Pharmacology of Anticancer Drugs
18.1 GENERAL PRINCIPLES OF PHARMACOLOGY
In this chapter, general principles of pharmacology relevant to anticancer drugs are presented. The specific properties of the most important anticancer drugs in clinical use are reviewed, with emphasis on their structure, mechanism of action, pharmacokinetics, and toxicity. In systemic cancer therapy, it is common to combine several drugs. Drugs in such combinations generally have different mechanisms of action, and different toxicity profiles, so that each drug can be administered at close to its maximally tolerated dose. Combination therapy may overcome tumor resistance to an individual drug, and is generally more efficacious than a single drug. It is also common to combine surgery, radiation, and chemotherapy in treatment. Adjuvant chemotherapy and/or radiation are usually given after the definitive management of the primary cancer through surgery. The purpose of adjuvant chemotherapy is to eradicate micrometastatic disease, and reduce the risk of tumor recurrence. Adjuvant chemotherapy is given for a defined period of time, generally 4 to 6 months. Neoadjuvant chemotherapy refers to chemotherapy given before the definitive management of the primary cancer, and is given to reduce the size of the primary tumor for better cosmetic and functional outcomes.
18.1.1 Pharmacokinetics and Pharmacodynamics
Pharmacokinetics is the study of the time course of drug and metabolite levels in different body fluids and tissues, including absorption, distribution, metabolism, and elimination. The study of the relationship between drug effect and its concentration is known as pharmacodynamics. Alterations in pharmacokinetic properties of a drug may result in different drug concentrations over time at tissue levels. Understanding the pharmacodynamics of the drug can help to account for subsequent differences in drug effect or response.
Although most anticancer drugs are administered intravenously and drug absorption is not a therapeutic concern, many newer agents are given orally. Oral drug administration is convenient for patients, but it requires patient compliance and depends on efficient absorption from the gastrointestinal tract. For an orally administered drug, only a proportion may be delivered to the systemic circulation intact and become available for potential therapeutic effects. The term bioavailability refers to the amount of a drug that is available after oral administration compared to that after intravenous administration. Factors influencing the bioavailability of a drug include patient compliance, disintegration of a capsule or a tablet, dissolution of drug into gastrointestinal fluid, stability of the drug in the gastrointestinal tract, absorption through the gastrointestinal mucosa, and first-pass metabolism in the liver. Problems seen in cancer patients, such as changes in gastrointestinal motility, mucosal damage from cancer therapy, and the use of other medications, can also affect bioavailability. Absorption of a drug may vary among patients receiving the same treatments, or within 1 patient from one course of treatment to another. This variability can account for some differences in toxicity, and possibly in tumor response. In most cases, such variations in drug bioavailability are not detected clinically because routine pharmacokinetic measurements are not made in patients.
Once in the blood, the distribution of a drug within the body is governed by factors such as blood flow to different organs, protein and tissue binding, lipid solubility, diffusion, and carrier-mediated transport. In general, drugs with extensive protein–tissue binding or with high lipid solubility will tend to exhibit prolonged elimination phases because the release of bound drug from tissues is slow.
Metabolism of drugs takes place primarily in the liver and consists of oxidative, reductive, and hydrolytic reactions via the superfamily of cytochrome P450 (Phase I) and conjugation (Phase II) enzymes. Phase I reactions can produce metabolites that retain therapeutic activity or convert an inactive prodrug (eg, cyclophosphamide) to an active moiety. Phase II reactions generally produce inactive metabolites that can be eliminated from the body by biliary or renal excretion. Many anticancer drugs have active metabolites, and this introduces additional complexity into understanding the relationship between drug pharmacokinetics and pharmacodynamics. There are genetic polymorphisms that can affect the activity of many of the drug-metabolizing enzymes (Paugh et al, 2011). As well, acquired changes as a result of hepatic impairment or the use of other medications may affect their activity. Unfortunately, simple tests of liver function, such as serum levels of bilirubin or transaminases, have not proved useful in predicting hepatic metabolic activity because the decline in activity of metabolizing enzymes varies in the setting of hepatic dysfunction.
Most drugs are eventually eliminated from the body by the kidney or through the biliary tract. Renal excretion can either be of the active drug or of metabolites. Impairment of renal function will influence drug clearance and may enhance toxicity for drugs that are eliminated unchanged in the urine, such as carboplatin and methotrexate. Dosage reductions proportionate to the decline in creatinine clearance (a common measure of kidney function) are usually required. Several chemotherapy drugs are also toxic to the kidney (eg, cisplatin and high-dose methotrexate), and combinations of these drugs with others that are eliminated by the kidney require extra caution and maintenance of a high urinary output.
Cancer patients frequently take multiple other medications for relief of pain, nausea, and other symptoms. Interaction between drugs may influence each of the processes of absorption, metabolism, distribution, and excretion. For example, patients receiving warfarin (to prevent blood clotting) require increased monitoring if they are also receiving fluoropyrimidines because of such interactions. Possible interactions between anticancer drugs, and between such drugs and other medications, are common (Riechelmann et al, 2008), but have not been investigated extensively.
The concentration of most anticancer drugs can be measured in the blood or in the tissue of a patient. If a drug is measured in blood or tissue over time, then a curve relating drug concentration to time can be defined. The area-under-the-concentration-time curve (AUC) is a commonly used measure of total systemic drug exposure. For drugs like cisplatin, their effect in killing of tumor cells or toxicity to normal tissues is related directly to the AUC; whereas for other drugs, such as taxanes, the duration of exposure above a threshold concentration may be more important than the AUC.
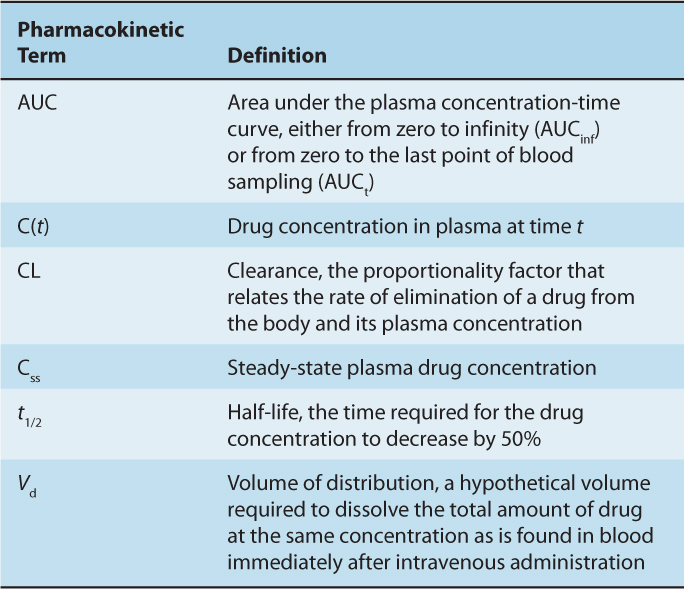
The concentration-time curve (Fig. 18–1) can be modeled mathematically. Table. 18–1 lists some important terms derived from such modeling. Clearance (CL) is the proportionality factor that relates the rate of elimination of a drug from the body and its plasma concentration:
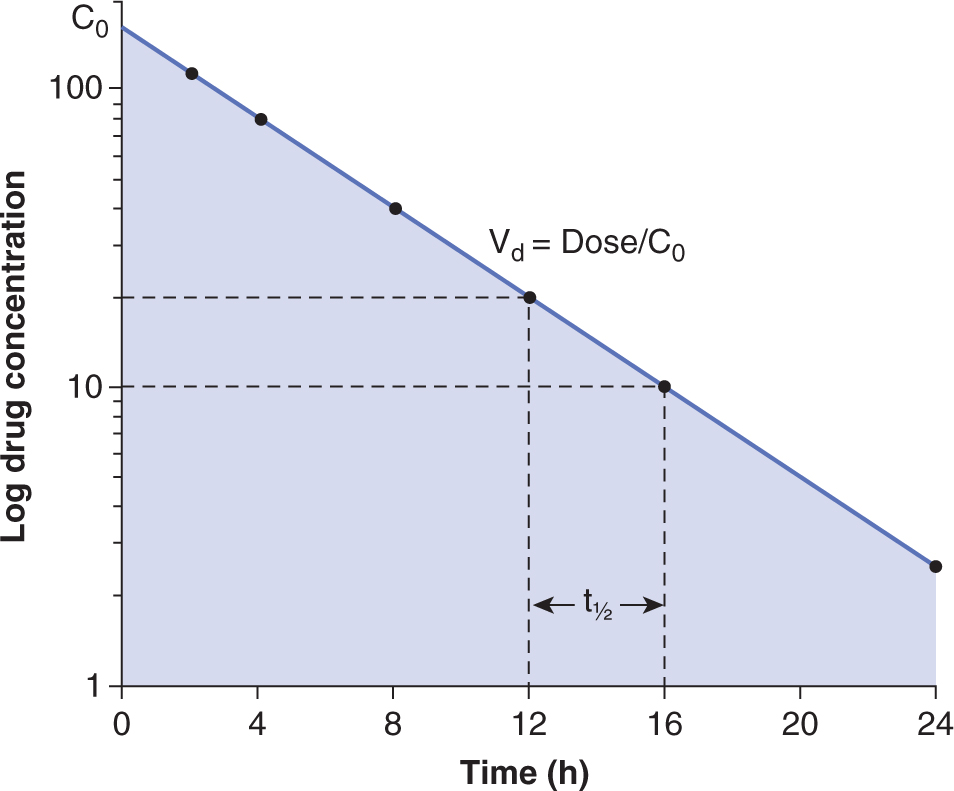
FIGURE 18–1 The plasma time-concentration curve after the intravenous bolus administration of a drug. Drug concentrations are measured every 2 hours. In this case, the drug is eliminated via a first-order process with the elimination half-life (t1/2) being 4 hours. The shaded area is the AUC. Volume of distribution (Vd) can be calculated by dividing dose by the concentration at time 0 (C0) which can be obtained by extrapolating the curve to time 0.
TABLE 18–1 Glossary of terms used commonly in pharmacokinetics.
Rate of elimination = CL × plasma concentration
Clearance can be represented by a volume from which the drug is totally eliminated in a unit of time, such as liters per hour. The AUC of a drug is related to its dose and clearance:
AUC = Dose/CL
Thus, if the clearance of a drug declines (eg, in the setting of renal or hepatic dysfunction), its AUC will increase without an adjustment in dose, resulting in increased toxicities. Similarly, individual variability in drug clearance will manifest as differences in AUC and in effect. Clearance is independent of dose unless there is saturation of drug metabolizing enzymes in which case the AUC will increase at a greater rate than dose and severe toxicities can result.
The volume of distribution (Vd) represents a hypothetical volume of body fluid that would be required to dissolve the total amount of drug at the same concentration as that found in blood immediately after intravenous administration. A large Vd (a value larger than the total volume of the body water is possible) represents extensive binding of drug in tissue (eg, vinca alkaloids). Curves relating drug concentration in blood with time after administration often have components that are approximately exponential (see Fig. 18–1). The half-life (t1/2) of a drug is the time required for the drug concentration to decrease by half, and different values of t1/2 (labeled α, β, and γ) can be used to characterize successive exponential components of the clearance curve; the last of these is commonly referred to as the elimination half-life. The elimination half-life is useful in estimating the time required for a drug to reach a steady state (where the amount of drug being eliminated from the body is equal to the amount being added after multiple administrations such that the plasma concentration remains constant).
18.1.2 Dosing of Chemotherapy in Individual Patients
Most anticancer drugs have a low therapeutic index (ie, a small difference between doses that cause anticancer effects and those that cause toxicity) and are given at close to the maximum tolerated dose (MTD) (see Chap. 22, Sec. 22.2.1). There is a need to reduce interindividual variability to have a consistent response and to minimize toxicity. Efforts in relating drug concentration to effects have generally been unsuccessful. The majority of chemotherapeutic agents are dosed on the basis of body surface area (BSA) by tradition on the premise that the factors relevant in pharmacokinetics, such as cardiac output, body fat, and creatinine clearance, are all related to body size. BSA dosing is useful in interspecies comparisons as MTD remains relatively constant between different animals when expressed as milligrams per square meter, and it may have value in pediatric patients where they may be large ranges in body size. Many reports have questioned the convention of calculating the dose of chemotherapy in adults on the basis of BSA (Ratain, 1998). Multiple studies show that there is no reduction in pharmacokinetic variability when compared to a standard dose or dose based on body weight. Furthermore, BSA based dosing makes drug administration unnecessarily complex and more subject to human error.
Factors within an individual that may account for variability in pharmacokinetics and pharmacodynamics are still poorly understood. The usual approach is to use a standard starting dose (in mg/m2) and then modify subsequent doses based upon the observed toxicity. An exception is the dosing of carboplatin, where a relationship between kidney functions and clearance of carboplatin has been demonstrated and carboplatin dose is calculated on the basis of the creatinine clearance (Calvert et al, 1989). The approach to dose modification will also be influenced by the goals of therapy. In a setting where treatment is curative (eg, testicular cancer), the drug doses are usually maintained despite severe toxicity, often with administration of G-CSF (see Chap. 17, Sec. 17.5.2), whereas in a palliative situation (eg, 5-fluorouracil [5-FU] for advanced colon cancer) dose reductions are appropriate for even moderate toxicity. The approach to dose reduction is usually empiric with fractional dose reductions of one or more of the drugs thought to be causing the toxicity.
Many of the newer anticancer drugs, especially molecular-targeted agents, are administered orally. Therapy is usually initiated at a fixed dose regardless of height or weight.
18.1.3 Pharmacogenetics
Pharmacogenetics refers to the study of how genetic features of the patient (and their tumor) will influence response and toxicity. In the broadest sense, pharmacogenetics seeks to explain phenotypic differences in response on the basis of differences in the activity of genes that are involved in drug metabolism and are related to specific mutations or polymorphisms (Paugh et al, 20011). In the past, this was done by defining a particular phenotype following drug exposure (eg, serious toxicity, second malignancy) and then looking for changes at the genetic level that might account for this phenotype. This could be a result of determinants of drug pharmacokinetics, such as the function of drug-metabolizing enzymes, to genetic factors that would influence pharmacodynamics or even to genetic polymorphisms that might relate to the development of cancers (see Chap. 3, Sec. 3.6.2).
An early example of the effect of pharmacogenetics on response and toxicity is the use of 5-FU in the treatment of colorectal cancer. Approximately 80% of intravenously administered 5-FU is catabolized to an inactive metabolite through the enzyme, dihydropyrimidine dehydrogenase (DPD) (van Kuilenburg, 2004; see Fig. 18–9). Although DPD activity is present in different tissues and organs, including the gastrointestinal mucosa, the liver is the major site of 5-FU catabolism. The DPD gene is present as a single copy on chromosome 1p22, and consists of 23 exons. Mutations and polymorphism can lead to a deficiency of DPD activity. Although this deficiency does not lead to any detectable problems under normal circumstances, patients with this deficiency are subjected to increased toxicity when they are exposed to 5-FU. The most common mutation identified in patients who experienced severe 5-FU toxicity is a G-to-A mutation in the invariant GT splice donor site, which results in skipping of exon 14 immediately upstream. It is estimated that approximately 25% of patients with grade 3/4 5-FU toxicity are homozygous or heterozygous for this mutation (van Kuilenburg, 2004). This mutation is commonly detected in northern European patients, but, it has not been reported in Japanese or African Americans.
There are several other examples of genetic polymorphisms leading to alterations in drug toxicity, most of them relating to individual differences in drug metabolizing enzymes. The topoisomerase I inhibitor, irinotecan, is partially metabolized to SN-38, an active metabolite, through carboxylesterase. SN-38, when excreted unconjugated in the biliary tract, can cause damage to the gastrointestinal mucosa, resulting in diarrhea. Polymorphisms of uridine diphosphate glucuronosyltransferase (UGT)-1A1, the enzyme involved in conjugation of SN-38, have been identified and those leading to a reduced rate of conjugation are associated with higher risk of irinotecan toxicity, such as neutropenia and diarrhea (Mathijssen et al, 2001, 2003).
Several drugs rely on enzymes to be converted to active metabolites. Reduced enzyme activity as a result of genetic polymorphism would lead to lower levels of metabolites, resulting in lower therapeutic effects. Tamoxifen, which has a major role in endocrine therapy for breast cancer, is converted to an active metabolite, endoxifen, through the cytochrome P450 family of enzymes, in particular, CYP2D6 (see Chap. 20, Sec. 20.5.2). So far, more than 100 genetic variants in CYP2D6 have been identified, and patients can be grouped into 4 groups based on CYP2D6 activity: ultra-high, normal, reduced, and no activity (Schroth et al, 2009). Some, but not all, studies suggest that patients with reduced CYP2D6 activity are at higher risks of failing to respond to tamoxifen, compared to those with normal CYP2D6 activity.
18.2 ALKYLATING AGENTS
Alkylating agents were the first drugs introduced for the systemic therapy of cancer. They are a chemically diverse group of drugs, but they all contain alkyl groups (eg, —CH2Cl). In vivo, the alkyl groups generate highly reactive, positively charged intermediates, which then combine with an electron-rich nucleophilic group, such as an amino, phosphate, sulfhydryl, or hydroxyl moieties, on intracellular macromolecules, such as DNA. Although nucleophilic groups occur on almost all biological molecules, alkylation of bases in DNA appears to be the major cause of lethal toxicity.
Alkylating agents may contain 1 or 2 reactive groups and are thus classified as monofunctional or bifunctional, respectively. Bifunctional alkylating agents have the ability to form cross-links between DNA strands and are the most clinically useful of these agents. Interstrand crosslinking of DNA, which prevents cell replication, unless repaired, seems to be the major mechanism of cytotoxicity for bifunctional alkylating agents, whereas the toxicity of monofunctional alkylating agents is probably related to single-strand breaks in DNA or to damaged bases. Mechanisms of resistance of alkylating agents include decreased transport across the cell membrane, increased intracellular thiol concentrations (eg, glutathione); such compounds react with alkylating agents and thus reduce the likelihood of interaction with DNA (see Chap. 19, Sec. 19.2.4), increased enzymatic detoxification of reactive intermediates, and alterations in DNA repair enzymes.
