Discovery and Evaluation of Anticancer Drugs
17.1 INTRODUCTION
Chemotherapy is used primarily as (a) the major treatment modality for a few types of malignancies, such as Hodgkin disease and other hematopoietic cancers, acute leukemia in children, and testicular cancer in men; (b) palliative treatment for many types of advanced cancers; and (c) adjuvant treatment before, during, or after local treatment (surgery and/or radiotherapy) with the aim of both eradicating occult micrometastases and of improving local control of the primary tumor. Such treatments usually involve a combination of drugs. The most important factors underlying the successful use of drugs in combination are (a) the ability to combine drugs at close to full tolerated doses with additive effects against tumors and less-than-additive toxicities to normal tissues, and (b) the expectation that drug combinations will include at least 1 drug to which the tumor is sensitive. Since the first documented clinical use of chemotherapy in 1942, when the alkylating agent nitrogen mustard was used to obtain a brief clinical remission in a patient with lymphoma, about 45 cytotoxic drugs or biological agents (excluding hormonal agents) have been licensed for use in North America, and several more are undergoing clinical trials. The pharmacology of many of these agents is described in Chapter 18. In recent years, new types of anticancer agents have been developed, including monoclonal antibodies (eg, rituximab, trastuzumab, and bevacizumab) that target cell-surface receptors, and small molecules that interact with various cell signaling pathways (eg, imatinib). These newer agents represent a substantial shift in emphasis in anticancer drug therapy. In contrast to conventional cytotoxic agents, which usually target proliferating cells and interact with DNA, the newer agents target specific metabolic pathways that interfere with various functions of the cell, including those that promote cell division (trastuzumab, imatinib) or contribute to immune-mediated cellular damage (rituximab). Other agents, such as those that inhibit angiogenesis (eg, bevacizumab; see Chap. 11, Sec. 11.7.1) act indirectly to inhibit tumor growth.
This chapter deals with the scientific basis of cancer drug discovery. It introduces the concepts of how cancer drug targets are identified and some of the approaches used to discover and design new drugs. The chapter also discusses the biological properties of important anticancer drugs, experimental methods used to determine their activity, their toxicity to normal tissues and the concept of therapeutic index, and the biological basis of using drugs in combination and with radiotherapy. Chapter 18 addresses the pharmacology of anticancer drugs and Chapter 19 describes the many causes of drug resistance.
17.2 STRATEGIES TO DEVELOP ANTICANCER DRUGS
17.2.1 Gene Expression and Identification of Potential Targets for Anticancer Drugs
Although many chemotherapy drugs were developed by observing the ability of compounds to kill cancer cells in culture and in experimental animals without knowledge of a specific molecular target, modern drug discovery begins typically with the identification of a therapeutic target. To identify novel targets, a variety of strategies can be employed, including a survey of the expression of genes within a cancer cell to identify dysregulated genes, gene families, or pathways. DNA microarray analysis can measure the expression of messenger RNA (mRNA) in samples derived from cell lines or primary tissues of patients (Fig. 17–1). Microarray analysis is described in detail in Chapter 2, Section 2.7. The essence of the procedure is that it detects the presence of mRNA transcripts and assesses their relative abundance in the cancer tissue analyzed. Using bioinformatics, computer software, and advanced statistical analysis, patterns of dysregulated genes and pathways can be identified that can be targeted to develop new cancer drugs. How drugs are developed to inhibit these new targets is discussed in Section 17.2.4.
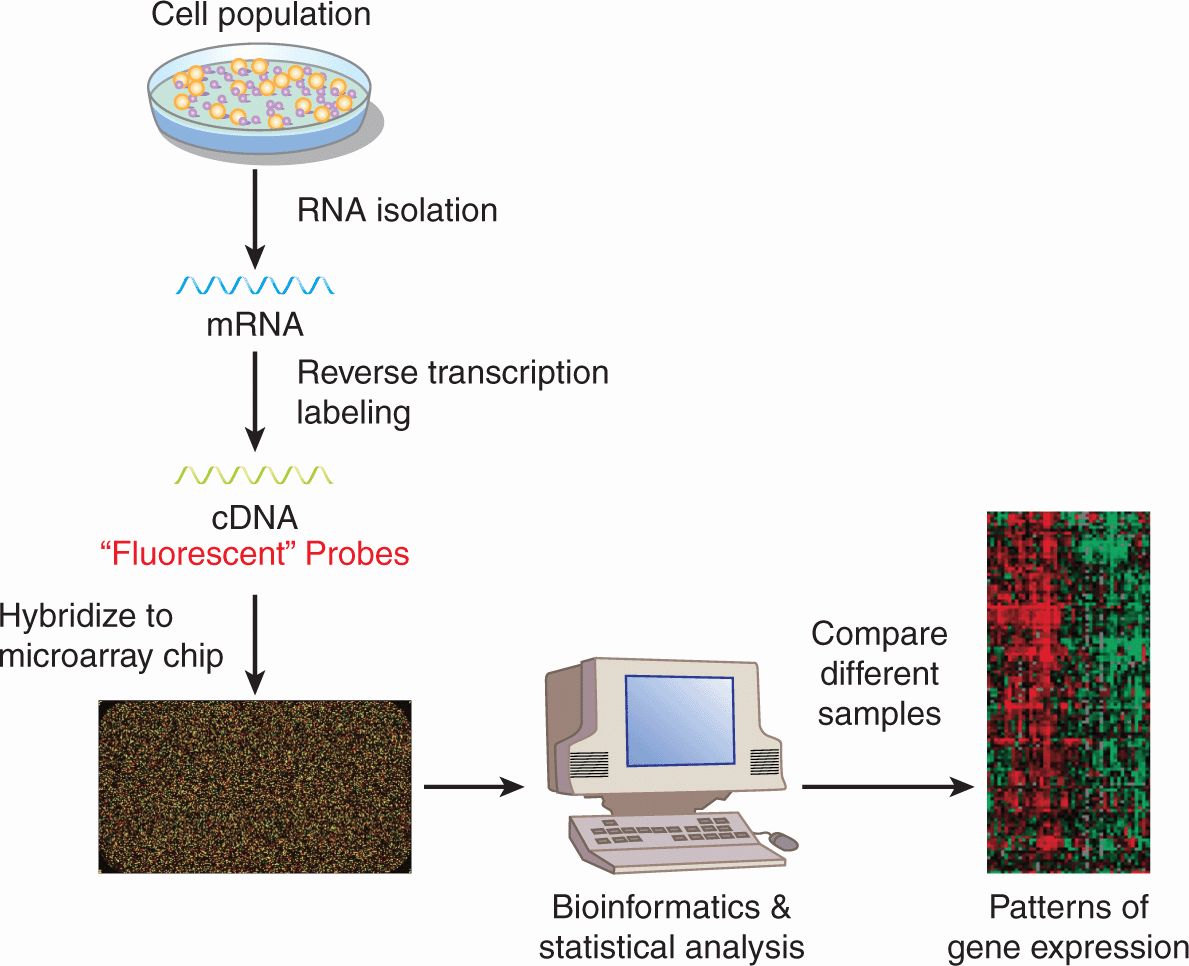
FIGURE 17–1 Microarray analysis to study gene expression. mRNA is isolated from cells and is reverse transcribed to complementary DNA (cDNA), which is fluorescently labeled. The cDNA is then hybridized to a microarray chip into which cDNA probe sequences representing tens of thousands of genes in the human genome have been embedded. After hybridization, the chip is washed to remove cDNA that has been bound nonspecifically, and fluorescent signal on each spotted gene is measured. The intensity of the fluorescent signal is proportionate to that amount of bound labeled cDNA and thus the amount of target mRNA in the cell. Using bioinformatics, computer software, and advanced statistical analysis, the abundance of mRNA sequences can be compared between groups of cells. These groups of cells can represent different cell lines, tissues, or treatments. As shown in this example, the mRNA from 1 sample is hybridized to 1 chip. Alternatively, 2 samples labeled with fluorescent probes of different colors can be hybridized to the same chip. See also Chapter 2.
