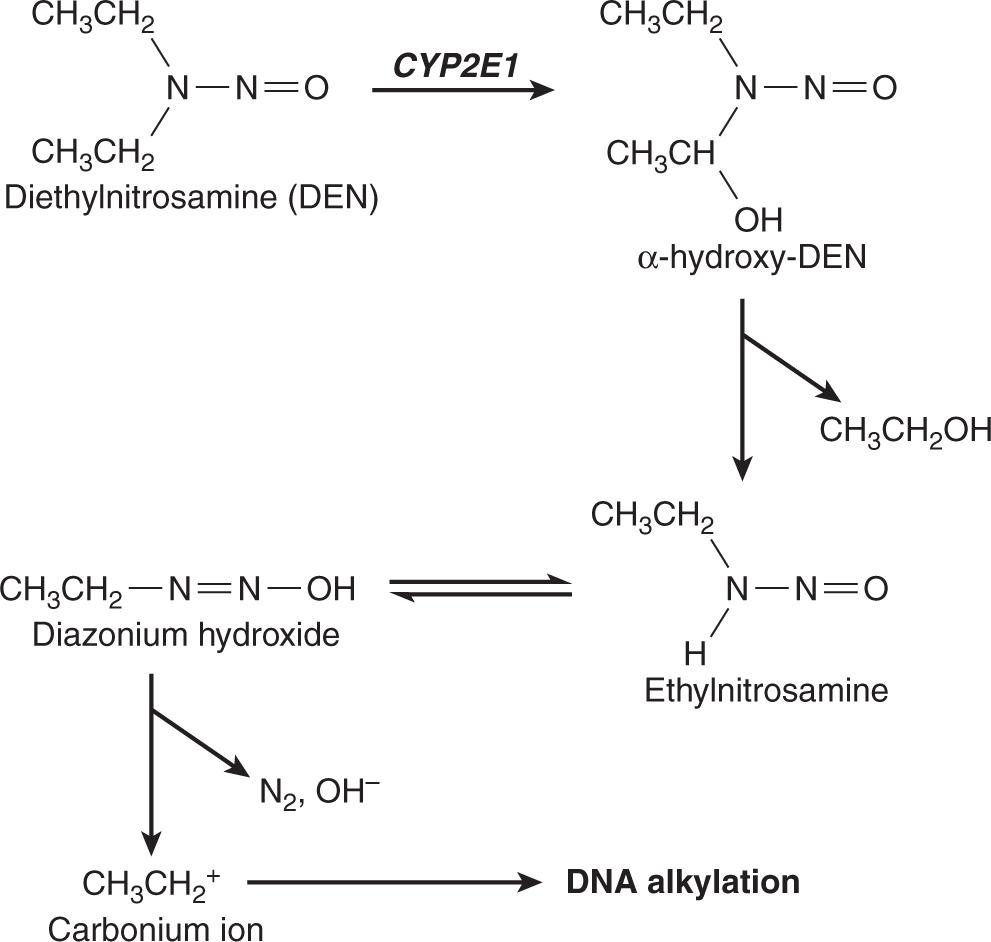
FIGURE 4–5 A proposed metabolic activation pathway for diethylnitrosamine.
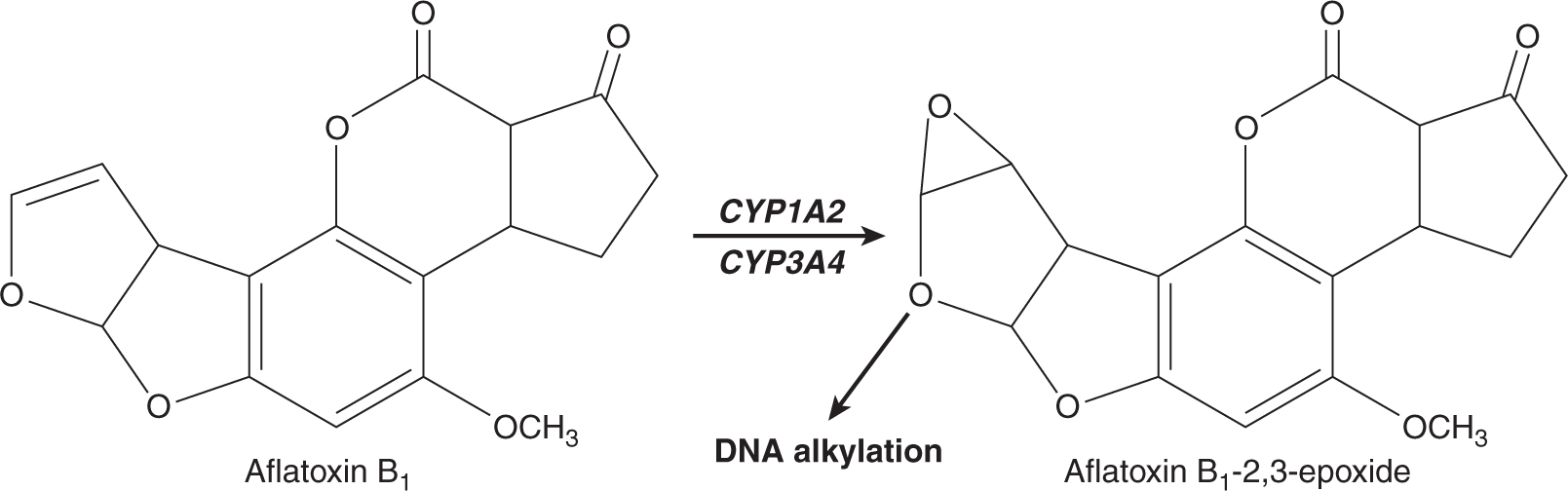
FIGURE 4–6 A proposed metabolic activation pathway for aflatoxin B1.
The pathways shown in Figures 4–3 to 4–6 illustrate the following principles of metabolic activation: (a) a central role is played by oxidative metabolism—often mediated by one or more CYP isozymes whose identities depend on the structure of the chemical—in initiating the activation process; (b) multiple enzymes may participate in producing the ultimate carcinogenic metabolite, either by acting sequentially on the chemical or by catalyzing the same reaction; and (c) the nonenzymatic, spontaneous chemical decomposition of unstable metabolites may contribute to activation. The pathways shown in Figures 4–3 to 4–6 represent only a subset of those that contribute to metabolic activation, and do not include many of the competing enzyme pathways that can produce stable, excretable metabolites and are thus protective. For example, although the initial step of βNA activation likely requires N-oxidation of its primary amino functional group into a hydroxylamine (Fig. 4–4), a competing reaction at the amino group is N-acetylation by NATs, which produces a stable and noncarcinogenic N-acetamide. This example also illustrates the potential duality of enzyme effects: the same NAT enzymes that may be protective by catalyzing N-acetylation on the parent molecule can participate in metabolic activation by catalyzing the O-acetylation of the hydroxylamine metabolite to the unstable acetoxy ester. Furthermore, O-conjugation of the hydroxylamine by either sulfotransferases or UDP-glucuronosyltransferases can also produce unstable oxy esters that ultimately generate the same nitrenium ion DNA-binding species.
The structures of the ultimate reactive electrophilic species that are produced by metabolic activation of carcinogens may vary widely. DNA damage may arise from the covalent interaction of its nucleotide bases with at least 11 different types of carbon, nitrogen and sulfur electrophiles on carcinogen molecules (Klaunig and Kamendulis, 2008). Of these, key examples are epoxides produced from the PAHs (see Fig. 4–3) and aflatoxin B1 (see Fig. 4–6), nitrenium ions derived from aromatic amines (see Fig. 4–4), and carbonium ions derived from nitrosamines (see Fig. 4–5).