Because alkylating agents bind directly to DNA, they lack cell-cycle specificity, although they may have greater toxicity for proliferating cells. Common toxicities include myelosuppression, immunosuppression, hair loss, nausea, and vomiting. Some alkylating agents have long-term effects, such as infertility and carcinogenesis caused by long-lasting DNA damage. Nitrogen mustard, melphalan, and nitrosoureas are associated with an increased incidence of acute myelogenous leukemia, whereas cyclophosphamide is associated with irritation of the bladder and rarely development of bladder cancer. Mechanisms of resistance to alkylating agents include decreased transport across the cell membrane, increased intracellular thiol concentration leading to reduced DNA interactions, increased enzymatic detoxification of reactive intermediates, and alterations in DNA repair enzymes such as guanine-O6-alkyltransferase (Pegg, 1990; see Chap. 19, Sec. 19.2.6).
18.2.1 Nitrogen Mustards, Nitrosoureas, and Others
The development of nitrogen mustard and related compounds as anticancer drugs originated from the observation of lymphoid aplasia in men exposed to the more reactive, but chemically similar, sulfur mustard gas during World War II. This family of drugs, all chemically derived from nitrogen mustard (mechlorethamine) contains several drugs in common clinical use. The structures of these drugs are shown in Figure 18–2; each is bifunctional, with 2 chloroethyl groups that form the reactive electron-deficient groups responsible for alkylation of DNA.
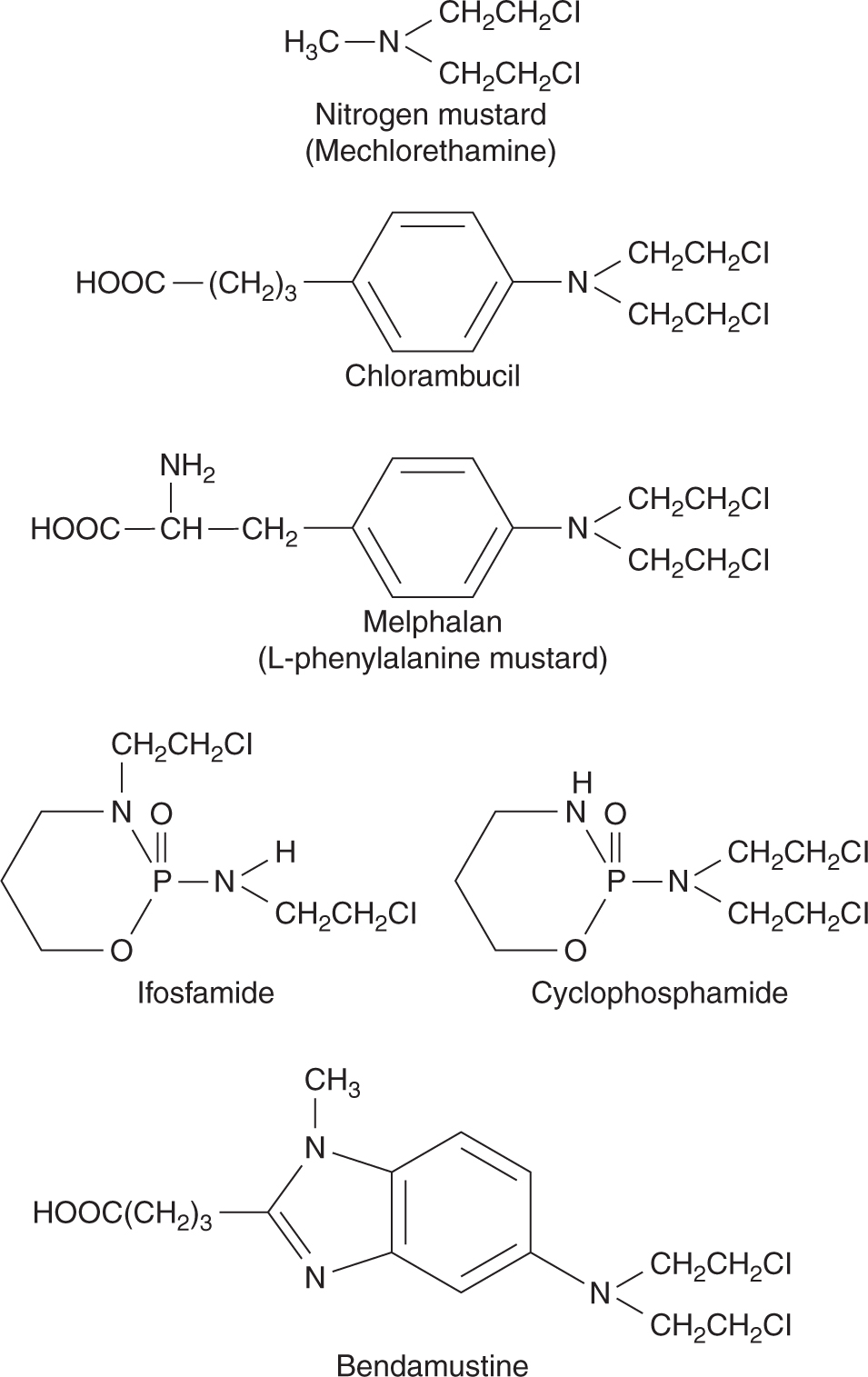
FIGURE 18–2 Structures of clinically used alkylating agents of the nitrogen mustard family.
The most common site of alkylation of DNA by nitrogen mustards is the N-7 position on the base guanine (Fig. 18–3). First, one of the chloroethyl side chains releases a chloride ion, resulting in a highly reactive, positively charged intermediate. This intermediate then binds covalently with the electro-negative N-7 group on a guanine base, resulting in alkylation. Alkylation of guanine leads to mispairing with thymine or to strand breakage. The second chloroethyl side chain of nitrogen mustard may undergo a similar reaction, leading to covalent binding with another base on the opposite strand of DNA and thus to formation of an interstrand crosslink.
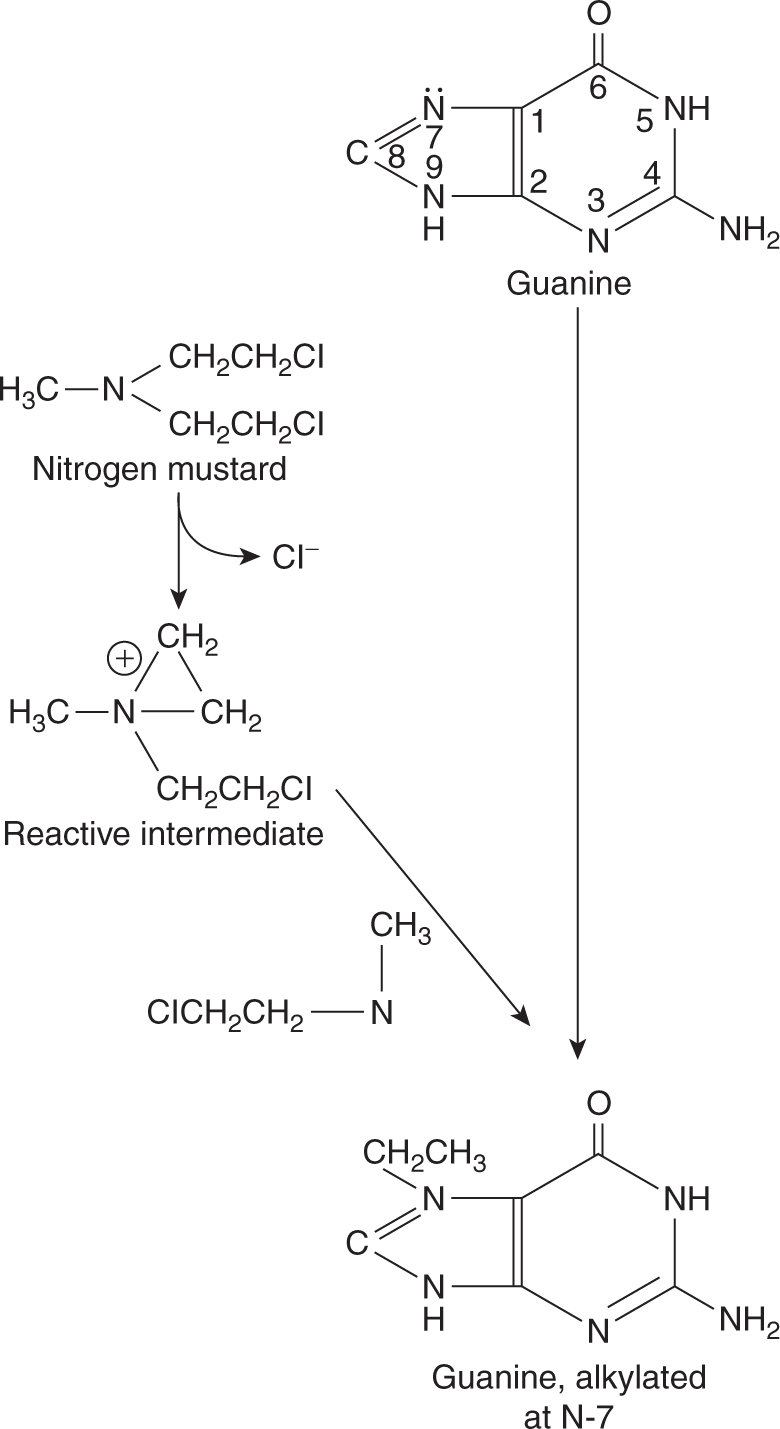
FIGURE 18–3 Reactions leading to alkylation at the N-7 position of guanine by nitrogen mustard.
Mechlorethamine was the first anticancer drug introduced to clinical use. However, it is chemically unstable, and undergoes spontaneous hydrolysis. A large number of analogs have been synthesized and 5 of them (chlorambucil, melphalan, cyclophosphamide, ifosfamide, and bendamustine) have largely replaced mechlorethamine, which is only used clinically as part of the MOPP protocol (mechlorethamine, Oncovin [vincristine], procarbazine, prednisone) for Hodgkin disease. The addition of ring structures to the nitrogen mustard molecule confers increased stability, such that these agents can be administered orally.
Chlorambucil is a well-absorbed oral drug with a narrow spectrum of activity, and is used mainly in slowly progressive neoplasms, such as low-grade lymphomas and chronic lymphocytic leukemia. Oral melphalan is used for treatment of plasma cell myeloma and in some high-dose bone marrow transplantation protocols. Absorption of melphalan is variable and unpredictable; some patients with no effect after oral administration may respond when melphalan is given intravenously. Both chlorambucil and melphalan are almost equally toxic to cycling and noncycling cells, and may lead to delayed and/or cumulative effects on bone marrow because of their toxicity to hematopoietic stem cells.
Cyclophosphamide is the alkylating agent in widest clinical use and is part of treatment protocols for breast, lymphatic, gynecological and pediatric tumors, in high-dose chemotherapy regimens, and for a number of autoimmune diseases. Cyclophosphamide is well absorbed after oral administration; however, the parent compound is inactive, requiring metabolism by hepatic mixed-function oxidases to form the alkylating intermediate phosphoramide mustard (Fig. 18–4). Hepatic microsomal enzymes metabolize cyclophosphamide to 4-hydroxycyclophosphamide, which exists in equilibrium with its acyclic isomer aldophosphamide. 4-Hydroxycyclophosphamide enters cells and spontaneously decomposes to form phosphoramide mustard and acrolein, or it is inactivated by aldehyde dehydrogenase. Elimination of cyclophosphamide and its metabolites is mainly by renal excretion. Cyclophosphamide induces cytochrome P450 enzymes, and hence its own metabolism with repeated administration. This alters the rate but not the absolute amount of phosphoramide mustard formation, so no dose adjustment is required.
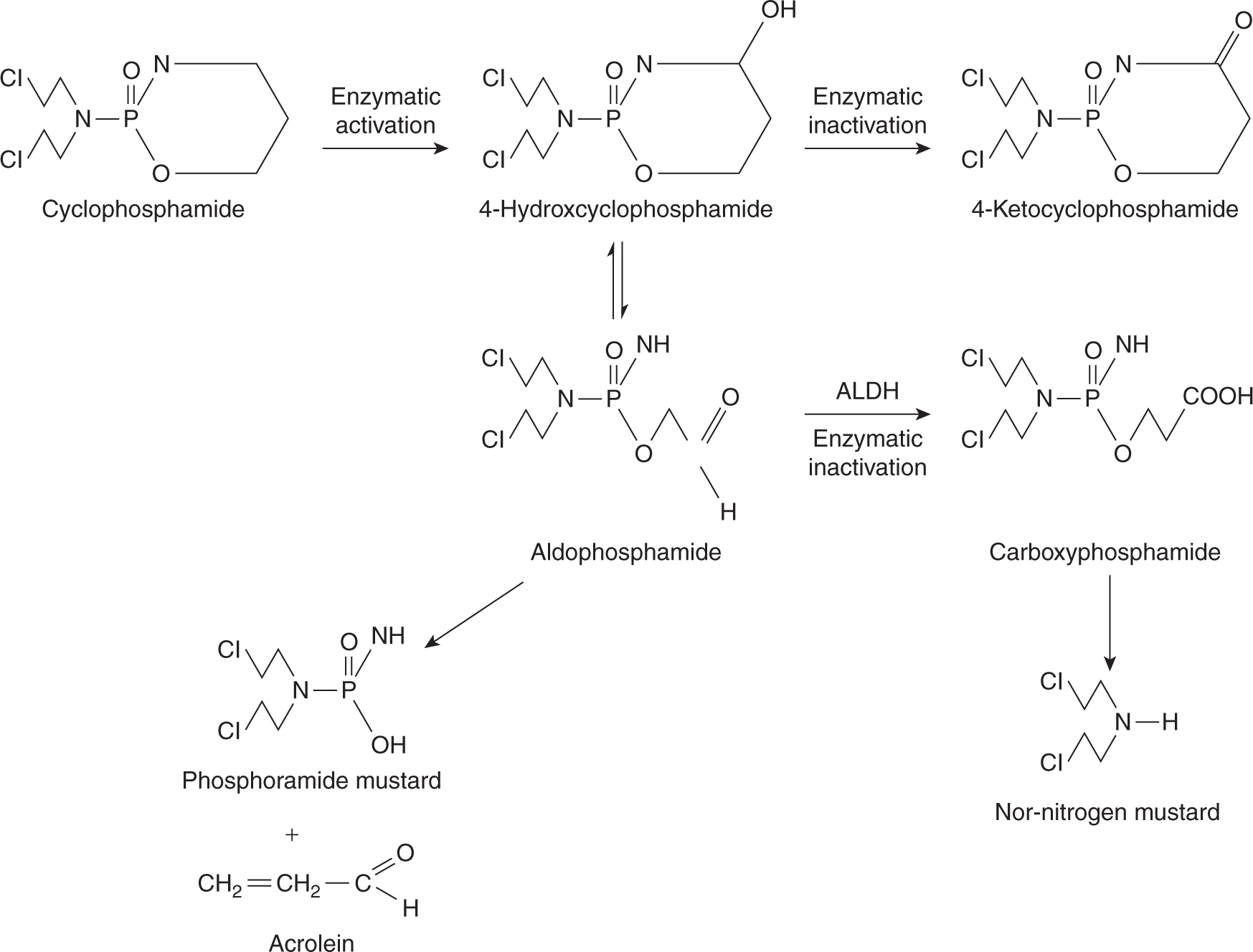
FIGURE 18–4 The metabolism of cyclophosphamide. ALDH, aldehyde dehydrogenase.
The dose of cyclophosphamide given to patients ranges from 100 to 200 mg/m2 per day given orally, to 600 to 1000 mg/m2 given intravenously every 3 to 4 weeks. Very high doses are used in preparation for bone marrow transplantation. The dose in this setting is limited by irreversible damage to the heart, which occurs with single doses greater than 60 mg/kg (approximately 2500 mg/m2). The usual dose-limiting toxicity is myelosuppression, and cyclophosphamide causes a fall in granulocyte count with rapid recovery by 3 to 4 weeks after administration (see Chap. 17, Sec. 17.5.2). There is relative sparing of stem cells and platelets, which may be a result of the higher concentrations of aldehyde dehydrogenase in early progenitor cells. Cyclophosphamide causes hemorrhagic cystitis with chronic use or at higher doses because of the direct irritative effect of acrolein on the bladder mucosa.
Ifosfamide is an analog of cyclophosphamide that differs in the presence of only 1 chloroethyl group on the oxazaphosphorine ring. It is used in the treatment of testicular cancer and sarcoma. Hemorrhagic cystitis is more common as a result of increased production of acrolein, such that all patients receiving ifosfamide should be given a sulfhydryl-containing compound, such as 2-mercaptoethane sulfonate (Mesna), which conjugates with acrolein in the urinary tract and renders it inactive. As Mesna dimerizes to an inactive metabolite in blood, and is hydrolyzed to its active form only in urine, it does not affect the cytotoxicity of cyclophosphamide or ifosfamide at other sites. Neurotoxicity, manifesting as changes in mental status including confusion, hallucination, cerebellar dysfunction, seizures, and coma, may occur with higher doses of ifosfamide, but not cyclophosphamide. Risks for ifosfamide neurotoxicity include low serum albumin, elevated creatinine and prior cisplatin treatment (Pratt et al, 1990; David and Picus, 2005).
Bendamustine is the newest bifunctional mechlorethamine derivative introduced into clinical practice. It is indicated for patients with chronic lymphocytic leukemia and indolent non-Hodgkin lymphoma.
The chloroethylnitrosoureas, BCNU (carmustine) and CCNU (lomustine), are lipid-soluble drugs that can penetrate into the central nervous system (CNS). These drugs have limited clinical application because they cause prolonged myelo-suppression and are leukemogenic, likely because of direct effects on bone marrow stem cells. Streptozotocin, a methylnitrosourea, is used mainly in the treatment of pancreatic islet-cell tumors; it has less hematological toxicity than other nitrosoureas.
Busulfan, an alkyl alkane sulfonate, has a different mechanism of alkylation from the nitrogen mustards. It reacts more extensively with thiol groups of amino acids and proteins, but its ability to crosslink DNA is uncertain. Busulfan has selective effects on hematopoietic stem cells, and is now used mainly in high-dose bone marrow transplantation regimens. Busulfan is eliminated via hepatic metabolism, and the higher doses of busulfan used in marrow transplantation may cause hepatic venoocclusive disease in patients who metabolize the drug slowly.