17.2.2 High-Throughput DNA Sequencing
Cancer drug targets can also be identified by high-throughput DNA sequencing of cell lines or primary patient samples (Hudson et al, 2010). Advances in DNA sequencing have increased the speed and throughput of sequencing and decreased the cost (see Chap. 2, Sec. 2.2.10). These advances have allowed sequencing of primary tumors and normal tissues from multiple patients with malignancy and have identified mutations in oncogenes or tumor-suppressor genes that are prognostic markers and/or potential therapeutic targets. For example, through a focused sequencing effort in patients with acute myeloid leukemia (AML), activating mutations in the FLT3 kinase have been identified. Activation of the membrane-based FLT3 tyrosine kinase is important for the expansion and proliferation of normal early hematopoietic cells. In AML, internal tandem duplication of the juxtamembrane domain results in constitutive activation of this kinase leading to increased proliferation of the leukemic blasts. FLT3 internal tandem duplications are found in 20% to 30% of adult patients with AML and are associated with a higher rate of relapse and worse overall survival (Kottaridis et al, 2001; Thiede et al, 2002). Specific mutations in the coding region of the FLT3 kinase in patients with AML have also been demonstrated to result in its constitutive activation (Frohling et al, 2007). Inhibitors of FLT3 kinase, such as midostaurin and AC220, are being evaluated in clinical trials for patients with AML. Patients with either FLT3 internal tandem duplications or mutations in the coding region are eligible for inclusion in these studies.
The International Cancer Genome Consortium (ICGC) is undertaking high-throughput sequencing of the entire genomes of approximately 500 tumors from 50 different types and subtypes of cancer, along with normal tissue controls, to identify tumor-specific mutations (Hudson et al, 2010). These studies are expected to identify mutations that contribute to (and are critical for) cancer development and/or growth, known as driver mutations, and mutations not associated with cancer development or growth, known as passenger mutations. Differentiating between drivers and passengers is a complex process and can require use of advanced statistical analysis, structural studies to investigate the impact of the mutation on the protein structure, and molecular studies to demonstrate transformation as a result of overexpression of the mutant gene (Torkamani et al, 2009). Driver mutations can then be targeted to develop new cancer drugs.
17.2.3 RNA Interference Screening
Potential targets of anticancer drugs can also be identified through RNA interference (RNAi) screening. The introduction of double-stranded RNA sequences leads to degradation of homologous host mRNA sequences with subsequent reductions in expression levels of the target protein (Fig. 17–2; Chap. 2, Sec 2.4.3) through mechanisms that have not been fully elucidated. However, it appears that upon entry of double-stranded RNA into cells, the endogenous enzyme Dicer digests that RNA into short sequences of approximately 21 nucleotides. These short sequences then interact with the RISC (RNA-induced silencing complex) that matches these sequences to complementary endogenous mRNA. Upon binding, the endogenous mRNA target is degraded leading to reduction in target protein levels. Using this approach, the functional importance of individual gene knockdown can be ascertained. When combined with high-throughput screening, the functional significance of knocking down thousands of genes in the human genome can be evaluated. For example, high-throughput screens have identified kinases critical for the growth of myeloma cell lines such as GRK6 (Tiedemann et al, 2010); inhibitors of such kinases can then be developed and evaluated in clinical trials for patients with multiple myeloma. Similar approaches can be used to identify synergistic therapeutic approaches. Here combinations of drug and RNAi can identify genes whose mutation or knockdown can sensitize cells to the drug of interest. For example, a genome-wide RNAi screen was performed to identify synthetic lethal interactions with the KRAS oncogene. The screen identified the kinase PLK1 whose depletion induced death preferentially in KRAS mutant cells. Thus, these results suggest that PLK1 inhibitors could be useful in KRAS mutant tumors.
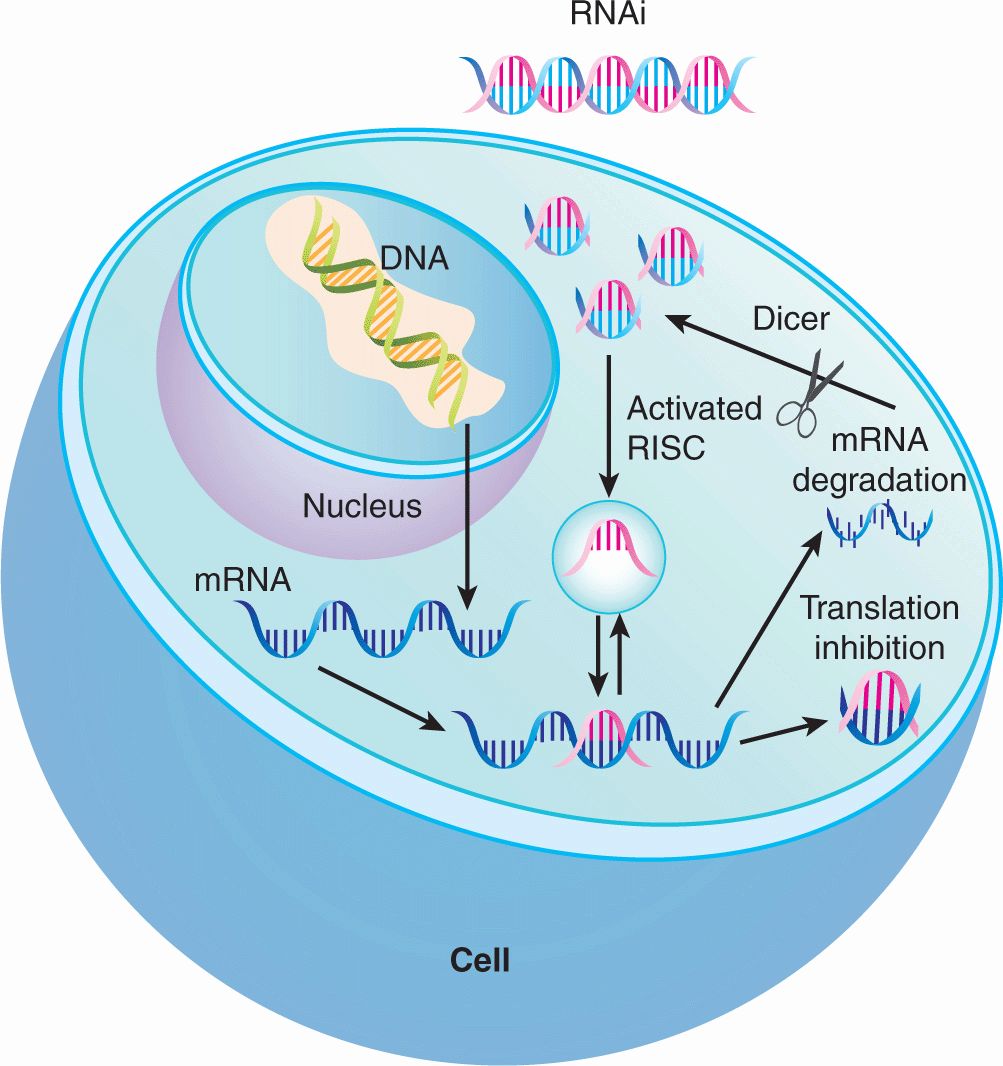
FIGURE 17–2 Proposed mechanism of action of RNAi to silence gene expression. Upon entry of double-stranded RNA into the cytoplasm, the endogenous enzyme Dicer cleaves the RNA into short sequences of approximately 21 nucleotides. These short sequences then interact with the RISC (RNA-inducing silencing complex) that matches these sequences to complementary endogenous mRNA. Upon binding, the endogenous mRNA target is degraded leading to eventual reduction in target protein levels or inhibition of translation by complementary base pairing to the mRNA transcript.