4.2.4 Nature and Consequences of DNA Damage
Different types of DNA-damaging chemicals tend to produce distinctive patterns of damage on the individual bases of DNA. In general, damage can consist of either a carcinogen adduct covalently bound to DNA, or of oxidative DNA damage. The site and type of adduct depends on the strength (charge) of the electrophile, the availability of nucleophilic sites (the unpaired O: and N: atoms) on DNA bases or the phosphodiester backbone, and the structure of the DNA relative to the size of the adduct. Strong electrophiles are capable of binding to a wide range of nucleophilic targets, whereas weaker electrophiles are only capable of binding to strong nucleophiles. Thus distinct chemical-selective patterns of nucleotide damage and the resulting mutations (see below) may be observed. However, it is likely that the persistence of particular adducts in particular genes is important in predicting carcinogenic risk, and the identification of these genes is important in monitoring DNA mutation profiles as biomarkers of either carcinogen exposure or cancer risk.
DNA may also be oxidized by hydroxylation of the nucleotide bases. Although there are a large number of possible oxidized forms of each of the 4 bases, 8-oxo-deoxyguanosine is one of the most prevalent, and it has been used extensively as a sensitive marker of overall oxidative DNA damage. Oxidative DNA damage can result from increased intracellular levels of reactive oxygen species, including hydrogen peroxide, hydroxyl radical, hydroperoxyl radical, and superoxide anions. These can be produced both by exogenous chemicals, often as a byproduct of CYP metabolism, and by endogenous processes. The latter include oxidative phosphorylation and inflammatory cell activation (Sec. 4.2.7).
Once DNA damage has occurred there are 2 possible cellular outcomes. Most probable is repair of the damage by DNA repair enzymes present in the cell, which have evolved to recognize a variety of types of DNA damage and replace damaged bases with intact ones (see Chap. 5, Sec. 5.2). If DNA repair has not taken place before the next cycle of DNA replication prior to cell division, 3 types of mutational events may take place: (a) error-prone replication resulting in a nucleotide substitution, whereby DNA polymerase incorporates the wrong complementary base (often adenine) in the nascent daughter strand opposite an adducted, apurinic, or apyrimidinic site; (b) frame-shift mutations (most commonly single-base deletions) that tend to occur when a carcinogen adduct is bound to a nucleotide base; and (c) DNA strand breaks resulting from either incomplete excision repair or alkylation and cleavage of the phosphodiester backbone.
Many of the relevant gene targets for mutagenesis by carcinogens include those classified as protooncogenes and tumor-suppressor genes, and mutations at specific sites in these genes have been detected in tumors. In general, when protooncogenes are activated by mutational events, signals for cell growth are increased, whereas for tumor-suppressor genes, which downregulate cell growth, loss of function abolishes this negative regulation (see Chap. 7). For example, in chemically induced rodent tumors, mutations commonly activate the ras family of oncogenes. Rat mammary tumors induced by exposure to nitrosomethylurea contain H-ras genes that have been activated by a single point mutation in codon 12 of the gene, while in mouse skin papillomas induced by 7,12-dimethylbenz[a]anthracene an activating mutation occurs in codon 61 of the same gene. The reasons for such DNA site selectivity are unclear, but they may include a combination of localized DNA accessibility in the context of chromatin packing, the nucleophilicity of particular bases within the exposed DNA region, the structure of the bioactivated chemical relative to the topography of the exposed DNA region, and the catalytic efficiencies (both substrate affinity and turnover rates) of DNA repair enzymes expressed within a given target tissue.
It has been estimated that mutations in the tumor-suppressor gene p53 are present in more than 50% of human tumors. The sites of mutation are not random but occur at discrete hotspots (see Chap. 7, Sec. 7.6.1). Aside from the possible physical and biochemical mechanisms that may explain the occurrence of mutational hotspots, it is likely that selective growth and retention of function-altering mutations also occurs during tumor promotion and progression. Thus different carcinogens may leave distinct mutational signatures in p53 and other genes. Lung tumors that develop in nonsmokers contain a different spectrum of p53 mutations than those in smokers, while tumors in ex-smokers retain the smokers’ pattern, indicating the persistence of molecular lesions that underlie the eventual manifestations of cancer (Hainaut and Pfeifer, 2001). Also, more than 50% of the liver tumors from aflatoxin B1-exposed populations in Africa and China have a G-to-T transition at codon 249 of the p53 gene, which is not present in tumors from patients with low aflatoxin B1 exposure (Shen and Ong, 1996). This mutation produces an amino acid change from Arg to Ser that alters the binding properties of the p53 protein to a hepatitis B viral antigen and confers a subtle growth advantage to initiated cells. Thus the codon 249 mutation in p53 is considered a molecular signature linking aflatoxin B1 exposure to the eventual development of hepato-cellular carcinoma by providing this selective growth advantage to cells rather than by an inherently greater frequency of its occurrence in DNA.
4.2.5 Exogenous Versus Endogenous Chemical Carcinogens
It is important to place the DNA damage produced by foreign electrophiles in the context of substantial damage that occurs within cells in the absence of exogenous chemical exposure. It has been estimated that more than 10,000 DNA-damaging events occur in every cell each day. This damage is produced not only by ambient ionizing radiation but also by reactive oxygen and nitrogen species and products of lipid peroxidation that are generated in the course of normal endogenous oxidative metabolism. Much of this high “background” damage may also be caused by continual exposure to low levels of the hundreds of natural and synthetic toxic chemicals and food constituents that enter the human body. The massive scale of this damage emphasizes the important role of cellular DNA repair pathways that ensure high efficiency, redundancy, and fidelity of repair for a broad range of DNA damage (see Chap. 5, Sec. 5.2). DNA damage that is detectable as a result of exposure to exogenous chemicals must be of sufficient magnitude to produce a signal above this high level of endogenous damage and very efficient repair. As discussed further in Section 4.3.2, this has important implications for the interpretation of dose–response relationships and thresholds for chemical exposure, as many tests for carcinogen potency make the assumption that cancer risk at low doses may be predicted by a linear extrapolation from experimental administration of high doses. Also, it is known that DNA damage may be necessary but not sufficient for tumor formation, and that additional tumor-promoting effects of chemicals on the cellular homeostasis and microenvironment of DNA-damaged cells contribute strongly to tumor formation (Secs. 4.2.6 and 4.2.7).
4.2.6 Chemicals as Modifiers of Cell Proliferation, Senescence and Death
Relative to normal cells, cancerous cells have impaired ability to control cell division, to age, and to undergo apoptotic (programmed) cell death. Thus any chemical agent that triggers or contributes to the impairment of these processes could promote progression to malignancy, tumor growth and metastasis, especially with chronic exposure. The nongenotoxic tumor promoters shown in Figure 4–2 are thought to function in this manner. TPA acts as a proinflammatory agent and inducer of oxidative stress to provide a microenvironment favoring the proliferation of initiated cells. TCDD appears to function by inhibiting the apoptosis of initiated cells (Schrenk et al., 2004). Phenobarbital influences both cellular proliferation and apoptosis by altering patterns of DNA methylation, thus modifying epigenetic control of gene expression (see Chap. 2, Sec. 2.3) in cancer cells (Phillips and Goodman, 2009). Chemicals may also promote the growth of initiated cells indirectly; for example, by producing acute cytotoxicity with resultant necrotic cell death in nearby cells that triggers the establishment of a chronic tumor-promoting inflammatory environment (Sec. 4.2.7).