A group of other compounds, with diverse chemical structures, that are also capable of forming covalent crosslink with intracellular macromolecules includes procarbazine, dacarbazine (DTIC), and temozolomide. Procarbazine is a synthetic derivative of hydrazine that was used in combination to treat lymphoma, including Hodgkin disease. It undergoes extensive metabolism to produce alkylating species, although details of its metabolism and mechanism of action remain unclear. It has largely been replaced by other alkylating agents. DTIC was synthesized originally as an antimetabolite to inhibit purine synthesis, but is believed to function through formation of methylcarbonium ions with alkylating properties. DTIC is metabolized by CYP450 enzymes to MTIC ([methyl-triazene-1-yl]-imidazole-4-carboxamide) which alkylates DNA at the O6 and N7 guanine positions (Marchesi et al, 2007). DTIC is sensitive to light, but is stable in neutral solutions away from light. Temozolomide is a prodrug that is stable under acidic conditions but undergoes rapid, spontaneous, nonenzymatic conversion to MTIC at pH levels greater than 7 (Marchesi et al, 2007); it is rapidly and completely absorbed after oral administration with a t1/2 of 1 to 2 hours. Temozolomide is indicated for patients with newly diagnosed or recurrent malignant glioma, and has also shown activity in melanoma.
18.2.2 Platinating Agents
The prototype agent is cisplatin (cis-diamminedichloroplatinum II; Fig. 18–5), a drug whose discovery followed an observation that an electric current delivered to bacterial culture via platinum electrodes led to inhibition of bacterial growth. The active compound was identified as cisplatin, and it was shown subsequently to exert broad cytotoxic activity. Cisplatin is one of the most useful anticancer agents and is part of first-line therapy for testicular, urothelial, lung, gynecological, and other cancers. It is also associated with substantial toxicity, which limits both the number of patients who are able to receive the drug, as well as the cumulative dose that can be given. There has been a major effort to identify other platinum analogs, either to reduce the toxicity while maintaining efficacy, or to expand the use of these compounds to tumors resistant to cisplatin. The 2 analogs in routine clinical usage are carboplatin and oxaliplatin (Fig. 18–5).
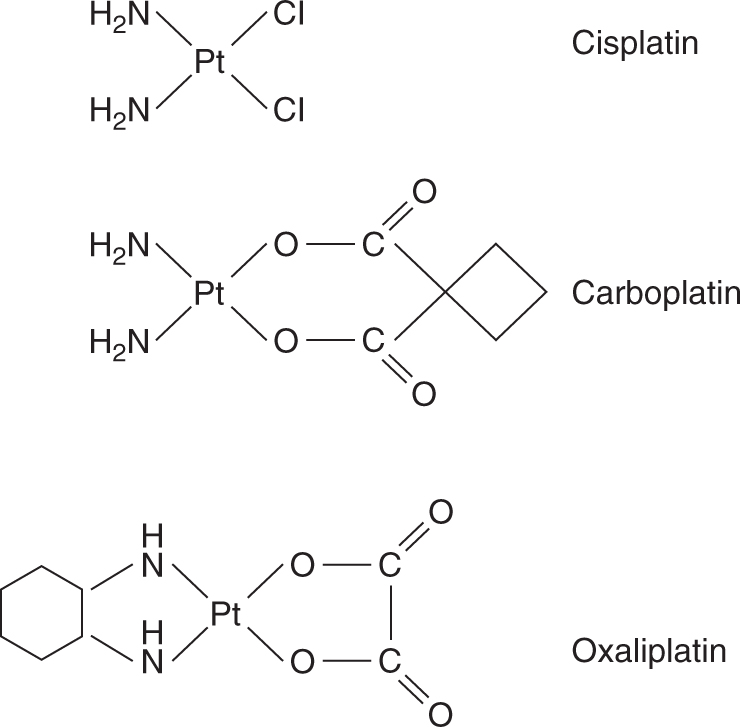
FIGURE 18–5 Structure of platinum agents.
Platinum drugs can exist in a 2+ (II) or 4+ (IV) oxidation state, with 4 or 6 bonds linking the platinum atom, respectively. All currently used platinum drugs are platinum II compounds that exhibit a planar structure and have 4 attached chemical groups. The nature of these groups dictates the efficacy and pharmacokinetic properties of these compounds. Two of the groups are considered carrier groups, and are chemically inert, whereas the 2 leaving groups are available for substitution and reaction with molecules such as DNA.
Cisplatin acts by a mechanism that is similar to that of classical alkylating agents. The chlorine atoms are leaving groups that may be compared to those of nitrogen mustards; these atoms may be displaced directly by nucleophilic groups in DNA or indirectly after chloride ions are replaced by hydroxyl groups through reaction of the drug with water. These reactions occur more readily in environments where the chloride concentration is low, such as within the cell. The preferred sites for binding of cisplatin to DNA are the 7 positions of the guanine and adenine bases. The fact that structurally similar analogs, such as transplatin, will produce DNA binding but are devoid of cytotoxicity suggests that the stereochemistry of the compound is critical. Cisplatin binds to 2 sites on DNA and 95% of the binding produces intrastrand crosslinkages, usually between 2 adjacent guanine bases or adjacent guanine and adenine sites, with the remainder being interstrand guanine crosslinkages. The binding of platinum compounds to DNA is responsible for their cytotoxicity, although the mechanism whereby this leads to cell death is not clear.
Carboplatin is an analog of cisplatin with substitution of cyclobutanedicarboxylate for the chloride leaving groups. This leads to a less-reactive compound that also has less toxicity but either comparable or slightly reduced efficacy to cisplatin. Oxaliplatin is one of a series of analogs with a substitution of a diaminocyclohexane (DACH) for the amine carrier groups (see Fig. 18–5). The DACH analogs have a different efficacy profile from cisplatin and have shown activity in tumors resistant to cisplatin, such as colorectal cancer. Carboplatin and oxaliplatin produce the same types of DNA adducts as cisplatin, although a higher concentration of carboplatin is required to produce a comparable number of adducts to cisplatin. Adducts formed by oxaliplatin are more likely to cause cell death, probably because of the different 3-dimensional structure that results from the DACH groups.
The pharmacokinetic differences between cisplatin and its analogs are a result of the differences in the leaving groups. Cisplatin is the most reactive and, following administration, it is rapidly and irreversibly bound to plasma proteins, with greater than 90% of free cisplatin lost in the first 2 hours. Total cisplatin (free and bound) disappears more slowly from plasma, with a prolonged t1/2 of 2 to 3 days. Cisplatin is excreted mainly via the urine, and 15% to 30% of the administered dose is excreted during the first 24 hours. Carboplatin is more stable in plasma and is excreted primarily unchanged by the kidney, with 90% of administered dose excreted within 24 hours. The clearance of carboplatin is predicated by creati-nine clearance; therefore, it is possible to determine the dose of carboplatin based on the desired carboplatin AUC and creatinine clearance (Calvert et al, 1989):
Carboplatin dose (mg) = AUC × (creatinine clearance + 25)
Similar to cisplatin, oxaliplatin also binds to plasma proteins, although at a somewhat slower rate; it is also excreted primarily by the kidney.
Cisplatin causes little toxicity to bone marrow as a single agent, but can add to the toxic effects of other drugs, and may lead to anemia. Its major dose-limiting toxicities are nausea and vomiting, and damage to the kidney, to nerves, and to the ear with resulting loss of hearing. Vigorous intravenous hydration and maintaining a rapid urine output during and after drug administration minimize the effects on the kidneys; there is no known method for reducing the auditory or neurotoxicity.
Carboplatin has comparable activity to cisplatin against ovarian and lung tumors but is less active against urothelial and testicular cancers. Carboplatin has a better overall toxicity profile, which may make it preferable in palliative treatment regimens. There is minimal nephrotoxicity, and the drug causes less nausea and vomiting than cisplatin, but bone marrow suppression, particularly thrombocytopenia, is the dose-limiting toxicity. Carboplatin is used in some high-dose regimens prior to stem cell transplantation because its toxicities other than myelosuppression are relatively mild.
Oxaliplatin is used mainly in the treatment of colorectal cancer; it has minimal renal toxicity, and causes less vomiting than cisplatin and no ototoxicity. Oxaliplatin causes a unique spectrum of sensory neurotoxicity, ranging from acute neuropathy (paresthesia, muscle spasm and fasciculations or muscle twitching) immediately after infusion with marked sensitivity to the cold, particularly in the oropharynx, to a late cumulative dose-limiting sensory neuropathy resulting in sensory ataxia and functional impairment. Coadministration of calcium and magnesium may delay the onset of neuropathy and reduce its severity (Gamelin et al, 2008).
Resistance to platinating agents can be a result of reduced platinum-DNA adduct formation, or of increased repair or tolerance of the platinum-DNA adduct. Decreased uptake or increased binding to intracellular scavengers can result in reduced platinum-DNA adduct formation (Koberle et al, 2010).
18.3 ANTIMETABOLITES
Antimetabolites are drugs that interfere with normal cellular functions, particularly the synthesis of DNA that is required for cell replication. Many of the clinically useful agents are purine or pyrimidine analogs that either inhibit the formation of the normal nucleotides or interact with DNA and prevent further extension of the new DNA strand, leading to inhibition of cell division. The antifolates (eg, methotrexate) are not nucleoside analogs; they prevent the formation of reduced folates, which are required for the synthesis of DNA.
Most antimetabolites are cell-cycle specific; their toxicity relates to effects on proliferating cells and is primarily seen in bone marrow and gastrointestinal mucosa. As they do not interact directly with DNA, they do not cause the later problems of carcinogenesis seen with alkylating agents. The effects of these drugs are dependent upon the schedule of administration. For many drugs the duration of exposure above a critical threshold required to inhibit an enzyme is more important than the peak concentration. Therefore, although large doses may be tolerated if the drug is given as a single intravenous injection, a much lower dose is required if the drug is given repeatedly or by continuous infusion.
18.3.1 Antifolates
Methotrexate is an analog of the vitamin folic acid (Fig. 18–6). Reduced folate is required for transfer of methyl groups in the biosynthesis of purines and in the conversion of deoxyuridine monophosphate (dUMP) to thymidine monophosphate (dTMP), a reaction catalyzed by thymidylate synthase. Reduced folate becomes oxidized in the latter reaction; its regeneration is dependent on the enzyme dihydrofolate reductase (DHFR) for reduction to its active form. Methotrexate competitively inhibits DHFR and prevents the formation of reduced folate (Fig. 18–7). The result of this inhibition may be cessation of DNA synthesis because of nonavailability of dTMP and/or purines, leading to cell death.
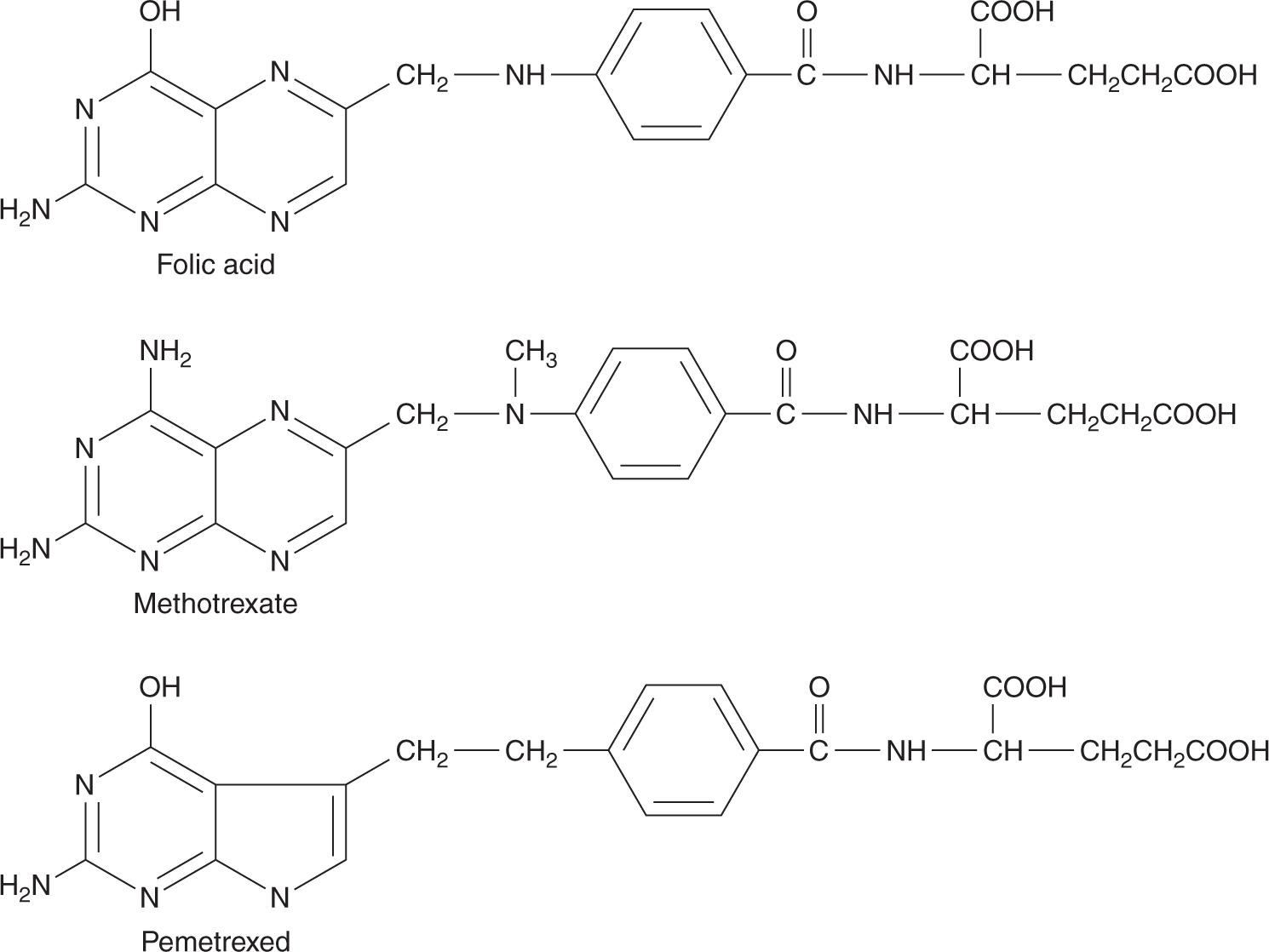
FIGURE 18–6 Structures of folic acid, methotrexate, and pemetrexed.
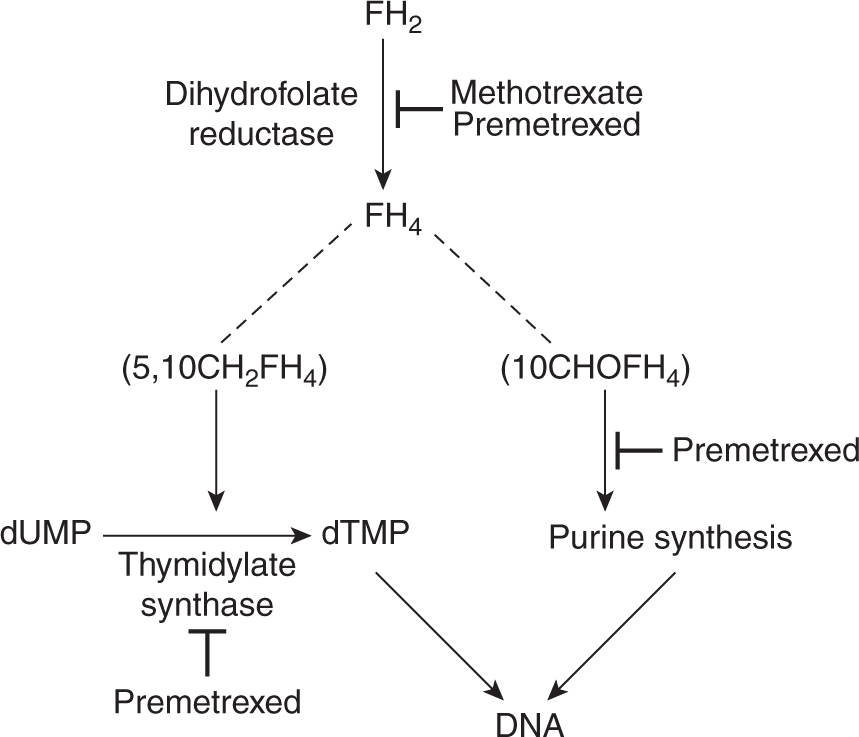
FIGURE 18–7 Influence of methotrexate on cellular metabolism. Methotrexate competitively inhibits DHFR and depletes the pools of reduced folates (FH4): 5,10-methylene tetrahydrofolate (5,10CH2FH4) and 10 formyltetrahydrofolate (10CHOFH4). Reduced folates are required in the conversion of dUMP to dTMP and for purine synthesis, respectively. Interruption of these processes leads to inhibition of DNA synthesis.
Methotrexate enters the cell primarily by active transport. However, its uptake may be by passive diffusion at high drug concentrations. Intracellular metabolism of methotrexate leads to addition of glutamic acid residues to the initial glutamate residue of the drug, a process known as polyglutamation. Methotrexate polyglutamates cannot be transported across the cell membrane, so their formation prevents efflux of the drug, and they appear to be more effective than methotrexate itself in inhibiting the activity of DHFR. The cytotoxic action of methotrexate depends critically on the duration of exposure of tissue to levels of drug above a certain threshold, rather than on the peak levels of drug in the tissue. For many tissues, the threshold concentration for cytotoxicity appears to be in the range of 10–8 to 10–7 M. Methotrexate has selective toxicity for cells synthesizing DNA, and prolonged treatment with the drug may cause toxicity to more cells as they enter the S-phase of the cell cycle.
The toxicity of methotrexate may be reversed by administration of thymidine and exogenous purines or by a source of reduced folate. These agents circumvent the effects of methotrexate by providing products of the interrupted metabolism (see Fig. 18–7). They have been used clinically to reverse the activity of methotrexate following a defined period of exposure (usually 24 to 36 hours) to methotrexate at high doses. Reduced folate in the form of 5-formyltetrahydrofolate (also known as leucovorin or folinic acid) has been used in many clinical protocols and has allowed the administration of doses of methotrexate that are increased by factors of 10 to 100 over conventional doses. The arguments put forward for such high-dose methotrexate treatment include (a) selective uptake by tumor cells, (b) better CNS penetration, and (c) lack of myelo-suppression. This type of protocol allows for frequent administration of methotrexate and retained therapeutic efficacy with little or no toxicity in many patients. However, responses to treatment are observed only rarely in patients who are refractory to conventional doses of methotrexate given without leucovorin rescue. Although toxicity is often lower with the use of high doses of methotrexate and leucovorin, an occasional patient may experience life-threatening toxicity, usually as a consequence of damage to the kidney or sequestration in fluid-filled spaces (eg, ascites, pleural effusions) and consequently delayed drug clearance. Methotrexate can cause renal dysfunction during infusion, and this is thought to be a result of precipitation of methotrexate and its metabolites in acidic urine. Therefore, most protocols of high-dose methotrexate mandate vigorous hydration and alkylation of the urine. Methotrexate infusion should not start until the urine output is more than 100 mL/h, and urine pH is 7.0 or higher.