17.2.4 Identification of Active Drugs
Once a therapeutic target has been identified several approaches can be used to develop or select for inhibitors of this target.
Chemical libraries can be used for high-throughput screening of large libraries of compounds to identify small molecule leads for novel anticancer drugs. These screens can identify chemical compounds that inhibit the desired target in cell-free assays, by using enzymatic methods or physical binding. Fluorescent polarization is a common high-throughput assay to evaluate drug–target binding; in this method, polarized light is used to measure the speed of rotation or “tumbling time” of a fluorescently labeled molecule. Under fluorescent polarization, the speed of rotation of the molecule is coupled to the emission of light in a polarized plane; small molecules rotate faster and larger molecules rotate more slowly. When the fluorescently labeled molecule (probe) interacts with its target protein, the molecule tumbles more slowly. Drug screening assays search for chemicals that disrupt the interaction between the fluorescently labeled probe and target protein thereby increasing the tumbling of the fluorescent probe. The lead compounds are then investigated for their effects on intact cells.
Alternatively, cell-based screens can be conducted to identify chemical compounds that alter a cancer-associated phenotype of the cells such as their viability, or their ability to migrate or invade in tissue culture models. Follow-up studies are then required to identify the targets and mechanisms of action of the lead compounds. The chemicals present in these large libraries are usually synthetic compounds that represent diverse types of chemical structures. Smaller and more focused chemical libraries can also be developed to provide more in-depth coverage of a specific type of chemical structure. These focused libraries are useful to help identify more active analogs of compounds identified from a larger screen.
Natural product libraries can also be fruitful sources of leads for novel therapeutic agents. Natural products from, for example, plants and marine life, are isolated initially as extracts containing multiple compounds. Later, the individual chemical compounds are identified and purified, and methods to isolate individual compounds from extracts have improved. In addition, advances in medicinal and synthetic chemistry permit modification of natural products to improve their utility as anticancer agents. Natural products offer some potential advantages over synthetic chemical compounds. For example, they may have unique physical and chemical structures that cannot easily be synthesized. Some extracts from natural products may be sufficiently active to permit their direct evaluation in clinical trials. Multiple chemotherapeutic agents in clinical use are natural products or are derived from them; for example, paclitaxel is a chemotherapeutic agent isolated from the bark of the yew tree. Likewise, the irreversible proteasome inhibitor NPI-0052 is derived from the marine bacterium Salinispora tropica and is being investigated in clinical trials for patients with advanced malignancies (Chauhan et al, 2008; Singh et al, 2010).
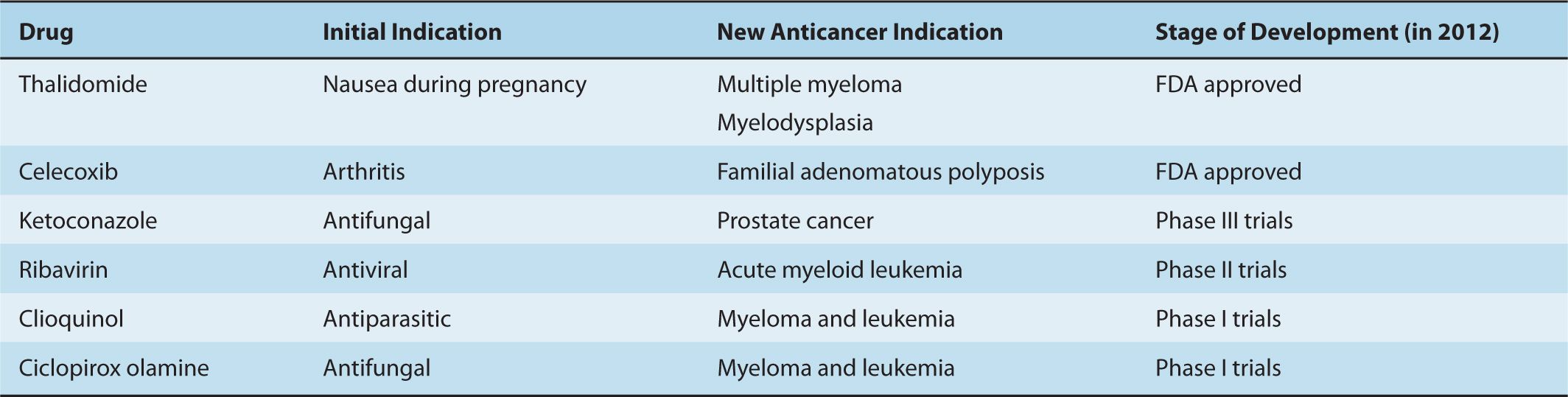
Libraries of known drugs are another potential source of new anticancer agents. Old drugs with previously unrecognized anticancer activity can be rapidly incorporated in cancer treatments by relying on their prior safety, pharmacokinetic, solubility, and stability data (Table 17–1). A classic example is the development of thalidomide as a novel antimyeloma therapy. Thalidomide was developed initially as a therapy for nausea during pregnancy, but was withdrawn from the market in 1961 because of teratogenicity. In 1999, thalidomide was reported to be active in multiple myeloma and produced a 32% response rate in patients with refractory disease (Sinha et al, 1999). Subsequently, thalidomide in combination with melphalan and prednisone was shown to prolong survival of older patients when compared to melphalan and prednisone alone (Palumbo et al, 2006). The success of thalidomide led to the development of the second-generation analog lenalidomide that also improves outcomes in patients with multiple myeloma as well as other hematological malignancies, such as myelodysplasia (List et al, 2005; Dimopoulos et al, 2007). The identification of old drugs with unrecognized anticancer activity has been largely serendipitous, but more recently, academics as well as industry have taken a systematic approach to their identification by compiling libraries of on-patent and off-patent drugs and screening these libraries for unrecognized anticancer activity.
TABLE 17–1 Examples of drug repositioning for new anticancer indications.
Rational drug design can be used to develop therapeutic agents if the 3-dimensional structure of a target is known. These structure-based studies can be used to follow-up on screens of available molecules or can be the starting point to develop the initial lead compounds. Guided by the crystal structure of a protein target, small molecules that bind the active site of the target can be synthesized. These initial compounds are then tested for activity in cell-free and cell-based assays and refinements are made to the chemical structure to improve potency, stability, specificity, and solubility. Thus, through an iterative process, potent and selective drugs are developed. Often, the selection of compounds can be aided by virtual modeling, where chemical structures are docked into target proteins using computer software that recreates 3-dimensional images of the molecule and its protein target. Million-compound libraries can be screened in virtual docking studies to identify “hits,” which can be validated in physical binding studies.