4.2.7 Chemicals as Modulators of Inflammation
Epidemiological and experimental evidence supports the concept that inflammatory cells and the innate immune response (see Chap. 21, Sec. 21.2) may play pivotal roles in carcinogenesis by facilitating both tumor-initiating and tumor-promoting events. Some of the most consistently observed associations between chronic inflammation and risk of human cancer include those between colitis and colon cancer, gastric acid reflux and esophageal cancer, hepatitis B or C infection and liver cancer, papillomavirus infection and cervical and head and neck cancer, and schistosomiasis infection and urinary bladder cancer. Recognition of the key importance of inflammation in cancer is reflected in a recent suggestion by Hanahan and Weinberg that the 6 hallmarks of cancer described in Section 4.2.2 may now be supplemented with 2 emerging hallmarks—modified energy metabolism and immune escape—and 2 enabling mechanisms—genomic instability and inflammation (Hanahan and Weinberg 2011).
Acute inflammatory responses may be elicited by infections, metabolic stresses, generation of reactive oxygen species, hypoxia or tissue injury, which can act by producing necrotic or autophagic cell death as opposed to the generally noninflammatory apoptotic cell death. With the exception of infection, exogenous chemicals may trigger any of the events listed above. Contents released from dead cells, such as the chromatin-associated protein high mobility group box 1 (HMGB1) (Campana et al, 2008), can trigger the activation of nearby resident macrophages to release proinflammatory cytokines such as interleukin-6 (IL-6) and tumor necrosis factor alpha (TNF-α), as well as oxidant-generating enzymes such as nicotinamide adenine dinucleotide phosphate (NADPH) oxidase in a transient “respiratory burst” that produces high levels of free radicals and other reactive oxygen and nitrogen species. Although designed to quickly kill invading pathogens, the respiratory burst can damage the DNA, RNA, proteins and lipids of neighboring cells (Ohshima et al, 2003), and activate signal transduction pathways involving pro-inflammatory transcription factors such as nuclear factor-κB (NF-kB), which drives the expression of genes whose products can result in chronic inflammatory conditions (Karin and Greten, 2005).
Some chemicals may increase cancer risk by more than one mechanism, whereas others may play chemopreventive roles. For example, DEN (see Figs. 4–1 and 4–5) is a potent liver carcinogen that produces not only liver DNA damage in mice, but also an acute hepatic necrosis associated with increased levels of reactive oxygen species (ROS) and release of intracellular damage-associated molecular pattern molecules (DAMPs); the net effect is an elevation in proinflammatory IL-6 and TNF-α levels, subsequent activation of NF-κB, and a compensatory cellular proliferation that promotes tumor growth. This suggests that DEN can act as a “complete carcinogen” because it serves both tumor-initiating and tumor-promoting functions (Maeda et al, 2005; Naugler et al, 2007). In contrast, administration of the antioxidant butylated hydroxyanisole to DEN-treated mice reduces tumorigenicity by preventing the accumulation of ROS that can both damage DNA and activate jun kinase-mediated cellular proliferation pathways (see Chap. 12, Sec. 12.3.4). These examples illustrate that some chemicals may result in the occurrence of tumors by multiple mechanisms, whereas others may be chemoprotective, as discussed in the next section.
4.2.8 Cancer Chemoprevention
The largest cancer chemoprevention trials to date have employed hormonal agents against breast cancer, such as tamoxifen and raloxifene, as discussed in further detail in Chapter 20, Section 20.4.1. There is also great interest in blocking carcinogenesis by other pharmacological means, especially in high-risk groups. Because the process of carcinogenesis is complex and multistage, chemopreventive agents could act by many different mechanisms. However, any chemical worthy of consideration to prevent cancer must pose a very low health risk itself, because it would require long-term administration. Candidates for chemopreventive therapy fall into 2 main categories: (a) the general population; and (b) those who may be at elevated risk because of genetic predisposition or heightened levels of carcinogen exposure. Epidemiological studies show consistently that diets rich in fruits and vegetables, which contain high levels of antioxidants, reduce risk for cancers at many sites. Also, increasing evidence suggests that obesity resembles a chronic inflammatory state, which is known to be tumor-promoting (Longo and Fontana, 2010). Thus for the general population, the most logical chemoprevention strategy is to encourage a healthy lifestyle that includes exercise and caloric moderation to maintain a healthy body weight, and the intake of diets that are rich in fruits and vegetables. More specific chemical interventions are being investigated for individuals at higher risk, and these may be aimed either at preventing DNA damage or reducing the likelihood that DNA-damaged cells will proliferate to form a malignancy.
As described in Section 4.2.3, many procarcinogens are activated to their DNA-binding metabolites by isozymes of the CYP superfamily. Chemicals that inhibit particular CYPs and/or induce the Phase II conjugating enzymes that facilitate excretion of CYP-produced oxidized metabolites could reduce metabolic activation of carcinogens. There is support for this concept from the dietary associations between intake of fruits and vegetables containing enzyme inducers and cancer risk: in experimental animals certain chemicals found in cruciferous vegetables can reduce the metabolism and covalent binding of carcinogens to DNA and subsequent tumorigenesis (Srinivasan et al, 2008; Takemura et al, 2010).
Indiscriminate inhibition of CYP enzymes would not be a safe chemopreventive strategy because CYP enzymes constitute the major pathway for elimination of many potentially harmful chemicals, as well as therapeutically administered drugs. Moreover, the catalytic promiscuity of CYPs is such that even isoform-selective inhibition is likely to alter the disposition of many chemicals entering the body, including therapeutically useful drugs whose pharmacokinetics would be modified. Furthermore, a particular CYP isoform may play dual, competing roles in both carcinogen activation and carcinogen elimination that may not be apparent from in vitro investigations. For example, experimental evidence implicates the CYP isoform CYP1A2 as a key first step in the bioactivation of aromatic amines into DNA-binding electrophiles (see Fig. 4–4). This evidence includes cellular and molecular studies of CYP1A2-dependent production of reactive metabolites from aromatic amines such as the cigarette smoke component 4-aminobiphenyl, covalent DNA binding of these metabolites, production of DNA mutations, and transformation of cultured cells. However, in mice the absence of CYP1A2 (achieved by gene knockout) paradoxically does not protect against either DNA damage or the formation of liver tumors following 4-aminobiphenyl exposure. It is presumed that the protective effect resulting from CYP1A2’s contribution to the efficient in vivo elimination of the chemical, hence reducing the overall exposure burden, outweighs its ability to produce DNA-damaging metabolites (Nebert et al, 2004).
A strategy of inducing expression of CYP enzymes so as to increase carcinogen elimination has similar challenges. High levels of Phase II drug-conjugating enzymes were thought to protect from chemical carcinogenesis. However, inhibition of a given Phase II enzyme may either be protective by preventing formation of an unstable metabolite (eg, acetylating, glucuronidating, and sulfonating enzymes can all produce very unstable oxyester metabolites of aromatic amines), or may be risk-enhancing if the enzyme’s predominant function in vivo is to produce a stable and readily excretable metabolite.
Whether either Phase I or Phase II drug-metabolizing enzyme induction or inhibition is beneficial or harmful depends upon which chemical agent is the main threat. Unfortunately, exposure often occurs to mixtures of structurally unrelated chemicals (cigarette smoke contains several different PAHs, aromatic amines, and nitrosamines), and manipulations that protect from one class of carcinogen might increase the risk from another chemical class, or even between individual chemicals of the same class.