Methotrexate can be given orally, intramuscularly, intravenously, and intrathecally (ie, into the cerebrospinal fluid that surrounds the brain and spinal cord). It crosses the blood–brain barrier but achieves cytotoxic concentrations in the CNS only with intrathecal or high-dose intravenous administration. It accumulates in fluid-filled spaces such as pleural effusions, from which it is released slowly. The parent compound and hepatic metabolites are excreted by the kidney. This excretion can be inhibited by the presence of weak organic acids such as aspirin or penicillin. Aspirin may also displace methotrexate from its binding sites on plasma albumin, and these 2 effects of aspirin can increase the toxicity of methotrexate. Methotrexate dose needs to be reduced in patients with renal dysfunction. The t1/2 of methotrexate ranges from 3 to 10 hours.
Methotrexate has a wide spectrum of clinical activity and may be curative for women with choriocarcinoma, a tumor derived from fetal elements. Its major toxicities are myelosuppression and inflammation of the oral and gastrointestinal mucosa; these toxicities are usually observed within 5 to 7 days of administration, earlier than for many other drugs. Rarer toxicities include damage to liver, lung, and brain, the latter occurring most frequently after intrathecal administration. In general, the drug is well tolerated compared with many other anticancer drugs.
Pemetrexed is a folate-based potent inhibitor of thymidylate synthase (TS) with glutamic acid at one end of the molecule (see Fig. 18–6). Similar to methotrexate, pemetrexed can be polyglutamated for increased retention in cells and increased potency of TS inhibition. In addition to targeting TS, pemetrexed inhibits DHFR and glycinamide ribonucleotide formyltransferase (GARFT); the latter is a folate-dependent enzyme involved in purine synthesis. Pemetrexed has activity in non–small cell lung cancer and mesothelioma; it is administered intravenously and renal excretion is the major route of elimination with a t1/2 of 3 to 4 hours. The main toxicities are myelosuppression, inflammation of the oral and gastrointestinal mucosa, and skin rash. Treatment with pemetrexed requires supplementation with folic acid and vitamin B12.
Raltitrexed is another inhibitor of TS with similar mechanisms of action to pemetrexed. Although it was initially thought to be as active as 5-FU in the treatment of colorectal cancer, subsequent studies showed increased mortality related to raltitrexed. Its use is limited to patients who are intolerant of 5-FU.
18.3.2 5-Fluoropyrimidines
5-FU is a drug that resembles the pyrimidine bases uracil and thymine, which are components of RNA and DNA, respectively (Fig. 18–8). It penetrates rapidly into cells, where it is metabolized to nucleosides by addition of ribose or deoxyribose; these reactions are catalyzed by enzymes that normally act on uracil and thymine. Phosphorylation then leads to the active fluorinated nucleotides 5-fluoro-uridine-triphosphate (5-FUTP) and 5-fluoro-deoxyuridine-monophosphate (5-FdUMP) (Fig. 18–9). 5-FUTP can be incorporated into RNA in place of UTP (uridine triphosphate); this leads to inhibition of the nuclear processing of ribosomal and messenger RNA and may cause other errors of base pairing during transcription of RNA. 5-FdUMP binds irreversibly to, and inhibits, the enzyme TS, leading to depletion of dTMP (thymidine monophosphate), which is required for DNA synthesis.
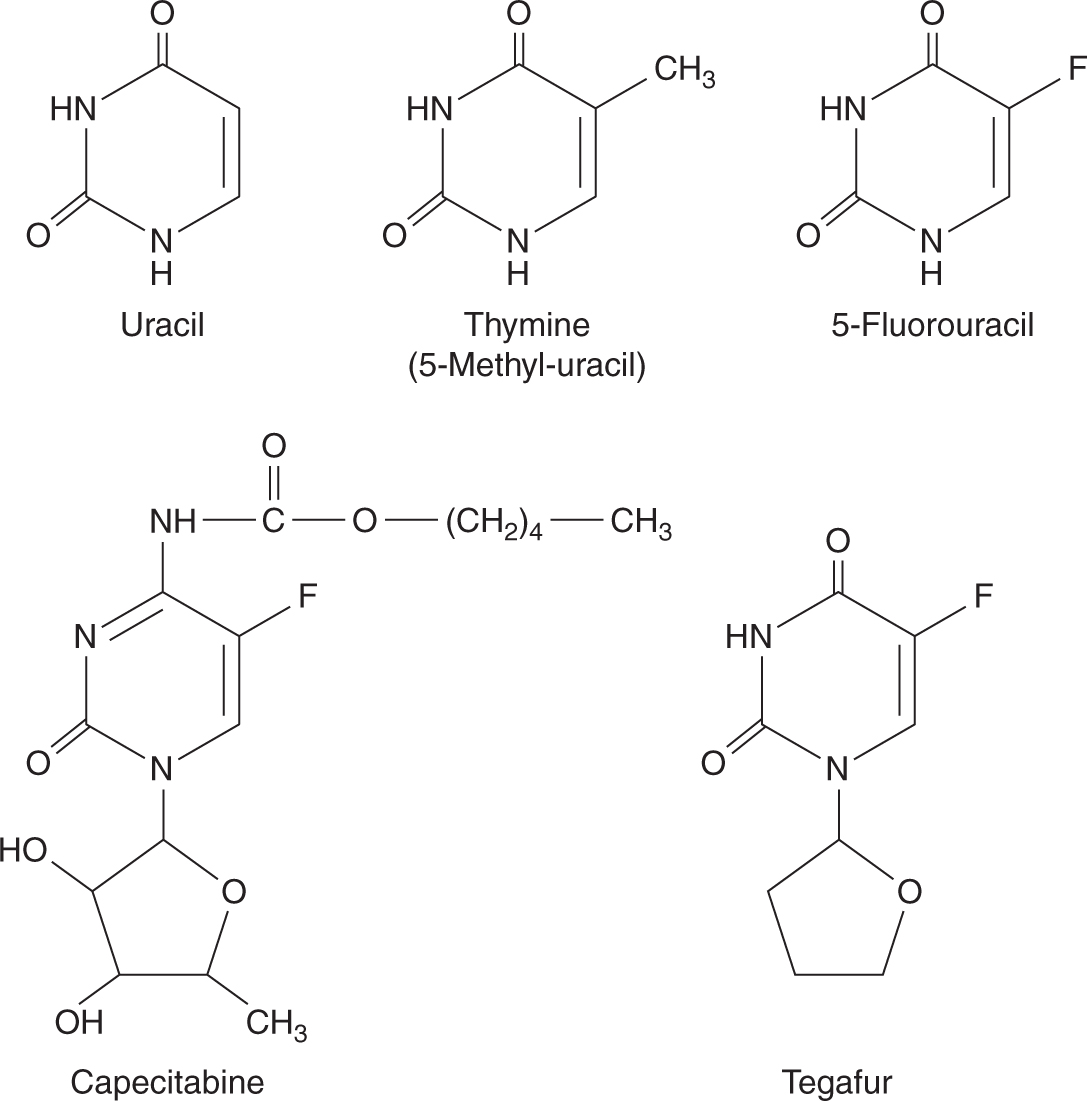
FIGURE 18–8 Structures of uracil, thymine, 5-FU, capecitabine, and tegafur.
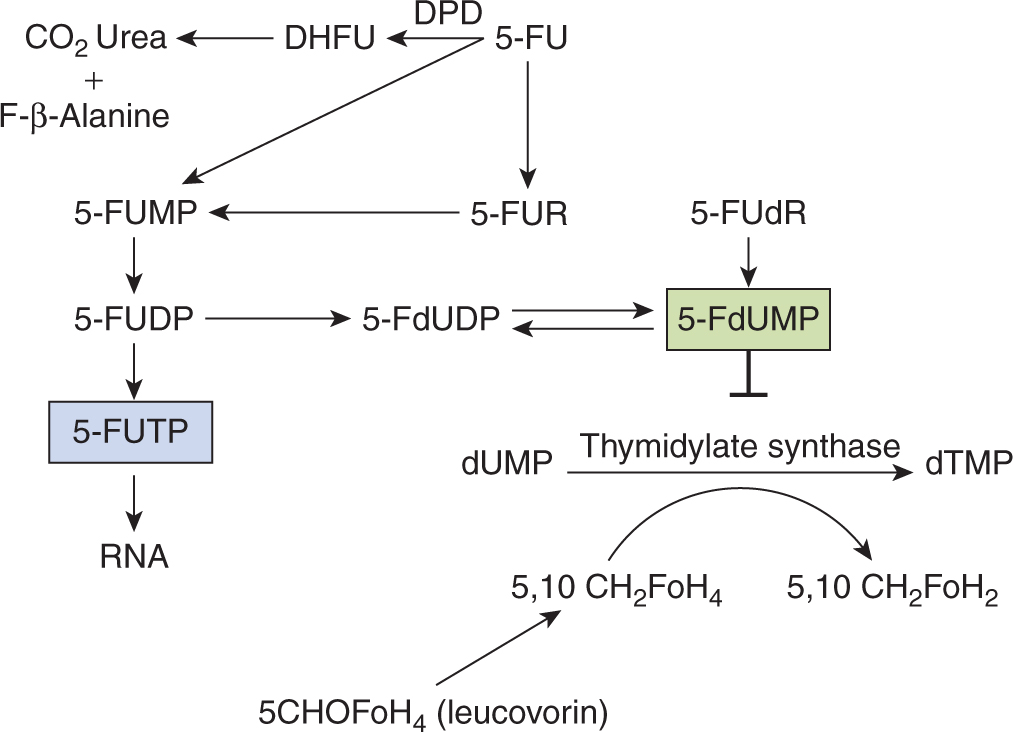
FIGURE 18–9 Metabolic pathways of 5-fluorouracil (5-FU). Fo is used here to distinguish ‘Folate’ from ‘F, Fluorine’. DHFU, dihydrofluorouracil; DPD, dihydropyrimidine dehydrogenase; 5-FdUDP, 5-fluorouridine diphosphate; 5-FdUMP, 5-fluorodeoxyuridine monophosphate; 5-FUDP, 5-fluorouridine diphosphate; 5-FUMP, 5-fluorouridine monophosphate; 5-FUR, 5-Fluorouridine.
Approximately 80% of 5-FU administered is catabolized to CO2, urea, and α-fluro-β-alanine, mainly in the liver. The rate-limiting step in 5-FU catabolism is mediated by the enzyme DPD. The catabolism of 5-FU appears to be an important determinant of normal-tissue toxicity. Although DPD deficiency does not lead to any detectable problems in healthy individuals, patients with a partial or complete deficiency of DPD are at risk for severe toxicity from the drug (see Chap. 3, Sec. 3.6.2 and Chap. 18, Sec. 18.1.3).
Inhibition of TS by FdUMP is dependent on the presence of the cofactor 5,10-methyl-N-tetrahydrofolate, which combines with TS and FdUMP to form a covalent ternary complex. The dissociation rate of this complex is decreased in the presence of excess cofactor, which led to studies showing that addition of the prodrug 5-formyltetrahydrofolate (5-CHOFH4, leucovorin or folinic acid) increased the cytotoxicity of 5-FU (see Fig. 18–8). Clinical studies demonstrate that this combination has greater activity in the treatment of patients with metastatic colorectal cancer than 5-FU alone.
5-FU is used most commonly for treatment of breast and gastrointestinal cancers. It is administered intravenously because bioavailability after oral administration is low and variable. 5-FU is eliminated rapidly from plasma with a t1/2 of a few minutes. This agent demonstrates nonlinear pharmacokinetics because of a saturation of metabolism at higher concentrations, which may be seen when it is given by bolus injection, but not when given by infusion. This difference in pharmaco-kinetic behavior under the 2 conditions of administration may explain why the dose-limiting toxicity is to bone marrow for bolus injection and to mucous membranes if the drug is given over 4 to 5 days by continuous infusion. Rarer toxicities include skin rashes, conjunctivitis, ataxia as a result of effects on the cerebellum, and cardiotoxicity. Prolonged low-dose infusion of 5-FU can be administered with a decrease in some of the above toxicities, but they are associated with changes in sensation as well as with redness and peeling of the skin on the palms of the hands and the soles of the feet, referred to as palmar-plantar erythrodysesthesia, or the hand-foot syndrome. There is limited evidence that this method of 5-FU administration results in improvement of antitumor effects when compared with 5-FU given by bolus injection.
Several oral fluoropyrimidine derivatives have been developed to provide a convenient route of administration and sustained drug exposure. Capecitabine is absorbed unchanged from the gastrointestinal tract, metabolized in the liver by carboxylesterase to 5′-deoxy-5-fluorocytidine (5′-DFCR), which is then converted to 5′-deoxy-5-fluorouridine (5′-DFUR) by cytidine deaminase, mainly located in the liver and tumor tissues. Further metabolism of 5′-DFUR to the cytotoxic moiety 5-FU occurs at the site of the tumor by thymidine phosphorylase, an enzyme present in higher concentrations in tumor cells, resulting in levels considerably higher in tumor tissues compared to normal tissues (Fig. 18–10; Miwa et al, 1998). The toxicity profile of capecitabine is similar to that of prolonged low-dose infusion of 5-FU, with lower frequencies of myelosuppression and stomatitis, but higher incidence of palmar-plantar erythrodysesthesia than intravenous bolus 5-FU. Capecitabine has demonstrated efficacy in breast and colorectal malignancies. Randomized Phase III trials comparing capecitabine to intravenous 5-FU plus leucovorin have shown equivalent efficacy in patients with metastatic colorectal cancer.
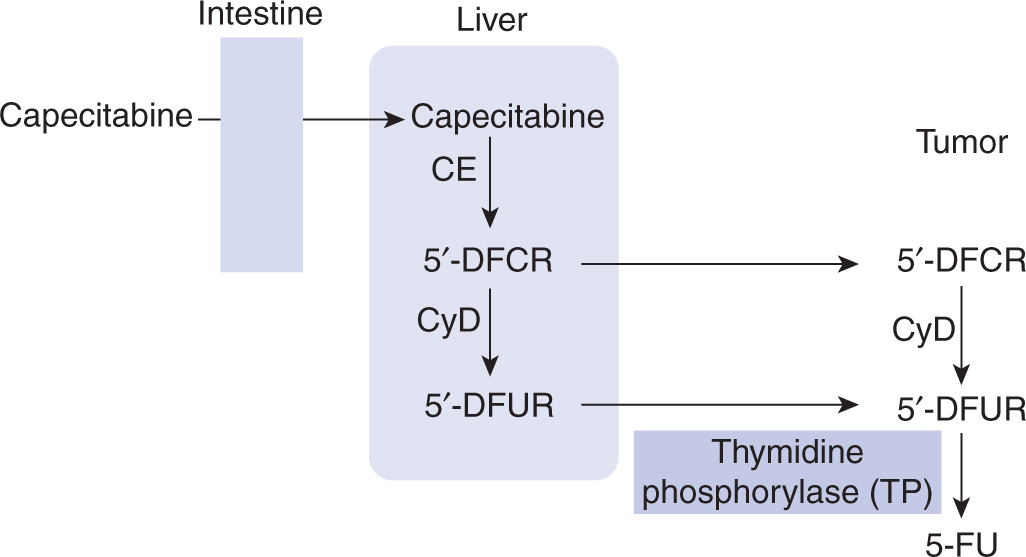
FIGURE 18–10 Enzymatic activation of capecitabine. CE, carboxyesterase; CyD, cytidine deaminase.
Tegafur is a prodrug of 5-FU. Although it is absorbed orally, tegafur needs to be administered at high doses to produce sufficiently high plasma 5-FU levels, and, unfortunately, high doses of tegafur are associated with a high incidence of CNS toxicity. UFT is a combination formulation of uracil and tegafur at a 4:1 ratio. The excess uracil competes with 5-FU for DPD, thereby inhibiting 5-FU catabolism and resulting in sustained and sufficiently high plasma 5-FU levels. S-1 is another oral combination formulation consisting of tegafur, 5-chloro-2,4-dihydroxypyridine (an inhibitor of DPD), and potassium oxonate (an inhibitor of orotate phosphoribosyltransferase, which phosphorylates 5-FU in the intestine resulting in diarrhea) in a 1:0.4:1 ratio. S-1 has similar efficacy, but a better toxicity profile compared to 5-FU (Ajani et al, 2010).
18.3.3 Cytidine Analogs
Nucleoside analogs compete with their physiological counterparts for incorporation into DNA and RNA, thereby exerting their cytotoxicity. Cytosine arabinoside (ara-C) differs from the nucleoside deoxycytidine only by the presence of a β-hydroxyl group on the 2-position of the sugar, so that the sugar moiety is arabinose instead of deoxyribose (Fig. 18–11). Ara-C penetrates cells rapidly by a carrier-mediated process shared with deoxycytidine and is phosphorylated to ara-CTP. Ara-CTP is a competitive inhibitor of DNA polymerase, an enzyme necessary for DNA synthesis, and has similar affinity for this enzyme to the normal substrate dCTP. When ara-CTP binds to this enzyme, DNA synthesis is arrested and S-phase cells may die. In addition, Ara-CTP is incorporated into elongating DNA strands, resulting in DNA chain termination.