Binding assays using nuclear magnetic resonance (NMR) can evaluate interactions between drugs and targets, and can identify compounds that bind with low affinity. Small libraries of approximately 10,000 low-molecular-weight compounds are incubated with isotopically labeled target proteins. Binding of fragments is assessed by NMR, which can detect weak interactions that require concentrations of the 2 components in the 20 to 100 μM range. By chemically linking 2 binding fragments, a molecule with much higher (nM) affinity can be generated (Fig. 17–3). Through this approach, small molecule inhibitors of BCL-2 were developed to promote apoptosis (see Chap. 9, Sec. 9.4.2); this strategy has proven to be capable of inducing regression in experimental models of cancer and one such inhibitor (ABT-263 or navitoclax) is being evaluated in clinical trials (Wilson et al, 2010).
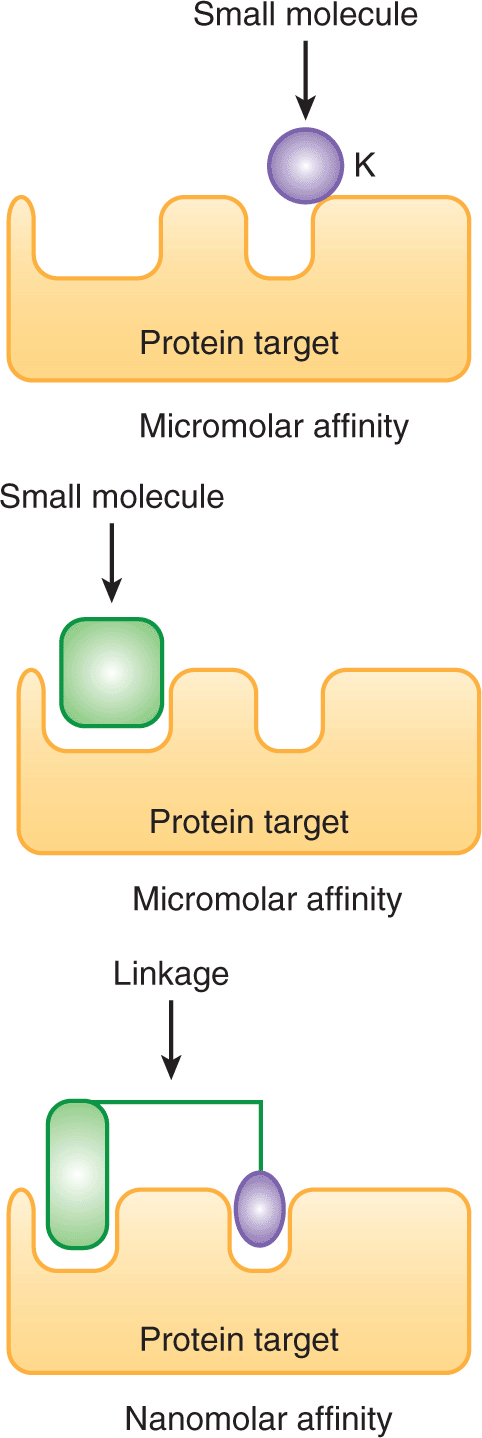
FIGURE 17–3 Methods for evaluation of interactions between a putative drug and its target. NMR binding studies can aid in the development of novel therapeutic agents for cancer. Using NMR, small fragment-based chemical libraries can be screened searching for small molecules that bind with micromolar affinity. When such fragments bind the target protein in close proximity, the molecules can be linked together chemically to create a new molecule that binds much more avidly with nanomolar affinity.
17.3 NEW STRATEGIES FOR CANCER TREATMENT
As discussed above, newer anticancer agents entering clinical use tend to be targeted to specific molecular targets in the cancer cell. These new therapeutic strategies include small molecules that target transmembrane and intracellular kinases, small molecules targeting other intracellular enzymes, antibodies that inhibit or activate cell-surface proteins, small molecules that disrupt protein–protein interactions, and gene knockdown with antisense oligonucleotides or RNAi.
17.3.1 Small Molecule Tyrosine Kinase Inhibitors
Multiple small molecules that target the adenosine triphosphate (ATP)-binding pocket of intracellular kinases have been developed. The first protein tyrosine kinase inhibitor to enter routine clinical use was imatinib, which was developed as an inhibitor of the ABL tyrosine kinase that is dysregulated as a result of the BCR-ABL fusion gene (Druker et al, 2001; see Chap. 7; Sec. 7.5.1). This agent, therefore, has activity in tumors that carry abnormalities in these genes and has found initial clinical application in the management of chronic myeloid leukemia (which expresses BCR-ABL) where it can induce remission in a large proportion of patients. Mutations in the active site of the ABL kinase can render chronic myeloid leukemia (CML) cells resistant to imatinib and lead to disease progression. Other drugs that inhibit ABL with greater potency than imatinib (eg, nilotinib), and drugs with a wider kinase inhibitory profile (eg, dasatinib) have been developed and approved for the treatment of imatinib-resistant CML (Hochhaus et al, 2007; le Coutre et al, 2008). These agents are superior to imatinib in their ability to induce a major molecular response at 12 months in previously untreated patients with CML, and as a result, this is now the front-line treatment of this disease (Kantarjian et al, 2010; Saglio et al, 2010). Imatinib also inhibits the tyrosine kinases of KIT, the product of the c-kit oncogene, and the platelet-derived growth factor receptor (PDGFR). Consistent with this activity, imatinib has activity against gastrointestinal stromal tumors, the majority of which express c-kit, and eosinophilic syndromes that have defects in PDGFR signaling.
Although drugs like imatinib highlight the attractiveness of highly specific kinase inhibitors, a growing trend has seen the development of kinase inhibitors that inhibit multiple targets. Examples of such multikinase inhibitors are sunitinib and sorafenib, which inhibit RAF, vascular endothelial growth factor receptor (VEGFR), PDGFR, and c-KIT kinases. These drugs have been shown to provide clinical benefit in the treatment of advanced renal cell and hepatocellular carcinoma (Escudier et al, 2007; Motzer et al, 2007; Llovet et al, 2008).
Small molecules have also been developed that target transmembrane kinases. For example, activation of the transmembrane epidermal growth factor receptor (EGFR) leads to cellular proliferation by activating RAS/RAF/MAPK (mitogen-activated protein kinase), PI3K (phosphatidylinositol-3 kinase)/AKT, and STAT (signal transducer and activator of transcription) signaling pathways (see Chap. 8, Secs. 8.2.4 and 8.2.5). EGFR is mutated or over-expressed in a variety of solid tumors, including non–small cell lung cancer, and this dysregulation contributes to pathogenesis and progression of the disease. Two inhibitors that interact with the ATP-binding pocket of the EGFR kinase have been developed and approved for the treatment of non–small cell lung cancer, gefitinib and erlotinib (Fig. 17–4; Grunwald and Hidalgo, 2003). Predicting which patients are most and least likely to respond to these newer therapies is an important component of the rational prescription of anticancer therapy. Studies demonstrate that tumors with mutations in KRAS have little chance of responding to EGFR inhibitors as the KRAS mutation leads to constitutive activation of the signaling pathway downstream of the EGFR receptor (Eberhard et al, 2005; Zhu et al, 2008). Thus, testing for KRAS mutations prior to treatment with EGFR inhibitors should help to identify patients most likely to respond.