Chemicals that reduce either the levels of ROS, inflammatory mediators, or inflammation-inducing pathogens have the potential to protect against the development of tumors. For example, research is focusing on the identification of effective antioxidants, such as flavonoids, polyphenols, isothiocyanates and phytoalexins found in foods, that may reduce cellular oxidant burden and thus the levels of oxidative DNA and protein damage. Agents that inhibit inflammation, such as nonsteroidal antiinflammatory drugs that inhibit prostaglandin synthesis by blocking cyclooxygenase (COX) enzymes, have also been studied as potential cancer chemo-preventive agents (Das et al, 2007; Lee et al, 2008). Although some randomized trials of selective COX-2 inhibitors have shown encouraging results, the possible cardiovascular side effects associated with large-scale use of these compounds limits their utility. Reducing the incidence of infection as a result of pathogens that cause chronic inflammatory conditions via immunization, such as papillomavirus vaccines for cervical cancer and hepatitis B/C virus vaccines for liver cancer, is effective in reducing cancer risk.
The relationships between exogenous chemicals and dietary/lifestyle factors to cancer risk and prevention are complex. For example, tumor production in rodents by potent genotoxic carcinogens such as aflatoxin B1 may be drastically reduced, even after initiation has occurred, by dietary manipulations such as reducing the percentage or even the source (ie, animal vs. plant) of dietary protein. It is likely that lifestyle-based cancer prevention—comprised of a combination of exposure avoidance and dietary measures—promises greater potential for overall population impact than targeted, chemical-based chemoprevention.
4.3 ASSESSING RISK FROM CHEMICAL CARCINOGENS
Establishing that a chemical is a human carcinogen is a challenging and protracted process. Humans are exposed to chemicals in foods, medicines, and the environment, and there is a long latent period between exposure and tumor appearance. The most productive approach has involved astute clinical observation followed by carefully designed epidemiological and laboratory studies in vitro and in vivo. Reduction of risk first requires that the factors contributing to risk be identified. These factors may be external or they may be intrinsic to the population at risk. A systematic, stepwise approach is employed by regulatory agencies such as the Environmental Protection Agency (EPA) in the United States, which, in order to determine risk, attempts to integrate the multiple factors that interact in human carcinogenesis (Fig. 4–7). The assessment relies heavily on data from laboratory animals despite the numerous caveats that apply in any attempt to extrapolate data from animals to carcinogenesis in humans. The EPA’s 2005 Guidelines for Cancer Risk Assessment use the following set of criteria, based on an integrated assessment of all evidence, to categorize chemicals according to their risk: (a) carcinogenic to humans; (b) likely to be carcinogenic to humans; (c) suggestive evidence of carcinogenic potential; (d) inadequate information to assess carcinogenic potential; and (e) not likely to be carcinogenic in humans. A “mode of action framework” is applied to the analysis, which uses criteria that take into account the chemical’s hypothesized mode of action and biological plausibility; identification of key events; the strength, specificity, and consistency of epidemiological associations; dose–response concordance and temporal relationship; consideration of other modes of action; support for the proposed mode of action in laboratory animals; relevance of the mode of action to humans; and population subgroups that may be particularly susceptible. Table 4–1 lists some of the methods that are used to determine the carcinogenicity of chemicals, which are described in further detail below.
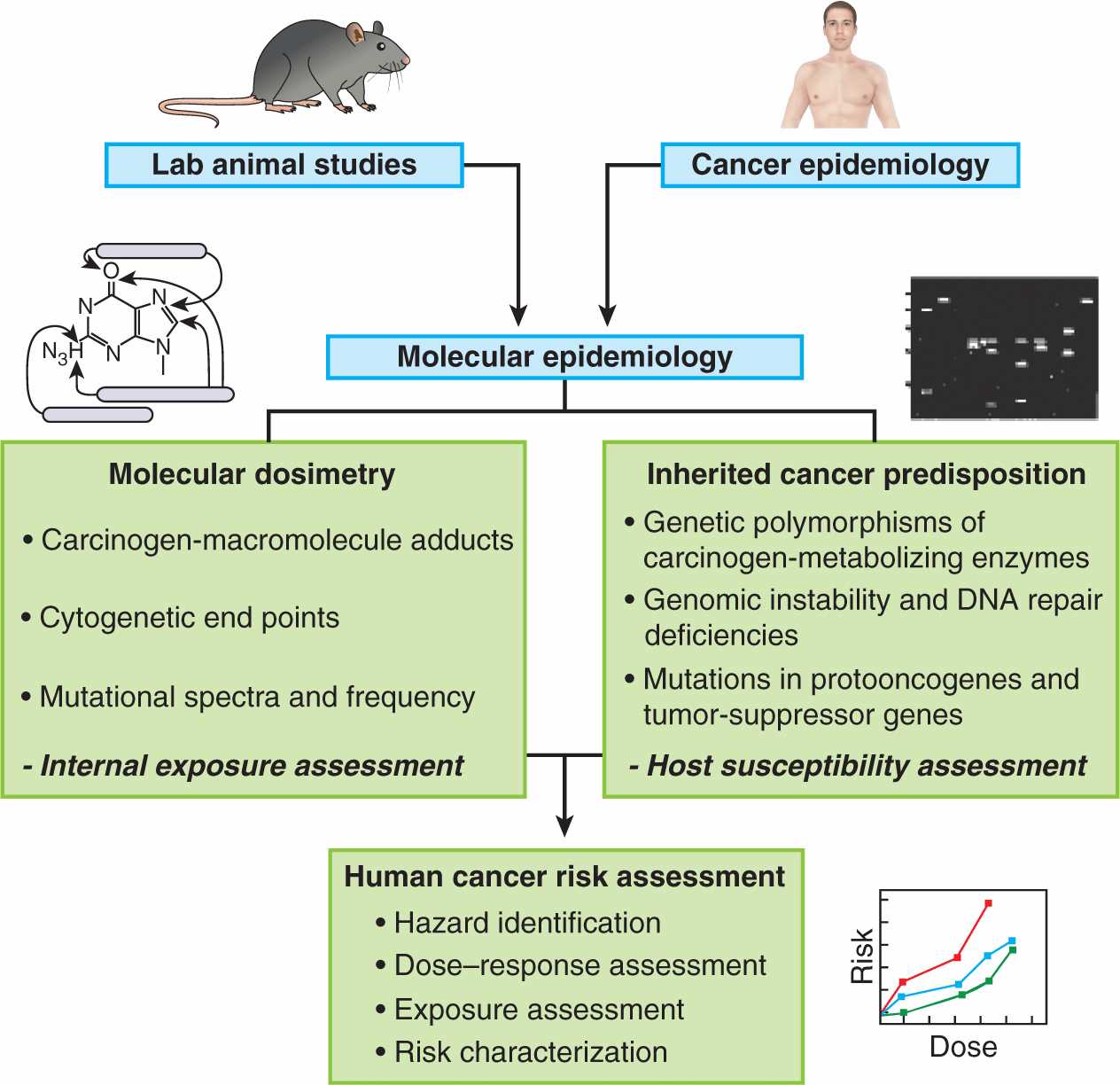
FIGURE 4–7 Strategy for using multiple approaches to assess chemical carcinogens for their risk of causing human cancer. (Adapted from Harris, 1993.)
TABLE 4–1 Assays for carcinogens.