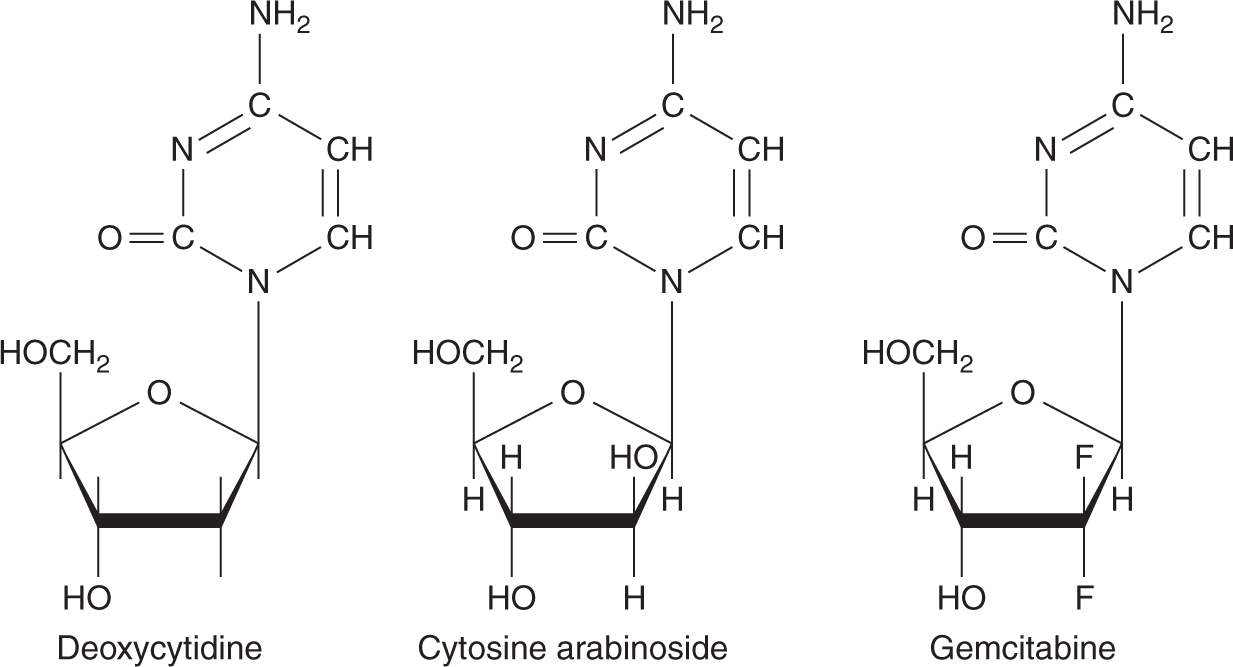
FIGURE 18–11 Structures of deoxycytidine and its analogs.
The availability of ara-CTP for cytotoxic activity depends critically on the balance between kinases that activate the drug and deaminases that degrade it. The activity of these enzymes varies greatly among different types of cells, leading to different rates of generation of ara-CTP. Resistance to the action of ara-C may occur by mutations that lead to deficiency in deoxycytidine kinase or to cells with an expanded pool of dCTP that competes with the active metabolite ara-CTP and regulates enzymes involved in activation and degradation of the drug. Ara-C is specific in its activity for cells synthesizing DNA. Because it is rapidly degraded in plasma with a t1/2 of 7 to 20 minutes, it must be given intravenously by frequent injections or by continuous infusion. The drug is used primarily for treatment of acute leukemia. Myelosuppression and gastrointestinal toxicity are the major side effects, but abnormal behavior and thought processes may also occur after high doses.
Gemcitabine (2′,2′-difluorodeoxycytidine) is a cytosine analog with structural similarities to ara-C (see Fig. 18–11). Unlike ara-C, gemcitabine has activity against a variety of solid tumors. Like ara-C, gemcitabine requires intracellular activation to its triphosphate derivative dFdCTP, which is then incorporated into DNA and inhibits further DNA synthesis. Although gemcitabine is less effective than ara-C in DNA chain termination, a favorable pharmacokinetic characteristic of gemcitabine is the prolonged retention of dFdCTP in cells, with a t1/2 as long as 72 hours. Gemcitabine has other intracellular effects that may contribute to its cytotoxic activity including inhibition of ribonucleotide reductase, stimulation of deoxycytidine kinase, and inhibition of cytidine deaminase. Through inhibition of ribonucleoside reductase, gemcitabine affects DNA synthesis by preventing de novo synthesis of the deoxyribonucleoside triphosphate precursors. Gemcitabine has activity against non-small cell lung cancer, pancreatic cancer, breast cancer, bladder cancer, and nasopharyngeal cancer. Toxicity is primarily myelosuppression, especially, thrombocytopenia.
18.3.4 Purine Antimetabolites
There are 2 adenosine analogs in clinical use (Fig. 18–12). Fludarabine is a derivative that is resistant to deamination and has activity against low-grade lymphoma, chronic lymphocytic leukemia, hairy cell leukemia, and Waldenström macroglobulinemia. After administration, fludarabine is rapidly dephosphorylated to 2-fluoro-ara-A, which then is transported into cells and converted to the active triphosphate derivative. Mechanisms of cytotoxicity include inhibition of DNA polymerase and termination of DNA and RNA replication. Because 2-fluoro-ara-A is excreted primarily unchanged in the urine, dose reduction is necessary in the setting of renal insufficiency. Fludarabine can be administered either intravenously or orally with a t1/2 of approximately 20 hours. The major toxicity of fludarabine is myelosuppression and immunosuppression. Rarely, fludarabine is associated with development of autoimmune disease, such as hemolytic anemia and autoimmune thrombocytopenia, and CNS toxicity.
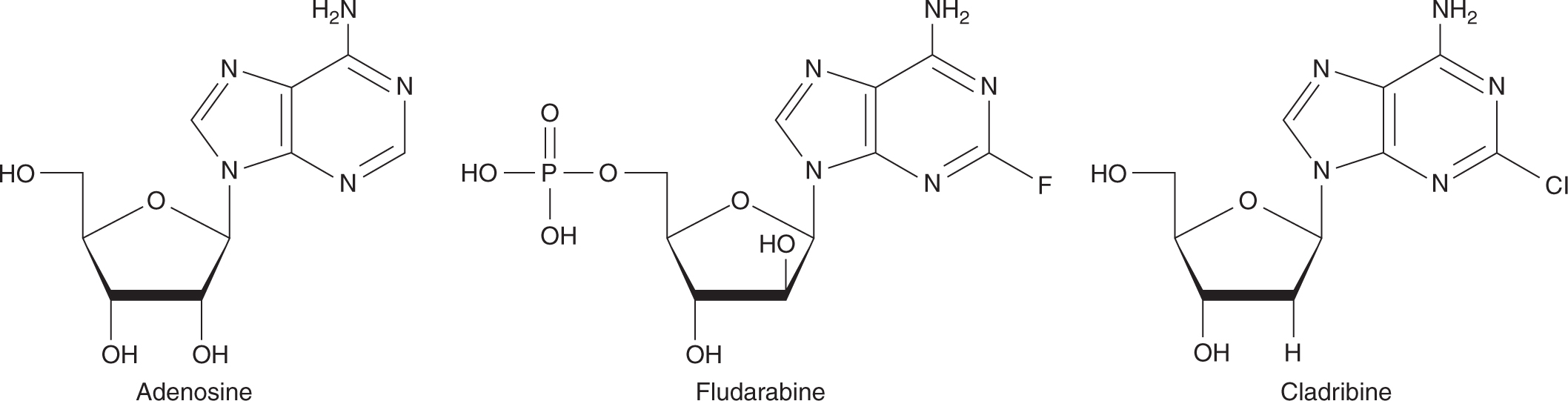
FIGURE 18–12 Structures of adenosine and analogs.
Cladribine (2CdA) is a potent chlorinated adenosine that is also resistant to deamination. It has a similar spectrum of clinical activity and toxicity to fludarabine. Cladribine is administered intravenously with a t1/2 of 5 hours.
18.4 TOPOISOMERASE INHIBITORS
DNA topoisomerases are ubiquitous nuclear enzymes that relax supercoiled double-stranded DNA to allow DNA replication and RNA transcription. Torsional strain is relieved via the formation of a single-strand nick (topoisomerase I) or a double-strand nick (topoisomerase II), followed by swiveling of DNA at the nick(s) and subsequent religation (Fig. 18–13; see also Chap. 19, Sec. 19.2.5). Topoisomerase inhibitors bind to and stabilize the DNA/topoisomerase cleavable complex, thus preventing the religation of DNA strands. Irreversible damage results when an advancing DNA replication fork encounters the drug-stabilized cleavable complex, ultimately leading to lethal double-stranded breaks and cell death.
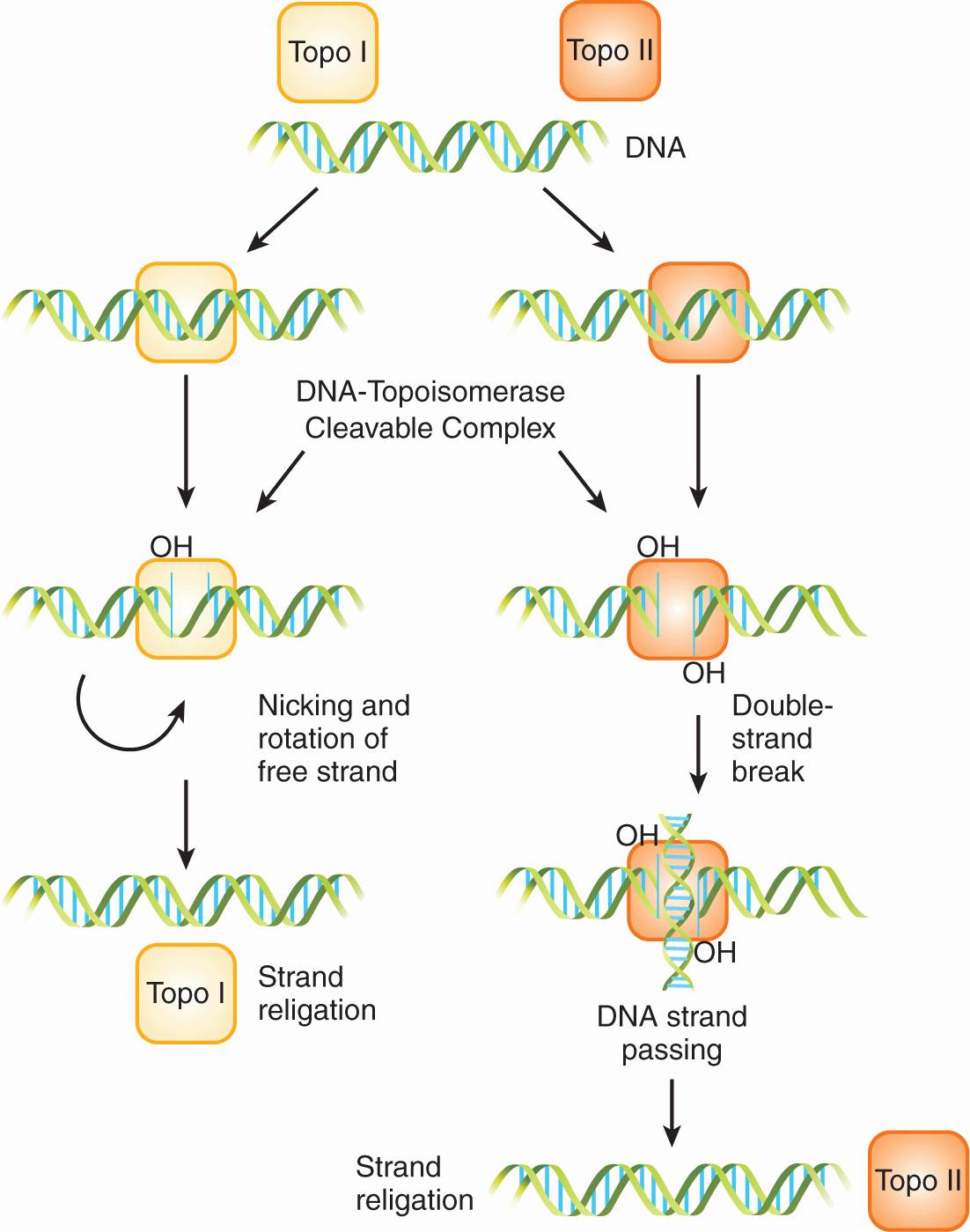
FIGURE 18–13 Topoisomerase (Topo) I and II enzymes form single-strand nicks and double-strand nicks, respectively. Swiveling of supercoiled DNA then occurs at the nick(s), followed by religation to relieve torsional strain.
18.4.1 Topoisomerase I Inhibitors
Camptothecin is an extract from the wood of the Chinese tree Camptotheca acuminate (Fig. 18–14). Camptothecin affects only topoisomerase I activity; cells in S-phase are very sensitive, possibly because the process of DNA replication requires topoisomerase I activity and because the topoisomerase-associated single-strand breaks are converted into double-strand breaks. Initial Phase I studies conducted in the early 1970s were terminated because of poor solubility and severe and unpredictable toxicity, mainly hemorrhagic cystitis and gastroenteritis. Several analogs of camptothecin have been synthesized to improve solubility and reduce toxicity after the elucidation of its mechanism of action. These analogs all have a basic heterocyclic 5-ring structure with a lactone moiety and a hydroxyl moiety on the E ring (see Fig. 18–14). Substitutions on the A ring tend to increase the aqueous solubility while retaining cytotoxicity. All camptothecins can undergo a rapid, reversible, pH-dependent, nonenzymatic hydrolysis of the closed lactone ring to yield an open-ring carboxylate form. The carboxylate form is more water soluble than the lactone; it predominates at physiological pH, but is much less active as an inhibitor of topoisomerase I.
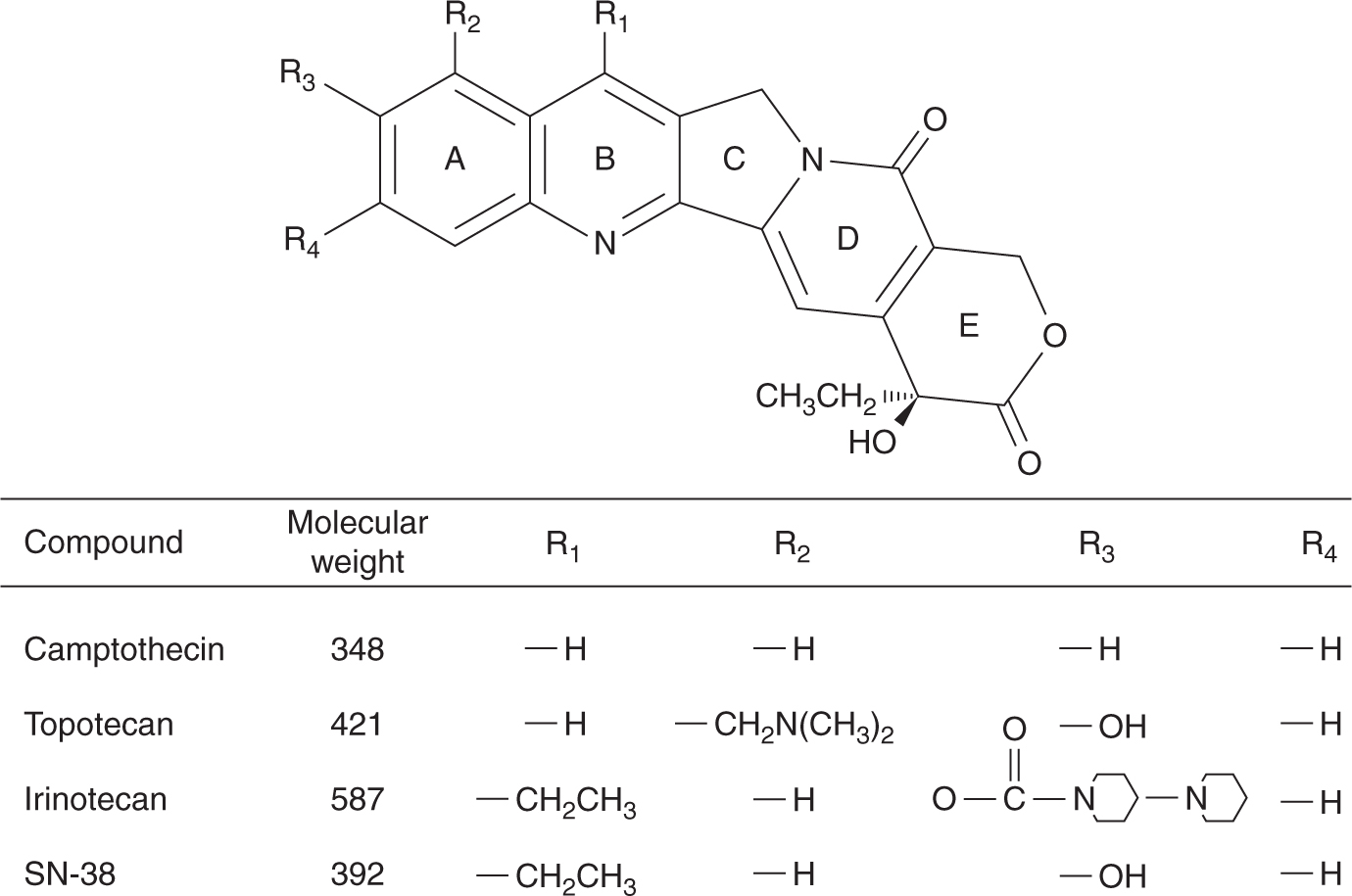
FIGURE 18–14 Structures of camptothecin and its derivatives.
Topotecan can be given orally or intravenously. It does not undergo any appreciable metabolism and is primarily eliminated unchanged by the kidneys. Therefore, dose reduction is required in patients with renal dysfunction. The dose-limiting toxicity is myelosuppression. It is used as treatment for ovarian cancer and small cell lung cancer.
Irinotecan (CPT-11) requires esterification by serum and tissue carboxylesterases to an active metabolite SN-38. SN-38 is subsequently inactivated through glucuronidation by the enzyme uridine diphosphate (UDP)-glucuronosyltransferase 1A1 (UGT1A1) into SN-38G, and excreted into bile and the intestine. Dose-limiting toxicity consists of myelosuppression and diarrhea. Irinotecan can produce an early cholinergic syndrome consisting of abdominal cramps, diarrhea, and diaphoresis that typically occurs acutely during or immediately after infusion, and prompt resolution can be obtained with intravenous or subcutaneous atropine. Patients who experience this reaction may benefit from prophylactic atropine prior to subsequent irinotecan infusions. A second distinct type of diarrhea is associated with irinotecan, typically with a delayed onset. This type of diarrhea tends to be more severe and protracted and is believed to be a result of damage to the gastrointestinal mucosa by SN-38. Severe late-onset diarrhea, especially in the setting of myelosuppression, can be life-threatening and can be managed with high-dose loperamide (Rothenberg et al, 2001). Genetic polymorphism in the UGT1A1 gene can result in reduced activity, and increased levels of SN-38. The most common variant is the UGT1A1*28 variant, which contains 7 TA repeats in the TATA repeat box instead of the normal 6 TA repeats. Patients who are homozygous or heterozygous for the UGT1A1*28 allele have reduced UGT1A1 activity, and are therefore at increased risk of neutropenia and diarrhea, and the irinotecan dose should be reduced (van der Bol et al, 2010). Irinotecan is approved for the treatment of advanced colorectal cancer, where it has been shown to improve survival when used in combination with 5-FU and leucovorin.