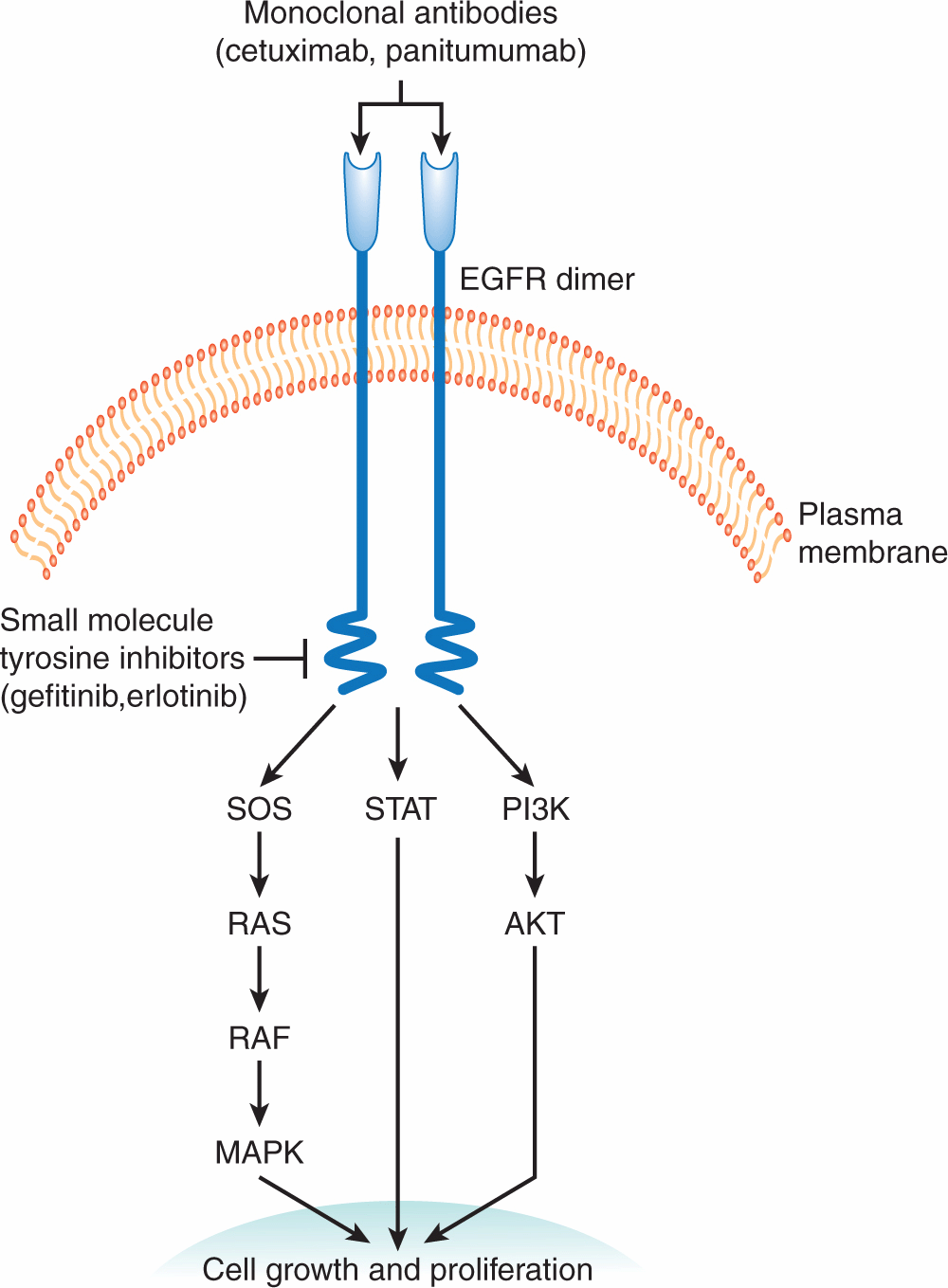
FIGURE 17–4 Cellular targets for anticancer drugs. Targeting the EGFR signaling pathway to develop novel therapeutic agents. Activation of the EGFR signaling pathway promotes cellular proliferation and survival by signaling through the RAS/RAF/MAPK, PI3K/AKT, and STAT pathways. Small-molecule ATP mimics (gefitinib and erlotinib) have been developed that bind and inhibit the enzymatic site of the EGFR kinase. Likewise, humanized monoclonal antibodies (cetuximab and panitumumab) bind the EGFR receptor and inhibit signaling through this pathway.
17.3.2 Enzyme Inhibitors
Small molecules that bind and inhibit the active sites of enzymes important for tumor growth and proliferation have also been developed and are in clinical use. Proteasome inhibitors are an example of this class of agent. The 26S proteasome has as its major function the degradation of cellular proteins, including damaged, misfolded, and regulatory proteins. Tumor cells require proteasome-dependent turnover of many cell-cycle proteins to complete mitosis successfully (see Chap. 9, Fig. 9–4B), and proteasome inhibitors may induce cell death. Such inhibition can lead to induction of endoplasmic reticulum (ER) stress with activation of the unfolded protein response, inhibition of the nuclear factor-κB (NFκB) inflammatory pathway, increased generation of reactive oxygen species, and activation of Caspase-8 and apoptosis (Hideshima et al, 2001; Chauhan et al, 2005; Bazzaro et al, 2006; Meister et al, 2007). Bortezomib is a covalently bound but reversible inhibitor of the β5-subunit of the proteasome that is used for the treatment of multiple myeloma and mantle cell lymphoma. Additional proteasome inhibitors are under development. Some of these newer proteasome inhibitors are oral agents, and some inhibit the proteasome through mechanisms distinct from bortezomib. For example, carfilzomib inhibits the active site of the protea-some irreversibly; this drug is more potent than bortezomib and can produce clinical responses in bortezomib-resistant patients (Kuhn et al, 2007; Parlati et al, 2009).
Histone deacetylase (HDAC) catalyzes the removal of acetyl groups from lysine residues of nucleosomal histones. The acetylation status of histones influences the regulation of transcriptional activity of some genes, and aberrant activity of HDAC is associated with the development of some malignancies. HDAC inhibitors have been developed that bind the zinc ion in the active site of HDAC, thereby inhibiting its enzymatic function. The HDAC inhibitors vorinostat and romidepsin are used for the treatment of cutaneous T-cell lymphoma (Olsen et al, 2007). These and other HDAC inhibitors are under investigation for the treatment of various malignancies, including AML and myelodysplasia.
Inhibition of the enzyme poly (ADP)-ribose polymerase (PARP), which is involved in DNA repair (see Chap. 5, Sec. 5.8) represents an approach to the development of agents that lead to synthetic lethality—2 genes or pathways are in a synthetic–lethal relationship if mutations or lack of function in either of them alone is not lethal but mutations or lack of function in both of them produce cell death (Fig. 17–5; Table 17–2; Kaelin, 2005; Iglehart and Silver, 2009). Cancers in patients with BRCA1 and BRCA2 mutations develop from cells that undergo loss of heterozygosity at these sites. Cells bearing these mutations have impaired DNA repair because of defects in homologous recombination and become dependent on a second mechanism of DNA repair, base-excision repair, for their survival (see Chap. 5, Secs. 5.3.2 and 5.3.4). A key enzyme in this process is PARP, and when PARP inhibitors are given to these patients, both mechanisms of DNA repair are impaired, and cancer cells undergo cell death particularly if exposed to agents that cause DNA damage such as platinum compounds or irradiation (Fig. 17–5; Turner et al, 2004; Iglehart and Silver, 2009). In the clinical development of these compounds, short-interference RNA (siRNA) libraries were used to identify genes that mediate sensitivity to PARP inhibitors including BRCA1 and BRCA2 (Turner et al, 2008). The PARP inhibitor olaparib was also evaluated in transgenic mice that mimic basal-like human breast cancer, a type of tumor with alterations in DNA-repair mechanisms and with enrichment of BRCA1 mutations (Rottenberg et al, 2008). Promising results with these compounds have been reported in several tumor types with these molecular alterations. Recent experimental studies also suggest that exposure of cells to prolonged hypoxia, as can occur in tumors, results in reduced DNA repair capacity and the potential for synthetic lethality with PARP inhibitors (Bristow and Hill, 2008).