4.3.1 Population Epidemiology of Chemical Carcinogens
Epidemiological observations provide the cornerstone for identifying cancer risk from chemicals (see Chap. 3, Sec. 3.5). Earliest observations were of associations between exposure to chemicals in industrial settings and the incidence of cancers at a variety of tissue sites. Some of these earlier observations led to drastic improvements in hygiene practices in the industrial workplace in the 20th century. The early associations were often striking because the exposure levels to particular chemicals were so high. Fortunately, such exposures and cancer clusters occur very rarely today, and it is much more challenging to establish unequivocally a causal association between cancer risk and exposure to chemicals at the low levels that occur in the environment or the cleaner workplace. Nonetheless, when evidence for a chemical–cancer association arises from epidemiological observations, it should prompt the conduct of dose–response and mechanistic studies using the experimental methods described below. These studies are then used to inform regulatory action designed to reduce exposure and risk where appropriate.
Even when epidemiological studies suggest that a particular chemical is carcinogenic, and it is shown to be DNA-damaging or mutagenic in laboratory animals or in vitro tests, several features of chemical carcinogens make it a challenge to establish unequivocally that a chemical does or does not cause cancer in humans. These are outlined in Table 4–2.
TABLE 4–2 Features of chemical carcinogens that cause difficulty in deciding that a chemical does or does not cause cancer in humans.

4.3.2 Animal Bioassays
Studies of carcinogenicity in rodents involve either the chronic repeated administration of high doses of a potential carcinogen, or the administration of the chemical at a particularly sensitive developmental stage. More recent developments include the use of genetically modified strains of mice that have been made more susceptible to carcinogens. Testing of putative carcinogens in animals is time-consuming, expensive, and requires relatively large numbers of animals to obtain valid results. Thus it is generally used to confirm results observed from short-term tests in bacteria or cell culture.
Studies in rodents are often criticized because they employ doses of suspected carcinogens that are far in excess of the probable human exposure. At high doses, acute cytotoxicity of the chemical (which would not be observed at lower doses) can cause necrosis, inflammation, and compensatory proliferation that contribute to tumor growth, and this might not occur at lower exposure levels. However, most chemicals that induce a high frequency of tumors at high doses will also induce some tumors at lower doses in studies using large numbers of animals. High doses are used for the practical reason of reducing the number of animals required, but such designs require the assumption that the dose–response relationship allows for valid extrapolation of risk from high doses to low doses. Most animal models can detect tumor incidence of approximately 5% but not as low as 1%, while in humans an increase in tumor incidence of 1% would be unacceptable.
The incidence of cancer in both rodents and humans caused by chemical agents generally increases with dose, but carcinogenic potency can differ between closely related compounds. Based on results from rodent studies, some generalizations concerning the quantitative relationship between exposure and response to carcinogens can be made:
1. A single exposure to some chemical carcinogens may be sufficient to induce tumors in a high proportion of animals. For example, a single dose of a polycyclic aromatic hydrocarbon can induce mammary carcinomas in more than 90% of female rats if the compound is administered as they are approaching sexual maturity. Similarly, 2 doses of DEN or 4-aminobiphenyl can lead to a high frequency of liver tumors in mice if the doses are administered on days 8 and 15 after birth (the so-called neonatal carcinogenicity dosing protocol), when the liver is still actively proliferating.
2. Tumor production is often enhanced and the latency period reduced if chemical exposure is repeated. Mechanistically, repeated exposures may either enhance the likelihood that a required set of mutations in key genes has occurred, or it may produce prolonged changes in the microenvironment that promote the growth of initiated cells.
3. Tumor susceptibility varies widely among animal species and even between strains of the same species after exposure to the same dose of the same chemical. For example, rats are much more susceptible to aflatoxin B1-induced liver tumors than mice, and hybrid offspring of inbred strains of mice (eg, the B6C3F1 offspring of a cross between C57BL/6 and C3H mice) are more susceptible to tumors than their inbred parents after exposure to many different chemicals.
4. Dose–response curves for carcinogenicity may vary in different tissues even within the same animals. For example, Figure 4–8 shows the results of a dose–response study of the aromatic amine-derived carcinogen 2-acetylaminofluorene. At lower doses liver tumors predominated, while at higher doses bladder tumors were more common, and the shapes of the dose–response curves in the 2 tissues differed markedly. The observed difference in tissue response is not related to differences in carcinogen pharmacokinetics, metabolism, or resultant DNA damage and mutation, but rather to increased rates of cell proliferation in bladder at higher doses of the chemical.
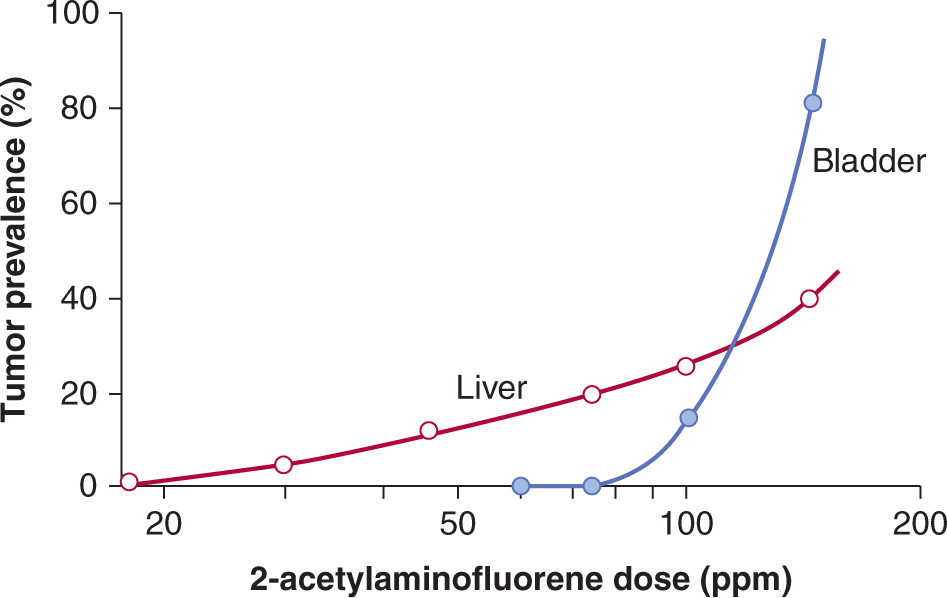
FIGURE 4–8 Dose–response curves for the production of liver and bladder tumors in female mice treated with 2-acetylaminofluorene. Tumors were observed after treatment for 18 to 33 months. PPM, Parts per million. (Data from Cohen and Ellwein, 1991.)
5. Not surprisingly, there is a very broad range of potencies among different chemical carcinogens. As shown in Figure 4–9, less than 1 μg/day of aflatoxin B1 is sufficient to produce tumors in 50% of rats after a lifetime of exposure, whereas compounds such as trichloroethylene or saccharin require more than 1 g/day to produce the same incidence of tumors. It is thus of key importance to relate tumor-producing doses of chemicals with the expected daily or lifetime exposures to the chemicals in humans.