18.4.2 Epipodophyllotoxins
Etoposide (VP-16) and teniposide (VM-26) are semisynthetic glycoside derivatives of podophyllotoxin, an antimitotic agent derived from the mandrake plant (Fig. 18–15). Although podophyllotoxin binds to tubulin and inhibits its polymerization, etoposide and teniposide act through inhibition of DNA topoisomerase II. These agents are substrates for P-glycoprotein, and drug resistance can thus be mediated by the multidrug-resistant mechanism (see Chap. 19, Sec. 19.2.3). Etoposide is a component of first-line treatment regimens in small cell lung cancer, testicular cancer, pediatric cancers, and malignant lymphomas. Teniposide has a more limited role and is used mainly in the treatment of childhood leukemia. The effectiveness of etoposide is markedly schedule dependent, with repeated daily doses providing greater activity than a single intravenous administration. Etoposide can be administered orally, with a bioavailability of approximately 50% but considerable interindividual variability. Following intravenous administration, etoposide is eliminated by hepatic glucuronidation, but approximately 40% of the drug is excreted unchanged in the urine. The toxicity of etoposide at standard doses is myelosuppression and hair loss, with other side effects being uncommon. This toxicity profile makes etoposide ideal for high-dose transplantation regimens, and at these higher doses, mucositis becomes dose-limiting. An association between the use of etoposide and a secondary leukemia with a characteristic 11q23 translocation has been described. In contrast to secondary leukemia arising from the use of alkylating agents, which occur with a latency of up to 10 years, those arising from etoposide tend to occur sooner, with a median latent period of approximately 2 to 3 years after drug administration. Most cases of etoposide-induced secondary leukemia are monocytic and myelomonocytic (FAB M-4 and M-5), with no antecedent pancytopenia before the development of frank leukemia.
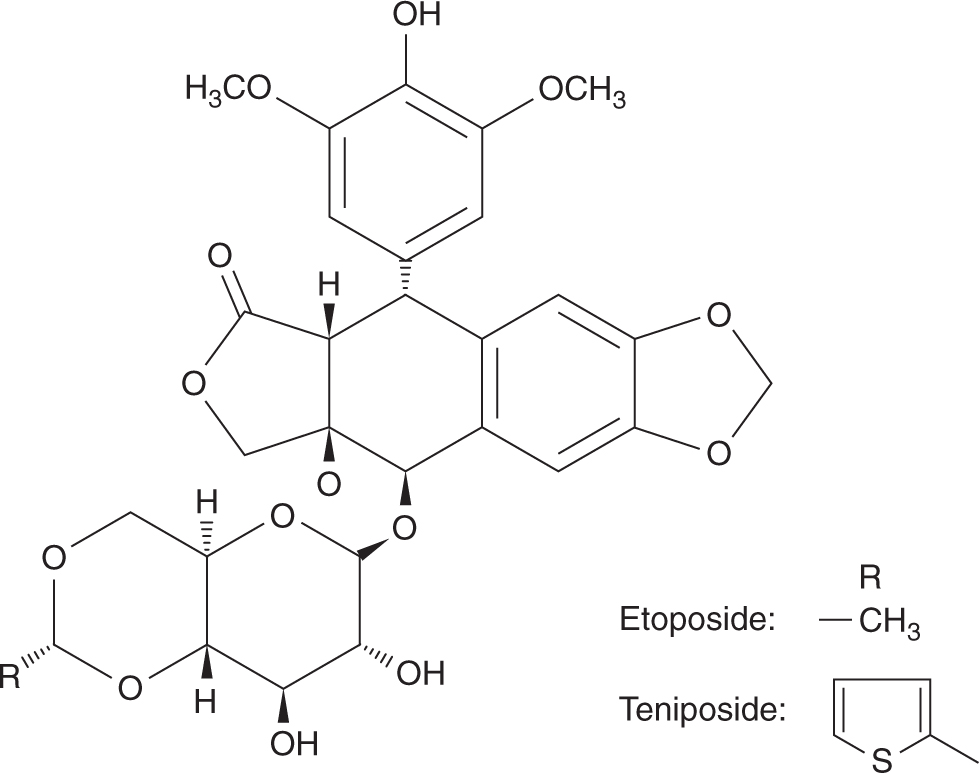
FIGURE 18–15 Structures of etoposide and teniposide. Solid and broken tapered arrows indicate bonds above and below the plane of the rest of the molecule, respectively.
18.4.3 Anthracyclines and Anthracenediones
The original anthracycline, daunorubicin, is a product of a Streptomyces species isolated from an Italian soil sample in 1958. The drug has a high activity against acute leukemia and remains a component of many current protocols for acute myelogenous leukemia. Modifications of the structure of daunorubicin led to the identification of doxorubicin, an analog with greater activity against many solid tumors and one of the most active anticancer drugs in clinical practice (Fig. 18–16). The success of doxorubicin led to synthesis of other analogs, but of the hundreds developed and tested, only 2 are used currently; both have only marginal advantages. Idarubicin can be given orally and has similar activity against acute leukemia. Epirubicin differs from doxorubicin only in its 3-dimensional configuration; it has equal activity and possibly less toxicity.
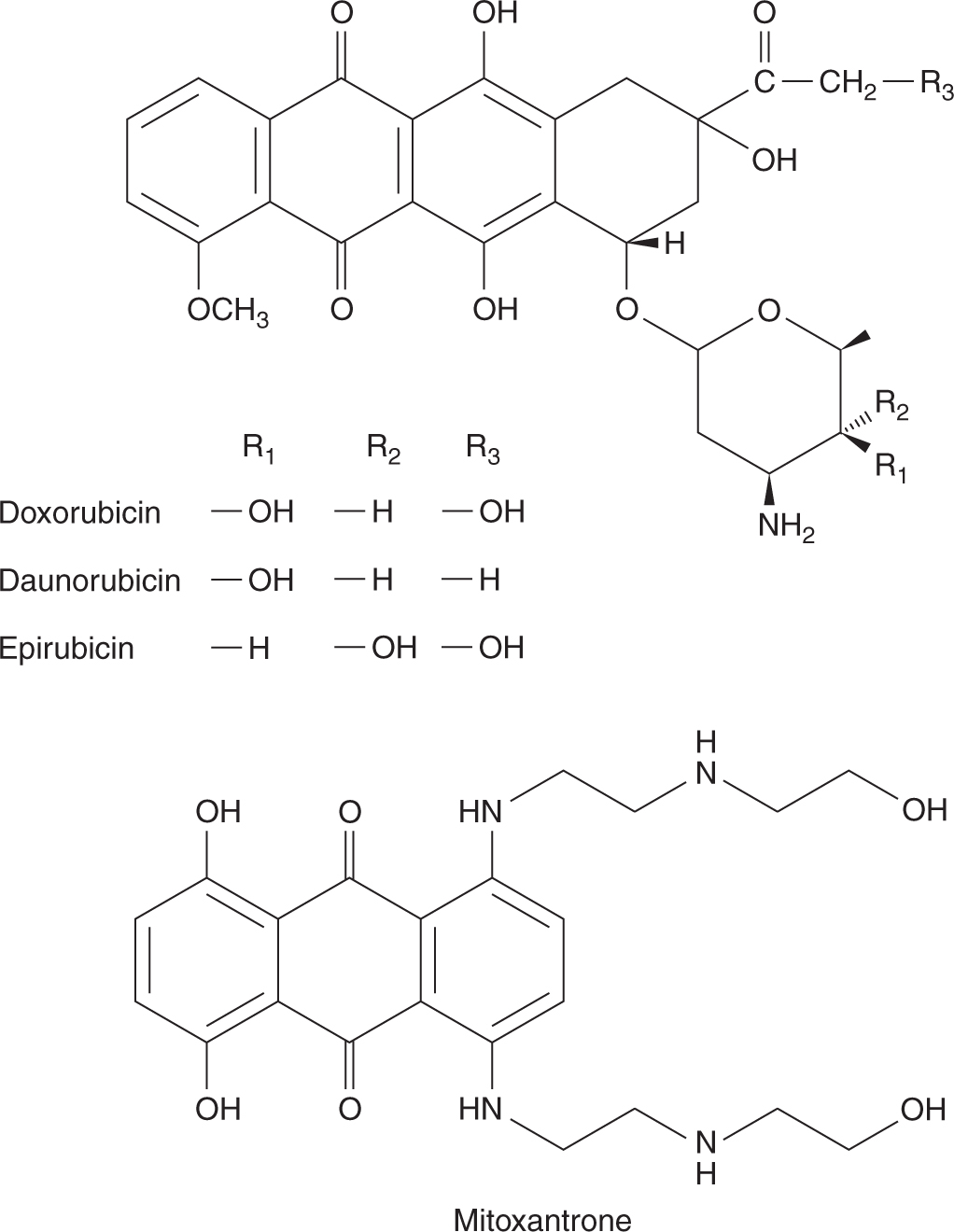
FIGURE 18–16 Structures of doxorubicin, daunorubicin, epirubicin, and mitoxantrone.
Several mechanisms may contribute to the cytocidal effect of doxorubicin and related drugs, including interaction with topoisomerase II, DNA intercalation, formation of free radicals, and effects on the cell membrane. Doxorubicin can interact with topoisomerase II by binding directly with the enzyme and preventing resealing of topoisomerase II-induced DNA cleavage, ultimately leading to cytocidal DNA breaks (see Fig. 18–13). Doxorubicin can also intercalate between base pairs perpendicular to the long axis of the double helix. However, much of the DNA is organized and folded into chromatin and may be protected from this type of drug interaction. Also the concentration of doxorubicin required to intercalate into DNA and to cause inhibition of DNA and RNA polymerase cannot be achieved in vivo without excessive toxicity.
Doxorubicin may undergo metabolism of its quinone ring to a semiquinone radical (ie, a group containing an unpaired electron) that, in turn, reacts rapidly with oxygen to yield superoxide, O2–. The superoxide radical is known to undergo several reactions that can lead to cell death, including oxidative damage of cell membranes and DNA (Gewirtz, 1999). There is evidence that free radical formation accounts for the cardiac toxicity of anthracyclines, but the contribution of free radicals to the killing of cancer cells is uncertain. Resistance to anthracyclines has been associated with an increase in the free radical scavenger system (glutathione and related compounds), but doxorubicin retains toxicity under hypoxic conditions, when superoxide radicals cannot be formed. Another mechanism of resistance is increased drug efflux caused by overexpression of P-170 glycoprotein and other multidrug resistant proteins (Broxterman et al, 2009; see Chap. 19, Sec. 19.2.3).
With the exception of idarubicin, anthracyclines are administered intravenously, because oral absorption is poor. They are widely distributed in the body, with significant binding to plasma proteins and tissue. Doxorubicin is metabolized in the liver to doxorubicinol, which retains some cytotoxic activity, and to several other metabolites; the drug and its metabolites are excreted via the bile. Thus, dosage reduction is required for patients with hepatic dysfunction or biliary obstruction.
The acute toxicities of doxorubicin include myelosuppression, total loss of hair, nausea, vomiting, mucositis, and local tissue necrosis following leakage of drug at the injection site. Repeated administration is limited by a chronic irreversible cardiomyopathy that occurs with increasing frequency once a total dose of approximately 500 mg/m2 has been given. The mechanism of cardiotoxicity is probably related to damage to sarcoplasmic reticulum mediated by the formation of free radicals within cardiac muscle. Patients with preexisting cardiac disease or those who have received mediastinal radiation are more likely to develop this problem. Cardiac toxicity appears to be more related to peak concentration of drug than to overall exposure, so that infusional or repeated lower-dose administration will reduce the chances of its occurrence. Dexrazoxane, an iron-chelating agent, reduces cardiac toxicity by binding free iron and prevents oxidative stress on cardiac tissues. Doxorubicin efficacy is not compromised when dexrazoxane is administered (Marty et al, 2006).
Pegylated liposomal doxorubicin is a formulation where doxorubicin is confined in liposomes that are coated with polyethylene glycol to resist degradation by the endoreticular system. As a result, it has a long half-life, approximately 70 hours, compared to less than 10 minutes when doxorubicin alone is administered intravenously. The long circulating half-life promotes preferential uptake into tumor tissues and reduces uptake by normal tissues. It has been shown that pegylated liposomal doxorubicin reduces cardiac toxicity, myelosuppression, and nausea, but maintains a comparable efficacy with doxorubicin. However, pegylated liposomal doxorubicin increases the risk of skin toxicity, with 20% of patients developing grade 3 hand-foot syndrome (Gordon et al, 2001).
Mitoxantrone is an anthracenedione that differs from the anthracyclines in lacking the sugar and the tetracyclic ring (see Fig. 18–16). It is a synthetic drug with 3 planar rings that intercalates into DNA, with a preference for guanine-cytosine base pairs. It may function as an alternative to anthracyclines in the treatment of acute myelogenous leukemia, breast cancer, and prostate cancer. Although generally less active than doxorubicin, it causes less nausea, vomiting, mucositis, and hair loss, and has found a role in the palliative treatment of cancers of the breast and prostate. Mitoxantrone can also cause cardiac toxicity leading to heart failure. Anthracyclines and mitoxantrone are also (like etoposide) associated with the rare development of secondary leukemia.
18.5 ANTIMICROTUBULAR AGENTS
18.5.1 Vinca Alkaloids
The vinca alkaloids, vinblastine, vincristine, and vinorelbine, are naturally occurring or semisynthetic derivatives from the periwinkle plant. These compounds bind to the protein tubulin and inhibit its polymerization to form microtubules. Microtubules have several important cellular functions, including formation of the mitotic spindle responsible for separation of chromosomes, and structural and transport functions in axons of nerves. Microtubules are in a state of dynamic equilibrium, with continuous formation and degradation from cytoplasmic tubulin. This process is interrupted by vinca alkaloids, and lethally damaged cells may be observed to enter an abortive metaphase and then lyse. However, experiments with synchronized cells have demonstrated that maximum lethal toxicity for vinblastine and vincristine occurs when cells are exposed during the period of DNA synthesis; presumably the morphological expression of that damage is observed in the attempted mitosis.
Vincristine and vinblastine are structurally similar, differing only in a substitution on the central rings (Fig. 18–17). Vinca alkaloids have large volumes of distribution, indicating a high degree of tissue binding, and are eliminated mainly by hepatic metabolism and biliary excretion. Consequently, dose reduction should be considered in patients with elevated bilirubin. The elimination half-lives of vinca alkaloids are approximately 20 hours.
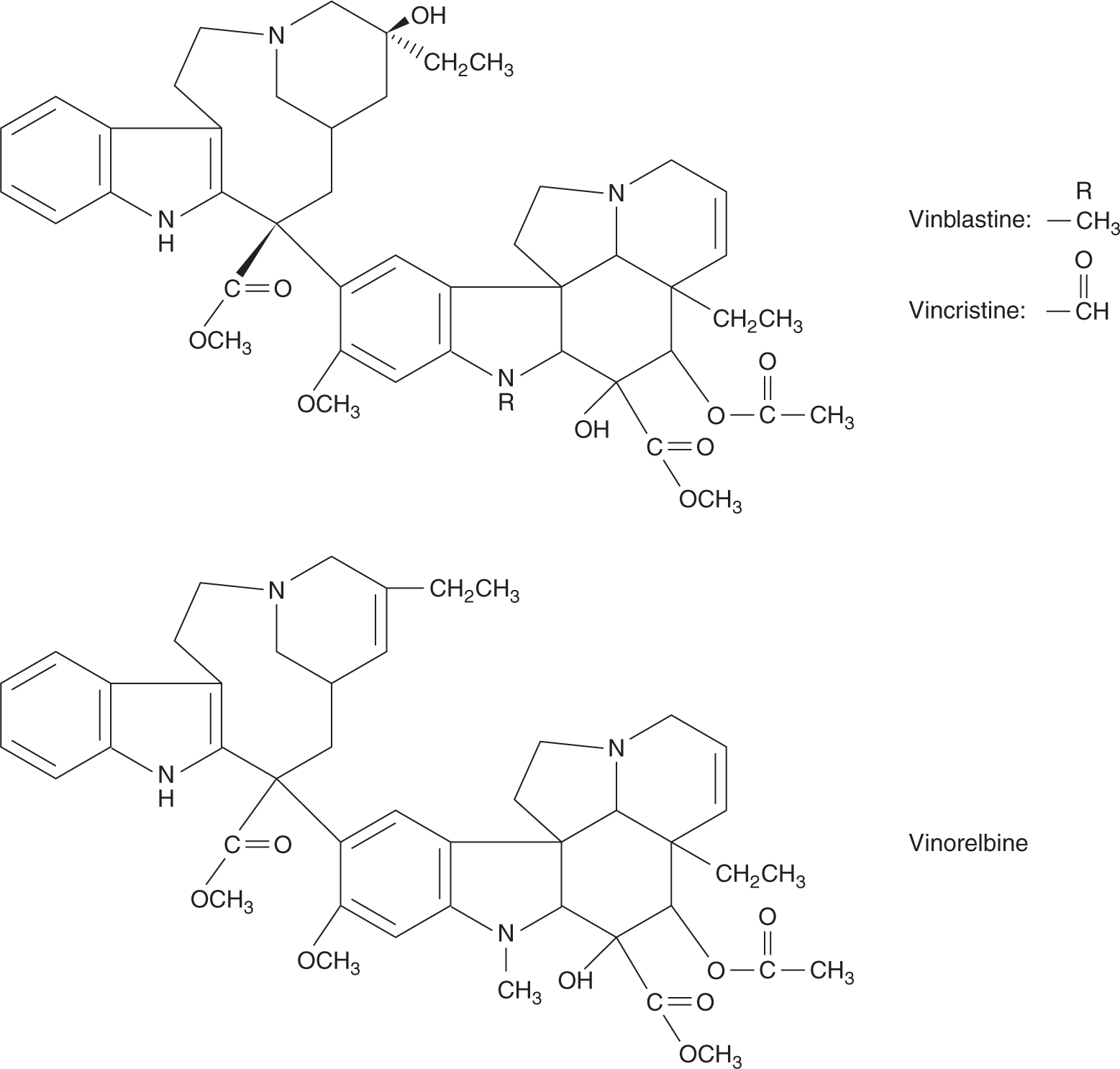
FIGURE 18–17 Structures of vinblastine, vincristine, and vinorelbine.
Despite similarities in their structures, these drugs differ in both their clinical spectra of activity and their toxicities. Vinblastine is an important drug in combination chemotherapy of testicular cancer, whereas vincristine is a mainstay of treatment for childhood leukemia. Both drugs have been combined with other cytotoxic agents to treat lymphomas or various solid tumors. Vinorelbine has activity against non–small cell lung cancer and breast cancer.