TABLE 17–2 Properties of tumor cells that could be exploited to provide therapeutic advantage from the combined use of two drugs or of radiation and drugs.

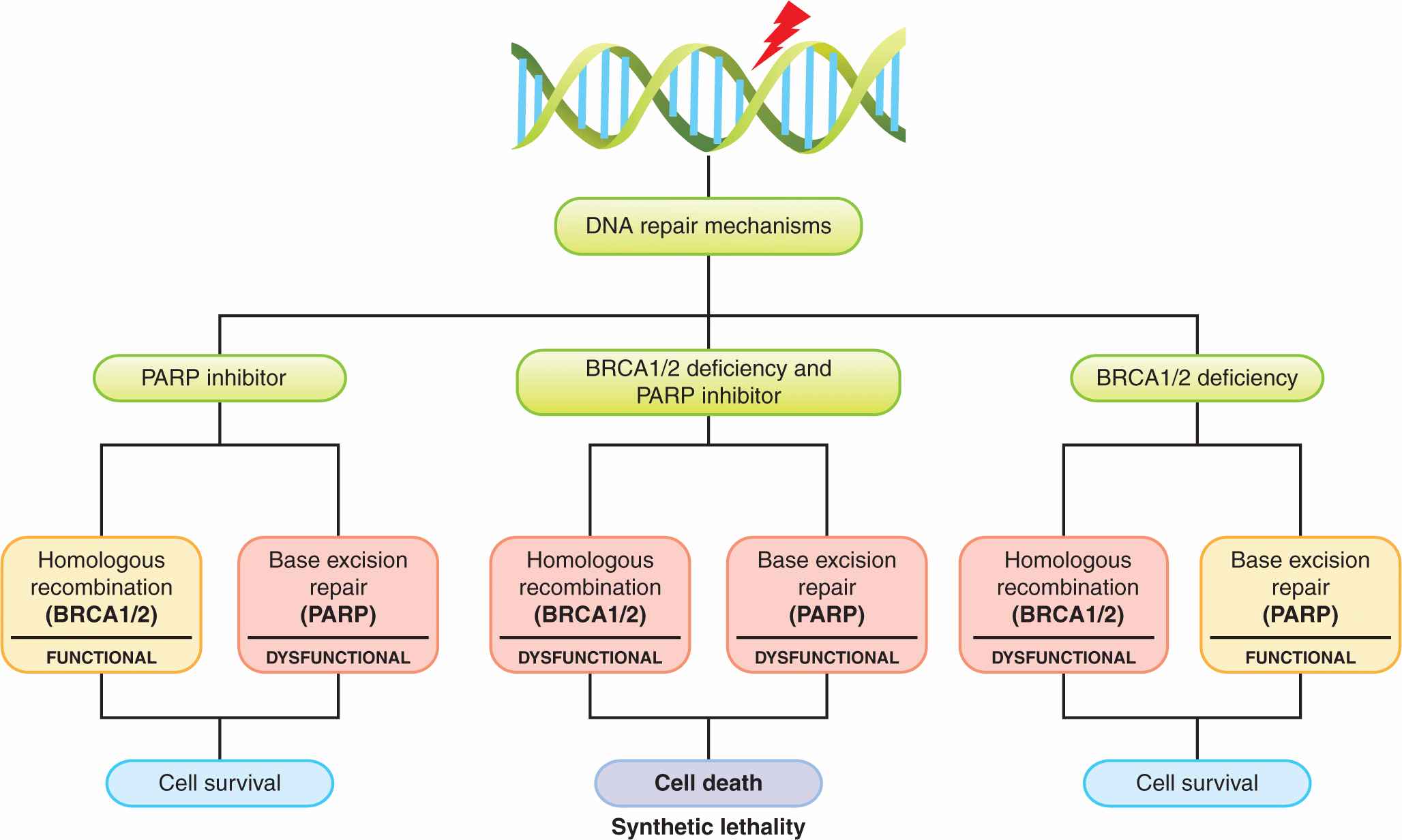
FIGURE 17–5 The concept of synthetic lethality. Cells with BRCA1 or BRCA2 deficiency lack the ability to repair DNA lesions by homologous recombination. Application of a PARP inhibitor, which inhibits the alternative pathway of base excision repair, is then selectively lethal to such cells, which are dependent on this pathway, especially after application of an agent that induces lesions in DNA such as chemotherapy with platinum-containing drugs or radiation. (Adapted from Amir et al, 2010 with permission.)
17.3.3 Antibody-Based Anticancer Therapies
Specific inactivating antibodies against the extracellular portion of a membrane-based receptor tyrosine kinase provide an alternative to small-molecule kinase inhibitors that inhibit signaling pathways. One such receptor is human epidermal growth factor receptor-2 (HER2), which does not have a known growth factor ligand, but whose constitutive activity provides intracellular signals leading to cell proliferation. HER2 is amplified or overexpressed in approximately 20% of breast cancers, and is associated with more aggressive disease and a worse prognosis (see Chap. 20, Sec. 20.3.3). In vitro experiments demonstrated that murine monoclonal antibodies directed against the extracellular domain of HER2 were able to inhibit the growth of HER2 overexpressing cell lines, but not of cells expressing normal amounts of the receptor. A humanized form of the most effective of these murine antibodies was developed, in an attempt to prevent the development of neutralizing antibodies, and thus allow long-term use in humans. This compound, trastuzumab, produces a prolongation in survival when used in conjunction with chemotherapy in women with HER2 overexpressing metastatic breast cancer (Slamon et al, 2001), and when used as adjuvant therapy after surgery (Piccart-Gebhart et al, 2005; Romond et al, 2005). To improve the efficacy of these antibodies, conjugates with chemotherapy have also been developed. For example, trastuzumab has been conjugated to the microtubule-disrupting drug DM1 and the conjugate (TDM1) has led to improved survival in women with HER2 positive breast cancer (Verma et al, 2012).
Structurally related to HER2 are the other EGFRs (HER1 or EGFR, HER3, and HER4; see Chap. 7, Sec. 7.5.3). The same approach as described above has been exploited in the development of cetuximab and panitumumab, which are humanized monoclonal antibodies that bind the extracellular domain of the EGFR receptor and prevent EGFR ligands from binding and activating the receptor (Fig. 17–4; Grunwald and Hidalgo, 2003). These antibodies are used for the treatment of advanced colon and head and neck cancer.
Other inhibitor antibodies include bevacizumab, which is a recombinant humanized monoclonal antibody directed against VEGF (Ferrara, 2004). Bevacizumab prevents the ligand VEGF from binding to its receptor, and thereby blocks its action in stimulating angiogenesis (see Chap. 11, Sec. 11.7.1). This drug has clinical efficacy in the treatment of colorectal and several other solid tumors, but clinical benefit is limited, probably because there are multiple alternative signaling pathways that can stimulate angiogenesis. Small molecule inhibitors of VEGF or its receptor such as sunitinib and sorafenib have also been developed and are in clinical use for treatment of kidney cancer.
Activating or agonistic antibodies are also being developed as anticancer agents. Activation of the death receptor pathway of Caspase activation with TRAIL (TNF-related apoptosis-inducing ligand) induces cell death by apoptosis (see Chap. 9, Sec. 9.4.4), and this effect may be greater in malignant cells where there is a high baseline level of cell death. Consequently, humanized monoclonal activating antibodies directed against the DR4 and DR5 TRAIL receptors have been developed and are being evaluated in clinical trials in combination with cytotoxic agents (Buchsbaum et al, 2007).