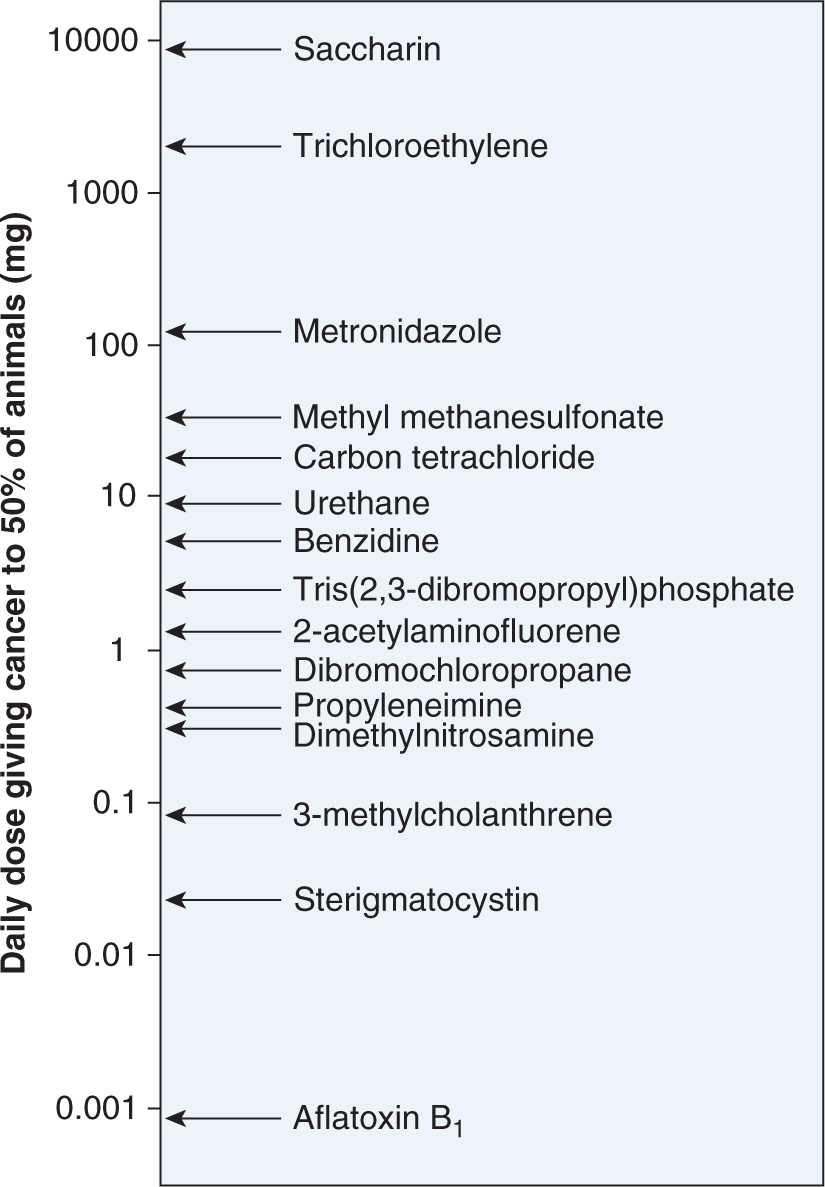
FIGURE 4–9 Range of carcinogenic potencies for various chemicals. (Data from Ames, as cited in Maugh, 1978.)
4.3.3 Short-Term Assays
The main advantages of using in vitro assays to predict the carcinogenic activity of chemicals lies in the drastically shortened time to obtain results and reduced cost compared to animal testing. Table 4–1 lists several commonly used classes of short-term assays.
The covalent binding of carcinogenic chemicals to DNA may be measured directly using a variety of techniques. The simplest involves the measurement of covalently bound radioactivity following the in vitro incubation of purified DNA with radiolabeled carcinogen in the presence of an appropriate source of metabolic activating enzymes, such as subcellular fractions of liver. Another method that is more physiologically relevant, involves exposure of cultured cells or living animals to a carcinogen, isolation of DNA, radioactive postlabeling of the adducted DNA species, and their separation by silica gel chromatography. An advantage of this method is that it allows for the identification and quantification of multiple specific adducts, but a major drawback of both of the above methods is the requirement for large quantities of potentially hazardous radioactivity. Technological advances now allow for DNA-carcinogen adducts to be detected and quantified, even in DNA isolated from humans exposed to carcinogens in the environment, using liquid chromatography coupled with mass spectrometry (see Chap. 2, Sec. 2.5). Finally, it is possible to raise antibodies that recognize particular DNA-adducted carcinogens, and these may be used in an immunoassay or with immunohistochemistry following exposure of animals or cultured cells to the carcinogen.
Other types of DNA damage may also be measured in short-term assays. Oxidative DNA damage following carcinogen exposure may be monitored by quantifying 8-oxodeoxyguanosine as a marker of oxidative damage using either liquid chromatography or an immunoassay. The comet assay provides a general measure of overall DNA damage in individual cells using single-cell agarose gel electrophoresis of cell nuclei to visualize fragmented DNA (see Chap. 5, Table 5.2). Indirect methods for monitoring levels of DNA damage include immunoblotting and immunohistochemical assays for the phosphorylated form of the histone protein H2AX (γ-H2AX; see Chap. 5, Sec. 5.6), which is produced in the chromatin microenvironment surrounding DNA doublestrand breaks, and an assay of unscheduled DNA synthesis, which is a marker for DNA repair occurring subsequent to carcinogen-induced DNA damage.
Various cytogenetic methods are also used for detecting gross chromosomal abnormalities caused by carcinogen exposure. These include breaks, terminal deletions, rearrangements and translocations; quantitation of damage-induced sister chromatid exchanges of differentially stained chromatids; and the frequency of micronuclei arising as a result of disruption of DNA distribution into nuclei during cell replication.
A potential drawback of many of the methods for detecting DNA damage is that they assume that DNA damage is correlated directly with DNA mutation—an assumption that does not take into account the importance of DNA repair. For this reason methods have evolved to measure the frequency of carcinogen-induced mutations in bacteria, mammalian cells, and whole animals. Perhaps the most widely used short-term mutagenesis assay is the Ames test (Fig. 4–10). It uses engineered strains of Salmonella typhimurium that have a mutation preventing them from synthesizing the amino acid histidine, thus they will grow only if histidine is added to the growth medium. Exposure of a culture of these bacteria to a mutagen can result in reversion of the mutation back to histidine-independent growth in a fraction of the cells, which can then grow on histidine-deficient agar plates. The number of revertant colonies that grow on these plates is a measure of the mutagenic potency of the test compound. Because most chemicals must be metabolically activated to be mutagenic or carcinogenic, these assays are usually conducted in the presence of a source of mammalian drug-metabolizing enzymes to generate the relevant reactive electrophile, or strains of Salmonella expressing particular recombinant human drug-metabolizing enzymes may be employed. An analogous method using mammalian splenic T cells monitors chemical-induced mutations in the endogenous hypoxanthine phosphoribosyltransferase (hprt) gene.