Vinblastine causes major toxicity to bone marrow, with some risk of autonomic neuropathy, leading to constipation. The dose of vincristine is limited by its toxicity to peripheral nerves, and this damage relates to the duration of treatment as well as to the total dose of vincristine used. This neurotoxicity probably occurs because of damage to the microtubules in axons. The dose-limiting toxicity of vinorelbine is myelosuppression. Neurotoxicity can occur but is less common than with vincristine, possibly because of a lower affinity for axonal microtubules.
18.5.2 Taxanes
Paclitaxel and docetaxel are plant alkaloids derived from the bark of the Pacific yew tree Taxus brevifolia, and the needles of the European yew tree Taxus baccata, respectively (Fig. 18–18). Paclitaxel was identified as an anticancer drug more than 30 years ago, but its clinical development was hampered by a limited supply, as the Pacific yew tree is relatively rare. Subsequent discovery of an intermediary from the relatively abundant needles of the yew tree made it possible to produce large quantities for clinical evaluation and application.
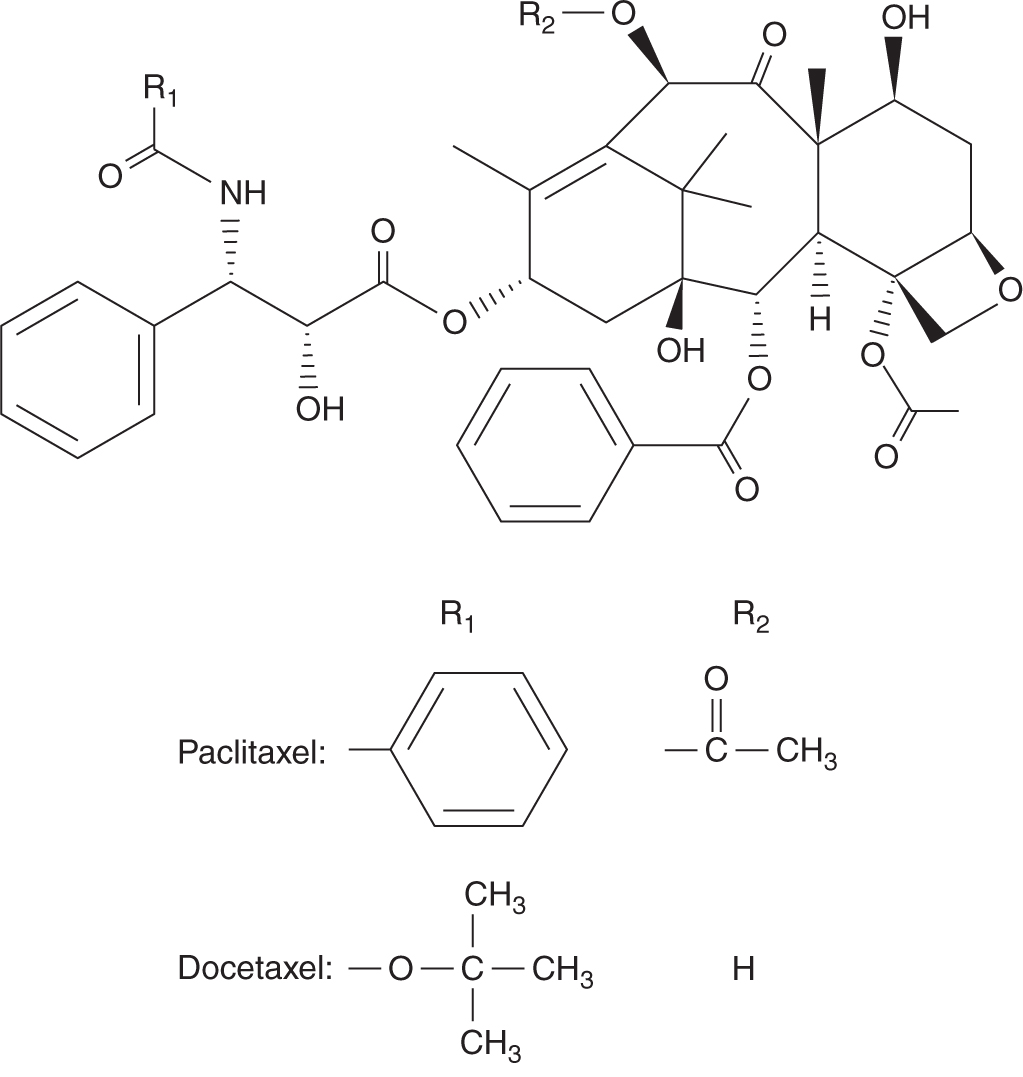
FIGURE 18–18 Structures of paclitaxel and docetaxel.
Taxanes bind to tubulin at a site different from that of the vinca alkaloids. In contrast to the vinca alkaloids, taxanes are believed to inhibit microtubular disassembly, which then prevents the normal growth and breakdown of microtubules that is required for cell division. However, the classic view of vinca alkaloids depolymerizing microtubules and taxanes stabilizing microtubules has been challenged. It has been suggested that both classes of agents may have a similar mechanism of action, involving the modulation of microtubule dynamics (Jordan and Wilson, 2004).
The pharmacokinetics of paclitaxel and docetaxel are characterized by a large volume of distribution with extensive tissue binding, elimination by hepatic metabolism, and elimination half-lives of 10 to 12 hours. As hepatic metabolism to inactive metabolites is mediated through cytochrome P450 enzymes, agents that influence cytochrome P450 can modify the clearance and toxicity of the taxanes; for example, patients on anticonvulsants have demonstrated increased clearance and reduced toxicity.
Paclitaxel and docetaxel share many common toxicities. The dose-limiting toxicity is myelosuppression, mainly neutropenia. For paclitaxel, the severity of neutropenia correlates best with the duration that plasma concentration exceeds a critical threshold level ranging from 0.05 to 0.1 μmol/L. Both drugs can cause hypersensitivity reactions with bronchial constriction, allergic skin reactions such as urticaria, and hypotension. This problem has been reduced substantially by prophylactic treatment with steroids and histamine antagonists. The vehicles in which the taxanes are formulated have been implicated as possible causes of the hypersensitivity reactions, but different vehicles are used for paclitaxel (Cremophor EL) and for docetaxel (polysorbate 80). A sensory peripheral neuropathy can occur with repeated or high-dose administration. Docetaxel can also cause fluid retention and skin and nail changes over time. These drugs have activity against ovarian, breast, lung, and prostate cancers. Alterations of microtubule structures as a result of mutations can result in altered microtubule dynamics or impaired binding of taxanes to tumors, leading to resistance. Increased expression of the multidrug-resistance gene, MDR1 can also result in resistance (Trock et al, 1997).
Nab-paclitaxel is a novel formulation of nanometer-sized albumin-bound paclitaxel particles. It allows preferential delivery of paclitaxel to tumor tissues by taking advantage of the increased delivery of albumin to tumors through receptor-mediated transport and enhanced passage of these particles through larger gaps between capillary endothelial cells. The absence of Cremophor EL reduces toxicity. Nab-paclitaxel is indicated for the treatment of advanced breast cancer.
18.6 MISCELLANEOUS CYTOTOXIC DRUGS
Bleomycin consists of a family of molecules with a complex structure; it is derived from fungal culture, the dominant active component being known as bleomycin A2. Bleomycin causes DNA double-stranded breaks through a complex sequence of reactions involving the binding of a bleomycin–ferrous iron complex to DNA. This binding leads to insertion of the drug between base pairs (intercalation) and unwinding of the double helix. A second step in the formation of DNA strand breaks may involve the reduction of molecular oxygen to superoxide or hydroxyl radicals, catalyzed by the bleomycin–ferrous iron complex. However, like doxorubicin, bleomycin retains some of its lethal activity under hypoxic conditions. Bleomycin may exert preferential toxicity in the G2-phase of the cycle, but also has toxicity for slowly proliferating cells in plateau-phase cell culture. Bleomycin is a large molecule that crosses cell membranes slowly. Once within the cell, it can be activated or broken down by bleomycin hydrolase; cellular sensitivity to bleomycin has been found to correlate inversely with the concentration of this enzyme.
After intravenous injection, most of the administered drug is eliminated unchanged in the urine with a t1/2 of 4 to 8 hours. The major use of bleomycin is in combination with other drugs for the curative therapy of testicular cancer and lymphomas. Bleomycin has little toxicity to the bone marrow but may cause fever, chills, and damage to skin and mucous membranes. The most serious toxicity is interstitial fibrosis of the lung leading to shortness of breath and death of some patients; its incidence is related to cumulative dose, age, renal function, and the use of other agents that may damage the lung, such as high oxygen supplementation or radiation therapy.
Mitomycin C is derived from a streptomyces species and is a quinine-containing compound that requires activation to an alkylating metabolite by reductive metabolism. Because of the requirement for reductive metabolism, the drug is more active against hypoxic than aerobic cells, at least in tissue culture. Mitomycin C causes delayed and rather unpredictable myelosuppression. More seriously, the drug can produce kidney failure through a hemolytic-uremic syndrome, which is usually fatal and is probably caused by small-vessel endothelial damage. Another potential lethal effect is interstitial lung disease with progression to pulmonary fibrosis. It is sometimes instilled into the bladder by a catheter to treat superficial bladder cancer and is also used with radiation therapy to treat cancer of the anal canal.
18.7 MOLECULAR-TARGETED AGENTS
Advances in molecular biology have provided a better understanding of intricate cellular pathways that are critical to tumor formation and growth. New classes of anticancer drugs have been developed that target aberrant expression or alterations in molecular pathways rather than DNA, as is the case for most drugs discussed above. Molecular-targeted agents offer greater specificity and different toxicity as these aberrant expressions or alterations are more common in tumors than in normal tissues (see Chap. 17, Sec. 17.3).
18.7.1 Inhibitors of Angiogenesis
A rate-limiting step in tumor growth is angiogenesis. Without neovascularization, tumor growth is limited and some cells in the tumor become hypoxic, resulting in increased production of proangiogenic factors (see Chap. 11, Sec. 11.4; Folkman, 1971). A dominant factor that stimulates angiogenesis is vascular endothelial growth factor (VEGF). There are 5 isoforms of VEGF, and they bind to 3 main subtypes of cell surface receptors (VEGFR). Binding of VEGF to VEGFR leads to dimerization, phosphorylation of intracellular tyrosine kinases, and subsequent activation of downstream signal cascades. There are 2 approaches to inhibit the VEGF signaling pathway: antibodies that bind to circulating VEGF to prevent its binding to VEGFR, and small molecules to inhibit intracellular tyrosine kinase activity.
Bevacizumab is a humanized monoclonal antibody against VEGF-A. It has limited anticancer effects when administered alone, but has shown activity when given together with cytotoxic chemotherapies in the treatment of advanced colorectal cancer, and other cancers (Hurwitz et al, 2004; Escudier, Pluzanska et al, 2007). It is hypothesized that agents such as bevacizumab cause a temporary normalization of poorly formed and more permeable tumor vasculature, leading to increased blood flow to tumor and reduced interstitial pressure. As a result, there may be enhanced delivery of cytotoxic chemotherapies to tumor sites. Bevacizumab is administered at 2- to 3-week intervals because the elimination t1/2 is approximately 20 days. Adverse events associated with bevacizumab include hypertension, bleeding, proteinuria, would healing complications, thromboembolic events, and perforation of the gastrointestinal tract. A rare syndrome of reversible posterior leukoencephalopathy has been reported in patients receiving bevacizumab (Marinella and Markert, 2009).
Several inhibitors of membrane-based tyrosine kinases have activity against human cancer (Fig. 18–19; see Chap. 17, Sec. 17.3.1). Sunitinib inhibits multiple receptor tyro-sine kinases, including those associated with VEGFR, platelet-derived growth factor receptor (PDGFR), and c-kit (Mendel et al, 2003). It is an oral agent and is usually administered daily for 4 out of 6 weeks, although its t1/2 ranges from 40 to 60 hours. Sunitinib undergoes hepatic metabolism via cytochrome P450 system, mainly CYP3A4; hence, the dosage of sunitinib requires adjustment in patients taking strong CYP3A4 inhibitors. Common adverse effects of sunitinib include fatigue, diarrhea, hypertension, thyroid dysfunction, and hand-foot syndrome. Sunitinib is approved for the treatment of advanced renal cell carcinoma and gastrointestinal stromal tumors (GISTs) (Demetri et al, 2006; Motzer et al, 2007).
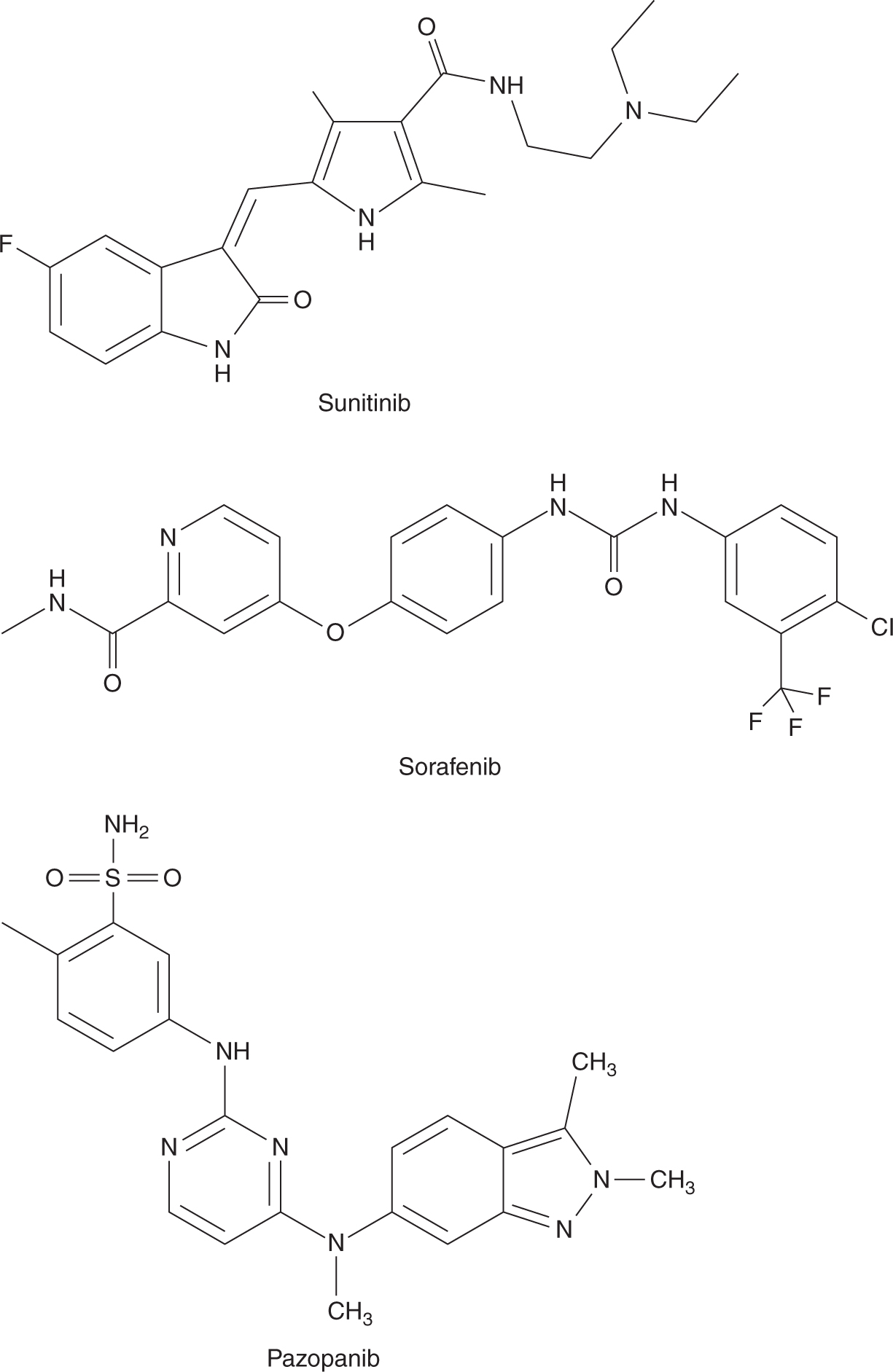
FIGURE 18–19 Structures of sunitinib, sorafenib, and pazopanib.
Sorafenib inhibits tyrosine kinases associated with cell-surface receptors, including VEGFR, PDGFR, and c-kit. It also inhibits intracellular raf kinases. Approximately 40% of sorafenib is absorbed when administered orally, and its bio-availability is further decreased when it is taken together with a high-fat meal. Sorafenib is administered twice daily on a continuous basis, even though its t1/2 is approximately 40 hours. Adverse events caused by sorafenib are similar to those associated with sunitinib. Sorafenib is used in treating advanced hepatocellular carcinoma and renal cell carcinoma (Escudier et al, 2007; Llovet et al, 2008).
Like sunitinib and sorafenib, pazopanib inhibits tyrosine kinases associated with multiple cell-surface receptors. It is given orally and its bioavailability is increased when given together with food or if the tablet is crushed. Pazopanib is administered once daily continuously and its t1/2 is approximately 30 hours. Pazopanib is also used for treating advanced renal cell carcinoma (Sternberg et al, 2010).
Thalidomide (Fig. 18–20) was first developed in 1950s as treatment for pregnancy-associated nausea and vomiting. However, it was soon withdrawn from market with the recognition that thalidomide use was associated with teratogenicity leading to a defect in the development of limbs. The interest in thalidomide and related drugs was rekindled in the 1980s because of anecdotal reports of activity in a number of conditions such as erythema nodosum leprosum (ENL). Several studies confirmed that thalidomide has activity in advanced multiple myeloma (Singhal et al, 1999). Although thalidomide is thought to inhibit angiogenesis, its precise mechanism of anticancer effects is not known. Because of its potential teratogenic effects, thalidomide can only be prescribed under strict guidance, particularly for female patients who are premenopausal and male patients whose partner is of childbearing age. Thalidomide is orally administered and undergoes nonenzymatic hydrolysis to form multiple inactive metabolites. Side effects include fluid retention, fatigue, skin toxicities, thromboembolic events, and peripheral sensory neuropathy.