Other antibody-based therapies target cell-surface proteins that are overexpressed by the tumor cell, but do not activate or inhibit the targets. For example, rituximab is a chimeric monoclonal antibody that binds to the CD20 protein found on the surface of chronic lymphocytic leukemia (CLL) and lymphoma cells, and has clinical benefit when used as a single agent or in combination with chemotherapy. Binding of rituximab to CD20 does not impact the function of this protein, but rather leads to elimination of the malignant cells through a variety of mechanisms, including promotion of antibody- and complement-mediated cytotoxicity (see Chap. 21, Sec. 21.5.2) and induction of apoptosis. Based on the success of rituximab, antibodies directed against other cell-surface proteins have been developed, including alemtuzumab that targets CD52, which is approved for the treatment of T-cell lymphoma and CLL. These antibodies can also be conjugated to radioisotopes to improve their ability to kill the tumor cells.
17.3.4 Disrupting Protein–Protein Interactions
Protein–protein interactions play a critical role in most cellular functions and the development of inhibitors of these interactions for critical proteins is an attractive therapeutic strategy. Vinblastine and vincristine are established cancer therapies that work by this mechanism as they disrupt the interaction between α- and β-tubulin. The relatively large surface area involved in protein–protein interactions has made the development of specific inhibitors of such interactions challenging, but when critical contacts can be localized to small areas of the protein, the development of these inhibitors can be successful. For example, binding assays identified the small molecules ABT-263 and ABT-737 (see Sec. 17.2.4), which binds BCL-2 and its family members in the BH3 pocket and prevents these antiapoptotic proteins from binding to and inhibiting their proapoptotic binding partners (see Chap. 9, Sec. 9.4.2; Kang and Reynolds, 2009). This agent and obatoclax mesylate, a compound with similar activity, are being evaluated for anticancer activity in clinical trials. Small molecules that bind the caspase inhibitor XIAP (X-linked inhibitor of apoptosis) and prevent it from binding to and inhibiting caspases 3 and 9, which are important mediators of apoptosis (see Chap. 9, Sec. 9.4.2), have also been developed and have advanced into clinical trial (Rajapakse, 2007).
17.3.5 Antisense Oligonucleotides and RNAi
Antisense oligonucleotides and RNAi (see Chap. 2, Sec. 2.4.3) act by binding to specific target mRNAs in cancer cells and promoting their degradation. Although RNAi is more effective than antisense oligonucleotides at knocking down protein expression, antisense technology was discovered first and is more advanced in clinical development. Antisense molecules such as oblimersen sodium that target BCL-2 are being evaluated in late-stage Phase III clinical trials for both solid tumors and hematological malignancies (Kang and Reynolds, 2009), but as yet none has been approved for routine clinical use. RNAi therapeutics are also being advanced into clinical trials, but are at an earlier stage of development.
17.3.6 Directed Drug Delivery
A major limitation to the use of nontargeted chemotherapy is lack of selectivity for tumor cells. As new targets that are expressed selectively on tumor cells are characterized, a potential method for increasing the therapeutic index of conventional drugs is to link them to a carrier that may be targeted to tumor cells. Such carriers may include liposomes and other types of nanoparticles, as well as monoclonal antibodies.
A large body of research relates to the entrapment of anticancer drugs in nanoparticles (Petros and DiSimone, 2010), and several agents have now been approved for clinical use. For example pegylated liposomal doxorubicin improved the outcome of patients with Kaposi sarcoma compared to conventional chemotherapy (Northfelt et al, 1998), and it also reduced cardiac toxicity when compared to conventional doxorubicin for treatment of metastatic breast cancer (Batist et al, 2001). Nanoparticle albumin-bound paclitaxel (nabpaclitaxel) may also improve outcome for women with meta-static breast cancer as compared to conventional taxanes (Gradishar et al, 2009). These approved agents are not targeted specifically against molecules expressed selectively on cancer cells, but there are several mechanisms whereby drugs associated with liposomes or other nanoparticles might lead to improvement in therapeutic index relative to free drug: (a) slow, continuous release of the anticancer drug into the circulation, which may protect against organ-specific toxicity (eg, cardiotoxicity caused by doxorubicin) and/or lead to improvement in antitumor effects; (b) fusion of liposomes with cell membranes, leading to efficient internal delivery of drugs, which may overcome drug resistance caused by impaired uptake of free drug; and (c) selective deposition of nanoparticles in tumor tissue, because of their enhanced permeability and retention as a result of the abnormal vasculature of tumors (see Chap. 11, Sec. 11.5.1). Many laboratories are trying to improve on these therapeutic results through the incorporation of molecules that target human cancer cells, but at present this research remains largely preclinical (eg, Byrne et al, 2008; Hirsjärvi et al, 2011).
Several anticancer drugs, as well as potent toxins such as Pseudomonas exotoxin, diphtheria toxin, and ricin, have been linked to monoclonal antibodies directed against tumor-associated receptors or other antigens (Teicher, 2009). Several problems have slowed development of this approach, including use of mouse monoclonal antibodies that were immunogenic, insufficient potency because of limited ability to bind sufficient toxins or chemotherapy, and instability of the linkage leading to release of drug or toxin in the circulation with accompanying toxicity. However, trastuzumab emtansine (TDM1), in which the anti-HER2 monoclonal antibody trastuzumab is bound to the microtubule-disrupting drug DM1, has improved survival of women with HER2– positive metastatic breast cancer (Verma et al, 2012).
17.4 PRECLINICAL EVALUATION OF POTENTIAL ANTICANCER DRUGS
Compounds identified through any of the above drug-discovery platforms need to be evaluated in preclinical studies for both their ability to inhibit their putative targets and their potential anticancer effects. There are multiple steps in the evaluation of a potential anticancer drug, and only a small proportion of such agents will eventually enter clinical trials (Ocana et al, 2011; Fig. 17–6). These steps involve identification and verification of the target and mechanism of action, evaluation for activity against cultured tumor cells, and evaluation of activity and toxicity in various in vivo models. It is important to verify the specificity of the target molecule or pathway in animal models and in early clinical trials, as putative targeted agents may sometimes have unexpected toxic effects that are independent of inhibition of the target for which they were designed.