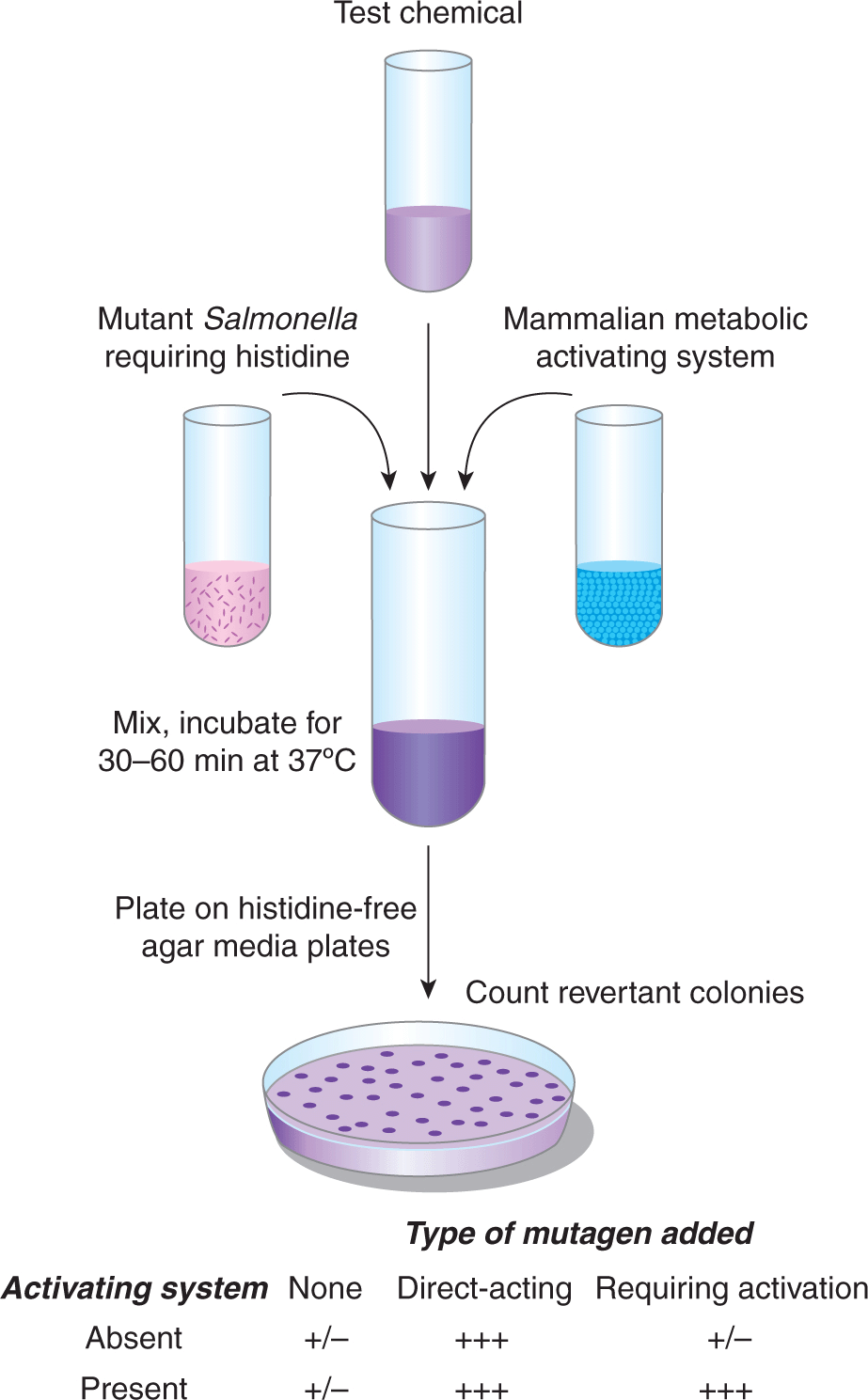
FIGURE 4–10 Detection of mutagenic chemicals in Salmonella typhimurium (Ames test). (Adapted from McCann, 1983.)
Transgenic animals carrying retrievable mutational target genes (eg, genes of viruses that infect bacteria—known as bacteriophages) have also been developed for in vivo testing of putative mutagens (Swiger et al, 1999). After in vivo exposure of the transgenic animal to a carcinogen and subsequent isolation of genomic DNA from a selected tissue, it is possible to retrieve the target transgene (such as the cII gene of a bacteriophage lambda) by in vitro packaging of the phage DNA contained in the mammalian host’s genome. Bacteria are then exposed to such bacteriophages under conditions that select for growth of only those bacteria infected by viruses harboring mutated target transgenes. Thus the mutation frequency in the carcinogen-exposed animal is proportional to the number of bacteriophage plaques observed on bacterial lawns growing on agar plates. Although such systems can provide valuable predictive information regarding mutation potential and tissue specificity, the expense of the assay is such that it is more commonly used following a primary screen in microorganisms.
The above assays that are used to detect potential carcinogens make the key assumption that rates of mutation are predictive of rates of tumor formation. Although most of the chemical agents that are carcinogenic in humans are also mutagenic in bacteria and cause cytogenetic changes in rodent bone marrow, a proportion of the chemicals that test positive in short-term assays or even in chronic rodent studies have not been shown to be carcinogenic in humans. Assessment of the carcinogenic potential of non-genotoxic carcinogens is even more challenging because there is no apparent common mechanism on which to base large-scale in vitro screening assays for chemicals that are not mutagenic.
4.3.4 Cellular Transformation Assays
Assays of malignant transformation in cell culture monitor the transformation of normal or immortalized cells into those that are capable of proliferating, independent of signals that would normally limit their growth, and that have tumor-forming potential. Although more time-consuming and expensive than the in vitro methods described above, these assays indicate whether cells exposed to a carcinogen exhibit an altered growth phenotype and can produce tumors when injected into recipient animals. Hence, cell transformation assays are probably the most relevant and predictive short-term tests for assessing risk of human cancer.
Conventional transformation assays may involve the plating of cells on soft agar plates and the counting of colonies that are able to grow after 3 to 4 weeks. Recent innovations allow assays to be performed in as little as 1 week with the use of microwell plates and intracellular dyes that permit automated colony counting using plate readers. One of the more widely used cell transformation assays is the Syrian hamster embryo (SHE) assay (Mauth et al, 2001), which counts the number of SHE cell colonies with an altered phenotype 7 to 8 days after carcinogen exposure based on distinctive morphological features such as cell piling, randomly oriented 3-dimensional growth, cell crisscrossing, and decreased cytoplasm/nucleus ratios. Advantages to this system are that SHE cells can be cryopreserved and that they maintain metabolic capabilities that eliminate the need for addition of an exogenous source of carcinogen-activating enzymes.
4.4 MOLECULAR EPIDEMIOLOGY OF CHEMICAL CARCINOGENESIS
Molecular epidemiology makes use of molecular markers that may relate quantitatively to either carcinogen exposure or to genetic markers of predisposition to cancer in human populations (see Fig. 4–7 and Chap. 3, Sec. 3.6). In estimating risk, molecular techniques allow for studies of human cancer incidence to be supplemented by surrogate markers and biological endpoints rather than only by cancer outcomes.
4.4.1 Biomarkers for Assessment of Carcinogen Exposure
The carcinogen dose received by individuals may be quantified by measuring biomarkers of exposure, such as carcinogen-DNA adduct levels or mutation frequencies in accessible cells. For example, Figure 4–11 illustrates a study showing that the level of B[a]P adducts in DNA samples from lungs of smokers was highly correlated with the activity of one of its key bioactivating enzymes, CYP1A1. CYP1A1 not only bioactivates B[a]P but its expression is also induced by it. In other studies induction of CYP1A1 has been correlated with the extent of exposure to cigarette smoke, which is a major source of B[a]P. Thus an increase in CYP1A1 activity may predict an increase in adduct formation and subsequent risk of lung cancer. Although this type of study is informative, it is limited by the requirement for lung biopsies to obtain tissue for analysis. In the Physicians Health Study, white blood cells were used as an easily accessible surrogate for lung tissue (Tang et al, 2001). It was found that current smokers who displayed elevated levels of carcinogen DNA adducts in their white blood cells were more likely to be diagnosed with lung cancer within 13 years than smokers who had lower adduct levels. Thus adduct levels in white blood cells of smokers appear to provide a predictive biomarker for risk of lung cancer.