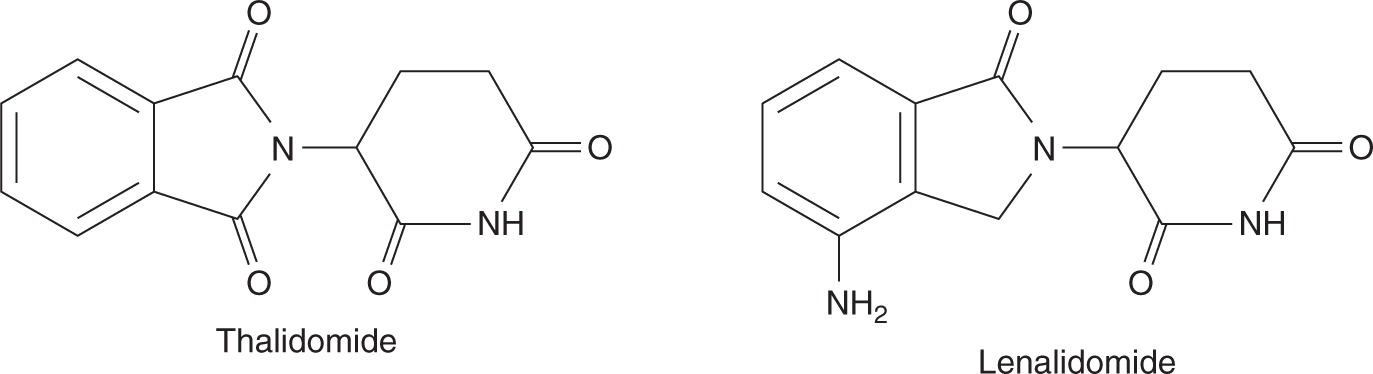
FIGURE 18–20 Structures of thalidomide and lenalidomide.
Lenalidomide is an analog of thalidomide (see Fig. 18–20), and also has activity in multiple myeloma (Badros, 2012). Unlike thalidomide which undergoes extensive metabolism, lenalidomide is excreted mostly unchanged in urine. Patients with renal dysfunction require dose reduction. The t1/2 of lenalidomide is approximately 3 hours in patients with normal renal function, and 9 hours in patients with moderate-to-severe renal impairment. It is readily absorbed after oral administration and is given daily for 21 days followed by 7 days of rest. Lenalidomide is also teratogenic, and its use must be strictly monitored in women of child-bearing potential similar to thalidomide. Lenalidomide is also associated with increased risks of thromboembolic events.
18.7.2 Inhibitors of Epidermal Growth Factor Receptors
The epidermal growth factor receptor (EGFR) is composed of an extracellular ligand-binding domain, a transmembrane region, and an intracellular domain with tyrosine kinase activity. Activation of the EGFR pathway initiates cascades of intracellular RAS-RAF-MAPK (mitogen-activated protein kinase) and phophatidylinositol-3 kinase (PI3K)-AKT signaling pathways, which, in turn, stimulate transcription of genes that promote cell proliferation and survival (see Chap. 8, Sec. 8.2). Aberrant signaling through the EGFR pathway induces tumorigenesis with cell proliferation, angiogenesis, invasion, and metastasis, as well as inhibition of apoptosis. There are 2 approaches to inhibit EGFR-related pathways: antibodies against the cell surface receptors, or small molecules to inhibit the intracellular tyrosine kinases. Inhibiting EGFR causes diarrhea and skin toxicities. Skin toxicities initially manifest as an acneiform skin rash involving the face and upper trunk, and over time progress to dry skin, fissures, and infection around nail beds (Lacouture et al, 2010).
Cetuximab is a chimeric monoclonal antibody against EGFR, whereas panitumumab is a fully humanized monoclonal antibody against EGFR. The half-lives of cetuximab and panitumumab are approximately 7 days, and they are administered intravenously weekly or every 2 weeks. Large, randomized trials have confirmed that cetuximab and panitumumab can prolong overall survival and progression-free survival in patients with advanced colorectal cancer compared to best supportive care. The Ras protein is involved early in the EGFR signaling pathway, and mutations in the Ras gene can result in constitutive activation, thereby abrogating blockage of the EGFR pathway. Up to 40% of patients with advanced colorectal cancer may carry a mutated Ras gene, and multiple studies have confirmed that anti-EGFR antibodies are only effective in patients with wild-type KRAS (Amado et al, 2008; Karapetis et al, 2008). Other mutations in the EGFR signaling pathway, such as BRAF, may affect therapeutic response, and these are active areas of investigation. Hypomagnesemia is a common side effect with EGFR inhibition since EGFR is also involved in regulating magnesium reabsorption in renal tubules. Cetuximab has a slight risk of hypersensitivity reaction, which can be prevented by antihistamines and corticosteroids. Recent data suggest that prophylactic measures, such as skin moisturizers, sunscreen, topical steroids, and oral antibiotics (doxycycline/minocycline) can reduce the severity of skin toxicities without affecting treatment efficacy.
Erlotinib and gefitinib are 2 small-molecule inhibitors of EGFR-associated tyrosine kinases (Fig. 18–21). Both agents are administered orally. Food increases the bioavailability of erlotinib from 60% to almost 100%, whereas gefitinib is absorbed slowly with peak concentrations at 3 to 7 hours after drug administration and the bioavailability is approximately 60%. Both erlotinib and gefitinib are extensively metabolized, primarily via CYP3A4, and metabolites are excreted through the biliary system. Their half-lives range from 30 to 40 hours, and both are administered on a continuous basis. Gefitinib is indicated for patients with advanced non–small cell lung cancer after progression on platinum or docetaxel therapies. A large, randomized study showed that gefitinib is superior to carboplatin-paclitaxel as an initial treatment for pulmonary adenocarcinoma among nonsmokers or former light smokers in East Asia (Mok et al, 2009). Furthermore, the presence in the tumor of a mutation of the EGFR gene is a strong predictor of a better outcome with gefitinib. In addition to non–small cell lung cancer, erlotinib provides a small survival advantage when used as first-line treatment for locally advanced or metastatic pancreatic cancer in combination with gemcitabine (Moore et al, 2007).
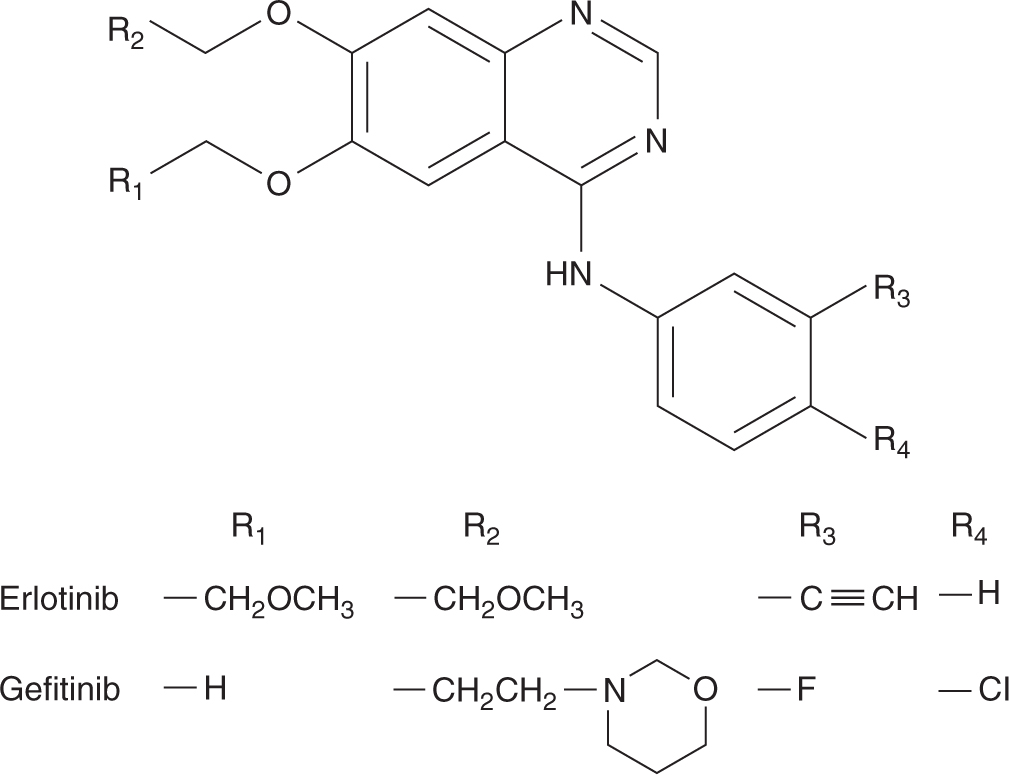
FIGURE 18–21 Structures of erlotinib and gefitinib.
The HER-2 oncogene encodes a transmembrane receptor with tyrosine kinase activity belonging to the EGFR family of receptors (King et al, 1985). Amplication of HER-2 or overexpression of its protein product occurs in approximately 20% of patients with breast cancer, and is a strong predictor of therapeutic response to anti–HER-2 agents (see Chap. 20, Sec. 20.3.3). Trastuzumab is a monoclonal antibody against HER-2, and is indicated as adjuvant therapy for HER-2 over-expressing early breast cancer or as therapy for metastatic breast cancer alone or in combination with other drugs (Baselga et al, 2005; Robert et al, 2006; Romond et al, 2005). Trastuzumab is associated with reduction in left ventricular ejection fraction and, less commonly, heart failure. Patients with preexisting heart disease, previous radiation, and concomitant administration or prior exposure to anthracyclines are at increased risks of heart failure with trastuzumab therapy. In addition, there is an increased risk of pulmonary toxicity. Other antibodies directed against HER-2, such as pertuzumab, and trastuzumab emtansine (TDM-1), in which the cytotoxic agent emtansine is linked to trastuzumab, may further improve therapy for HER-2-positive breast cancer (Baselga et al, 2012; Verma et al, 2012).
Lapatinib is a small-molecule inhibitor of tyrosine kinase activity associated with HER-2. It is administered orally and its bioavailability is improved with high-fat food (Ratain and Cohen, 2007). It undergoes extensive metabolism mediated by CYP3A4 and CYP3A5. It is indicated with capecitabine for patients with HER-2 overexpressing advanced breast cancer. Like trastuzumab, lapatinib causes cardiac and pulmonary toxicities. In addition, lapatinib is occasionally associated with severe hepatotoxicity.
18.7.3 Mammalian Target of Rapamycin Inhibitors
The mammalian target of rapamycin (mTOR) pathway is downstream of the PI3KAkt pathway which is regulated by PTEN (Wullschleger et al, 2006; see Chap. 7, Sec. 7.6.2). The mTOR pathway is aberrantly activated in many cancers. The first agent targeting the mTOR pathway, rapamycin (sirolimus), is a macrocyclic isolated from Streptomyces hygroscopicus in a soil sample from Britain. It binds to an intracellular protein, FKBP-12, and forms a stable complex to inhibit mTOR kinase activity. Sirolimus is an immunosuppressant and is used to prevent organ rejection in transplant recipients. Several analogs of rapamycin have been developed (Fig. 18–22). Temsirolimus is an intravenously administered rapamycin analog. It is metabolized via CYP3A4 to its major active metabolite, sirolimus. The dose of temsirolimus should be reduced in patients with liver dysfunction. Common side effects include skin toxicity; metabolic disorders such as hyperglycemia, hypercholesterolemia, and hyperlipidemia; gastrointestinal toxicities, such as mucositis, nausea, and vomiting; and hematological toxicities, mainly anemia and lymphopenia. Uncommon but potentially life-threatening toxicities include hypersensitivity reaction and pneumonitis. Temsirolimus is used in the treatment of advanced renal cell cancer (Hudes et al, 2007). Because of the relatively long t1/2 of sirolimus (approximately 50 hours), temsirolimus is given weekly.
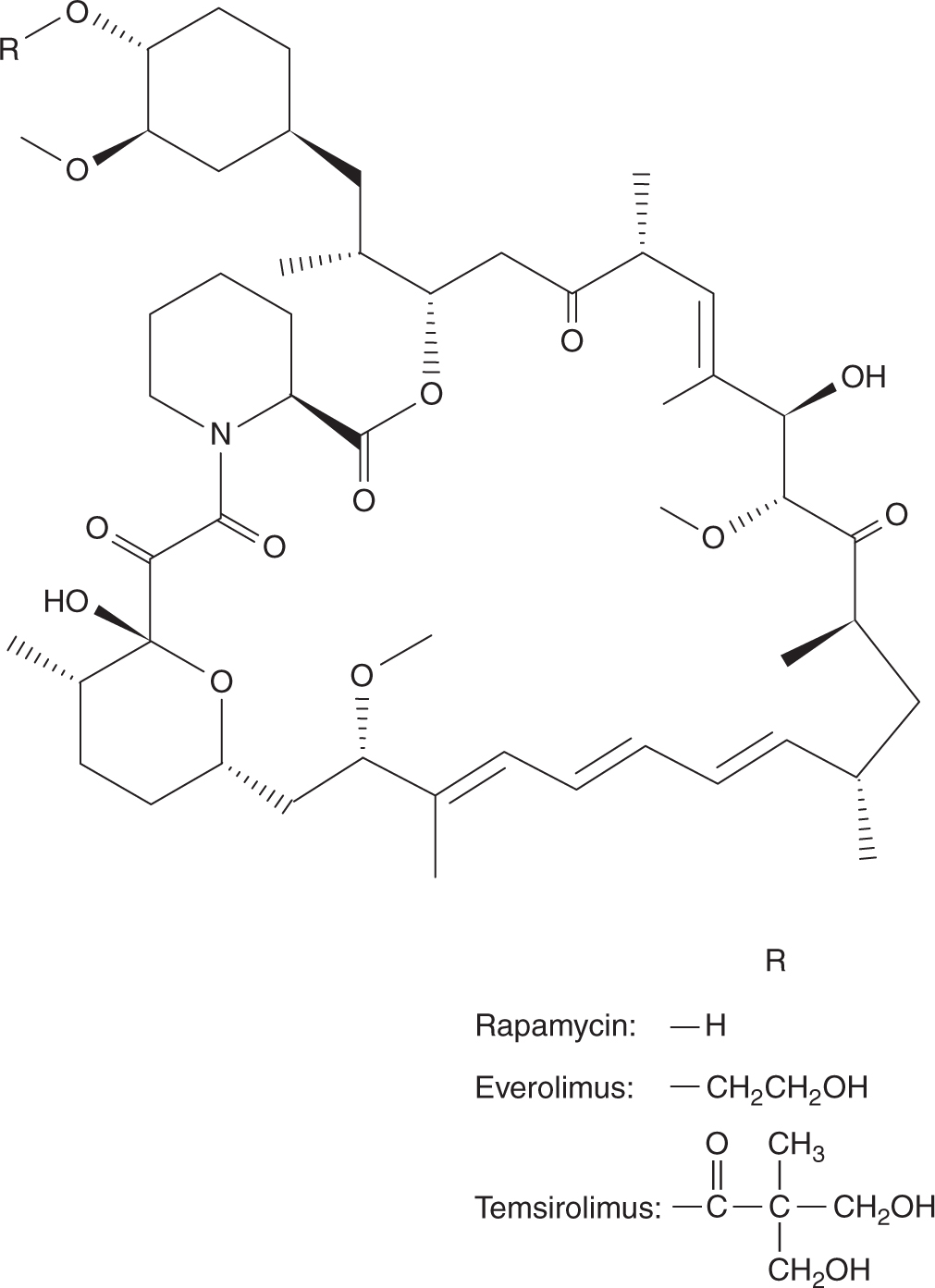
FIGURE 18–22 Structures of rapamycin and analogs.
Unlike temsirolimus, everolimus is rapidly absorbed orally with a bioavailability of approximately 30% and its t1/2 is approximately 30 hours. Everolimus is administered daily and its dose should be reduced in patients with moderate to severe hepatic dysfunction. In a Phase III study, everolimus significantly prolonged progression-free survival for patients with advanced renal cell carcinoma who had progressive disease on or shortly after therapy with VEGF tyrosine kinase inhibitors (Motzer et al, 2008). Common side effects include metabolic disorders (hyperglycemia, hypercholesterolemia, and hyperlipidemia), hematological toxicities (anemia and lymphopenia), and gastrointestinal toxicities, such as mucositis, nausea, and diarrhea. Everolimus is also associated with pulmonary toxicity.
18.7.4 Miscellaneous Agents
Imatinib is an orally available tyrosine kinase inhibitor. It inhibits the tyrosine kinase activity of the constitutively active fusion protein BCR-ABL arising from the Philadelphia (Ph) chromosome (see Chap. 7, Sec. 7.5.1) of chronic myelogenous leukemia (CML) through competitive inhibition at the adenosine triphosphate (ATP) binding site. The discovery and the successful treatment of CML with imatinib ushered in the era of cancer therapy with drugs targeting signaling pathways (Druker et al, 2001). Resistance to imatinib results from mutations that alter amino acids at the imatinib binding site or prevent BCR-ABL from achieving the inactive conformation that is required for imatinib binding (Shah et al, 2002). Imatinib also inhibits the tyrosine kinase activity of c-KIT (CD-117) which is overexpressed in 80% of GISTs and a related tyrosine kinase receptor, PDGFR. Treatment with imatinib leads to rapid and sustained clinical effects and has revolutionized the management of CML and GIST (Blanke et al, 2008). Although all GIST patients with KIT mutation are likely to respond to imatinib therapy, those with an exon 11 mutation have a more prolonged response than those with an exon 9 mutation. However, patients with exon 9 mutations can still respond with higher doses of imatinib. Imatinib is rapidly and completely absorbed, and undergoes hepatic metabolism via CYP3A4 enzymes. Imatinib is generally well tolerated with mild nausea, diarrhea, fluid retention, muscle cramps and fatigue.
Dasatinib inhibits BCR-ABL kinases among others, and has shown activity in patients who are resistant or intolerant to imatinib therapy (Apperley et al, 2009; Kantarjian et al, 2010). Dasatinib undergoes extensive metabolism and is mainly excreted through the biliary system.
Nilotinib is another inhibitor of multiple kinases, including BCR-ABL, c-KIT, and PDGFR kinases (Saglio et al, 2010). It is designed to fit into the ATP binding site of BCR-ABL with higher affinity than imatinib. It is also active in the presence of multiple mutations that lead to resistance to imatinib. Nilotinib is usually taken on an empty stomach because food may increase its bioavailability, and patients with hepatic dysfunction require dose reduction because nilotinib undergoes extensive metabolism. Nilotinib can cause changes in the electrocardiogram and electrolyte imbalance, such as hypokalemia and hypomagnesemia, which require correction before initiation of therapy. It is contraindicated in patients with long QT syndrome.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