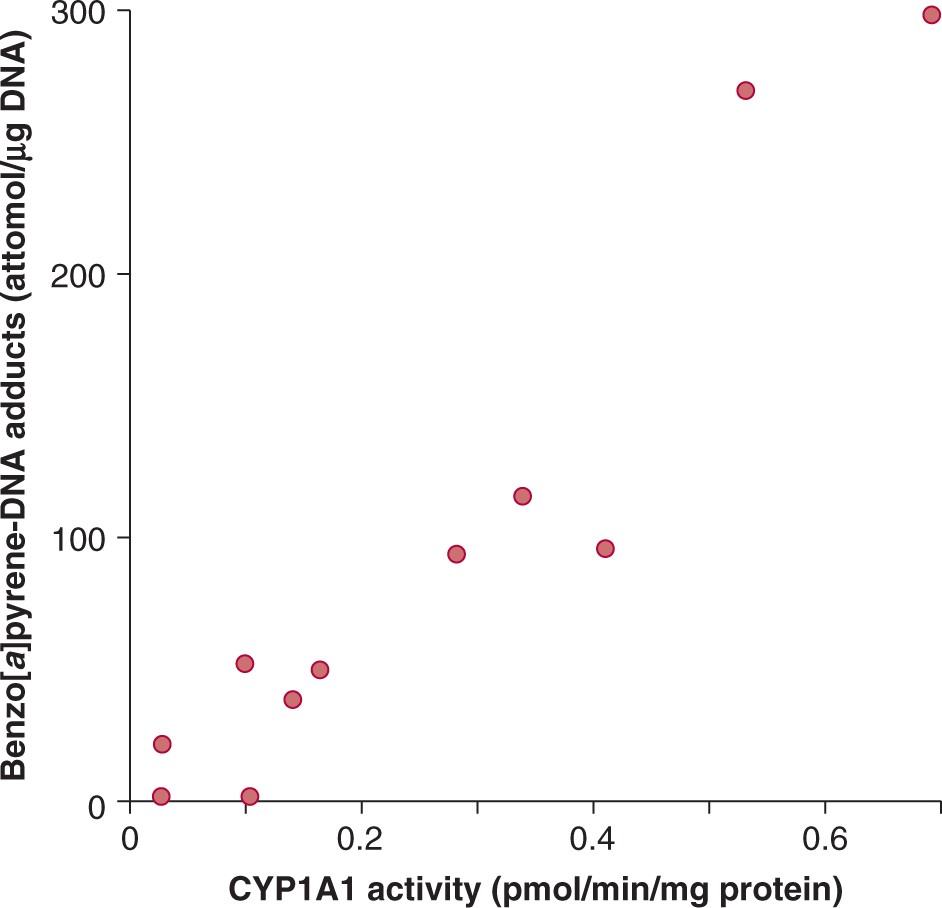
FIGURE 4–11 Correlation between cytochrome P450 isoform CYP1A1 activity and formation of benzo[a]pyrene DNA adducts in lung samples from human lung cancer patients. (Adapted from Alexandrov et al, 1992.)
4.4.2 Genetic Variation in Risk from Chemical Exposure
In populations where all members are exposed to the same carcinogens, some individuals develop cancer and others do not. For example, although cigarette smoking is a strong risk factor for lung cancer, some smokers develop lung cancer and a large number do not. One potential reason for this interindividual variation in risk for a given level of exposure is related to differences in the activities of the drug-metabolizing enzymes responsible for either detoxifying or activating pro-carcinogenic chemicals. Many epidemiological studies have provided evidence that genetic polymorphisms in drug-metabolizing enzymes may be associated (although usually weakly) with altered susceptibility to chemical carcinogens. For example, genetically based variations in the activity of arylamine N-acetyltransferase 2 (NAT2), which can play roles in either the detoxification or the metabolic activation of aromatic amines (see Fig. 4–4), are associated with altered risk for the occurrence of bladder cancer in populations that have been exposed to these agents (Hein et al, 2000). However, such associations are often inconsistent from study to study. For example, in one study individuals with an Ile462Val mutation in the PAH-metabolizing enzyme CYP1A1 were shown to have a 4.5-fold higher risk of lung cancer than those with the wild-type enzyme (San Jose et al, 2010), while other studies have shown no association between this variant and elevated risk of lung cancer. Such observations underline some of the challenges inherent in the conduct of epidemiological studies, particularly for genes related to risk of cancer that may be only weakly to moderately expressed.
In contrast, genetic variations in DNA repair enzymes are often associated with drastically increased predisposition to cancer. This is not surprising as cells repair a large amount of DNA damage on a daily basis. Thus any defect in the ability to repair DNA damage will result in the accumulation of mutations, and hence a greater probability that a cell will acquire mutations that drive its phenotype toward malignant growth. Examples of genetic defects in DNA repair that can significantly increase sensitivity to any DNA-damaging stimulus, including carcinogenic chemicals, include breast cancer (BRCA1/2), xeroderma pigmentosum (XP), Werner syndrome, ataxia-telangiectasis mutated (ATM), and Turcot syndrome (see also Chap. 5, Sec. 5.5). Each of these disorders has been studied extensively, and mutations in critical genes have been identified.
A major goal of molecular epidemiology is to identify individuals and populations who may have elevated cancer risk because of heritable predisposing factors so that preventive strategies can be implemented. In addition to mutations in DNA repair genes, mutation in other genes, such as the retinoblastoma gene, Rb, also confer a very high cancer risk in individuals who carry them (see Chap. 7, Sec. 7.6.4). However, such mutations are rare and thus do not constitute a major attributable risk to the population. A greater risk to the population may be caused by the common genetic traits that alter the balance of activation and detoxification of carcinogens. Particular combinations of polymorphisms can also act additively or synergistically to increase risk. For example, one study found no significant associations between risk for lung cancer and allelic variants in any of 10 postulated lung cancer susceptibility genes when considered individually, while a particular combination of 5 of these variants produced a highly significant 5.2-fold increase in overall risk, and an even higher 18-fold increased risk in females (Klinchid et al, 2009).
4.4.3 “Omics” Technologies and Molecular Signatures of Exposure and Risk
High throughput screening (see Chap. 2) can be used to study the structure and function of the genome (DNA sequences), the transcriptome (messenger ribonucleic acid [mRNA] expression levels), the proteome (proteins encoded by genes and transcripts), and the metabolome (small-molecule chemical fingerprints of intracellular processes) as they respond to toxic substances. The goal of such studies is to elucidate molecular mechanisms of toxicity and to derive molecular expression patterns that may better predict toxic events. Chemical carcinogenesis is particularly amenable to all of these approaches. Research has identified genomic signatures that associate with both risk for cancer (inherited alterations in DNA repair genes and carcinogen-metabolizing genes) as well as chemical-induced somatic mutations that are associated with tumor growth. Current research is studying the effects of exposures of experimental animals to genotoxic and nongenotoxic carcinogens on global gene expression profiles, protein expression patterns, epigenetic changes, and metabolic variables, and their consequences. For example, a recent study of the effect of chronic exposure of rats to the genotoxic carcinogen N-nitrosomorpholine demonstrated a concordance among altered gene, protein, and histopathological expression profiles for 8 candidate proteins in liver (Oberemm et al, 2009). Other proof-of-principle studies have compared the gene expression profiles of known genotoxic and nongenotoxic carcinogens to identify predictive multigene expression signatures for each class of chemical. Approaches to integrated systems biology that use computational modeling will be required to analyze the vast amount of data that can be generated by these techniques, and will ultimately contribute to enhancing our understanding of the complex impact of chemical carcinogens on the human genome and the cellular processes that it encodes.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