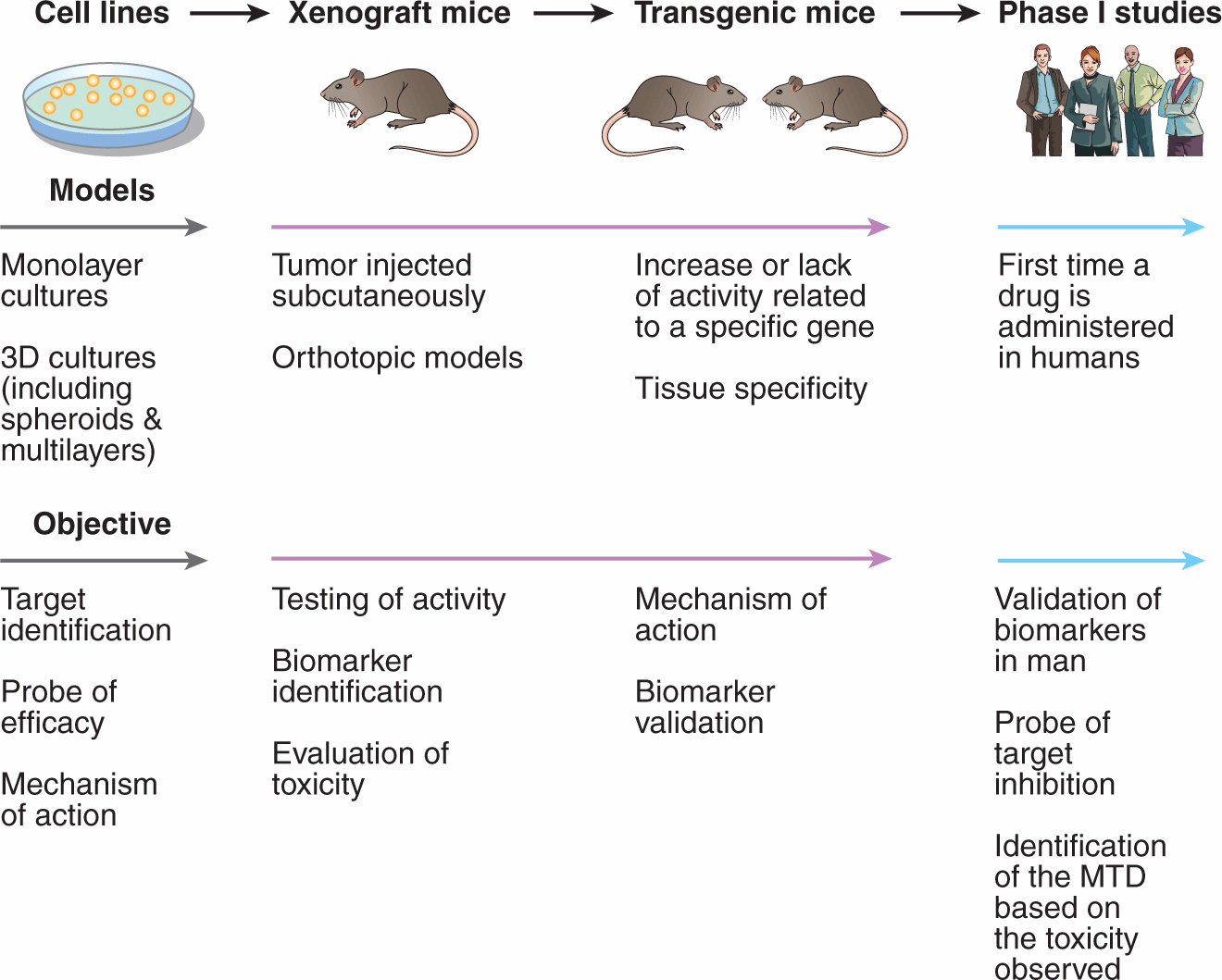
FIGURE 17–6 Preclinical to clinical evaluation of candidate anticancer drugs. Selected steps in development of anticancer drugs. Drugs are usually evaluated first for activity against cultured cells, followed by solid tumor models in order to reflect the microenvironment of solid tumors. This step is usually followed by demonstration of activity against xenografts generated from implantation of selected human tumor cell lines into immune-deficient mice; xenografts better reflect the properties of the corresponding human tumor if grown in the same tissue of origin (ie, implanted orthotopically). Use of genetically modified (transgenic or knockout) mice allows information about the relation between drug activity and expression of potential target genes. Finally, the drug is evaluated in Phase I studies in humans, which traditionally encompassed a simple evaluation of tolerance and pharmacokinetics and has expanded to provide further information about the molecular target and to identify and validate biomarkers that correlate with drug activity. MTD, Maximum tolerated dose. (Adapted from Ocana et al, 2011.)
17.4.1 Evaluation of Drugs for Activity Against Cultured Cells
An important phase of preclinical testing involves evaluating whether the candidate agent can either kill or inhibit the proliferation of cancer cells. Screening of candidate drugs requires an initial assay of activity that is rapid and can be automated. Some measures of cell damage that have been used employ dyes such as methyl thiazole tetrazolium (MTT), which depends on the reduction of a tetrazolium-based compound to a blue formazan product by living, but not dead, cells. The amount of reduced product is quantified in an automated system using multiple tissue-culture wells in which cells have been exposed to a range of doses of the drugs under test. Another dye that is useful in quantifying cell number is sulforhodamine B, a pink anionic dye that binds to basic amino acids of fixed cells such that dye intensity is linearly related to the number of cells. These tests assess the number of metabolically active cells in the culture at the time of analysis and hence largely track the effects of the treatment to induce rapid cell death and/or reduced proliferation. In general, they are useful for assessing changes in the number of metabolically active cells by a factor of 10 to 20 (ie, reductions down to 5-10% of control).
Following treatment with drugs, tumor cells may undergo programmed cell death or apoptosis (see Chap. 9, Sec. 9.4.2; Schimmer, 2007; Kang and Reynolds, 2009). Apoptosis can be quantified by various methods (eg, by annexin V and TUNEL [terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate-biotin nick-end labeling] staining) to provide an estimate of the number of cells undergoing apoptosis or an apoptotic index. This index generally increases after drug treatment of tumors, and the proportion of cells undergoing apoptosis may give a broad indication of drug effectiveness. However, there is evidence that radiation and many drugs often kill cancer cells by mechanisms other than apoptosis, and that assays of apoptosis correlate poorly with cell killing assessed by colony-forming assays (Brown and Attardi, 2005). Even where apoptosis is an important mechanism leading to drug-induced cell death, it is a dynamic process whose assessment can be very dependent on when measurements are made during or after treatment.
The important activity of an effective anticancer drug is to cause tumor cells to lose their capacity for indefinite proliferation, and therefore the ability to regenerate the tumor. The above assays, as well as other short-term methods for evaluation of drug effects, including induced morphological changes, or other properties of cancer cells, such as changes in DNA, RNA, or protein synthesis, all suffer from the fundamental limitation that they apply to an unselected population of cells and have limited correlation with loss of reproductive potential of the cells.
Colony-forming assays can be performed following exposure to potential anticancer drugs of established cell lines derived from tumors, primary malignant cells derived from patient samples or normal hematopoietic and other cells (see also Chap. 15, Sec. 15.3.1). In such assays, candidate cells are exposed to various doses of the agent of interest for a fixed time, or to a fixed dose (that is expected to be achievable in vivo) for varying times. The drug is then washed out and different numbers of cells are evaluated for colony formation in a new environment. If the assay is performed in tissue culture, the number of colonies per cell plated is called the plating efficiency, and the ratio of the plating efficiency from drug-treated cells to untreated cells is the surviving fraction. The surviving fraction (plotted on a logarithmic scale) can then be plotted against drug dose (for a fixed time exposure) or against time (for exposure to a fixed drug concentration) to produce a survival curve. This assay can usually assess cell surviving fractions down to 10–3 to 10–4 of control.
Figure 17–7 shows an example of a survival curve. The survival curve is analogous to survival curves generated to describe the dose response to radiation (see Chap. 15, Figs. 15–6 and 15–7), although there is rarely an exponential decrease in cell survival with increasing drug dose as may be observed for radiation. Many chemotherapy drugs (eg, anti-metabolites such as cytosine arabinoside and methotrexate) are relatively specific in killing cells in a given phase of the cell cycle (usually S-phase) so there is an initial steep fall in survival as the sensitive cells are killed, but then a flattening of the survival curve. Survival may be further decreased by more prolonged exposure to the drug, although such drugs often also retard entry of cells into the drug sensitive phase. Most drugs, including molecular-targeted agents, are more active against rapidly proliferating cells, even if they are not cell-cycle-phase specific. For these drugs, also, there will be a trend for cell-survival curves to be concave upward, as in Figure 17–7, as higher doses interact with progressively more resistant, slowly proliferating cells.
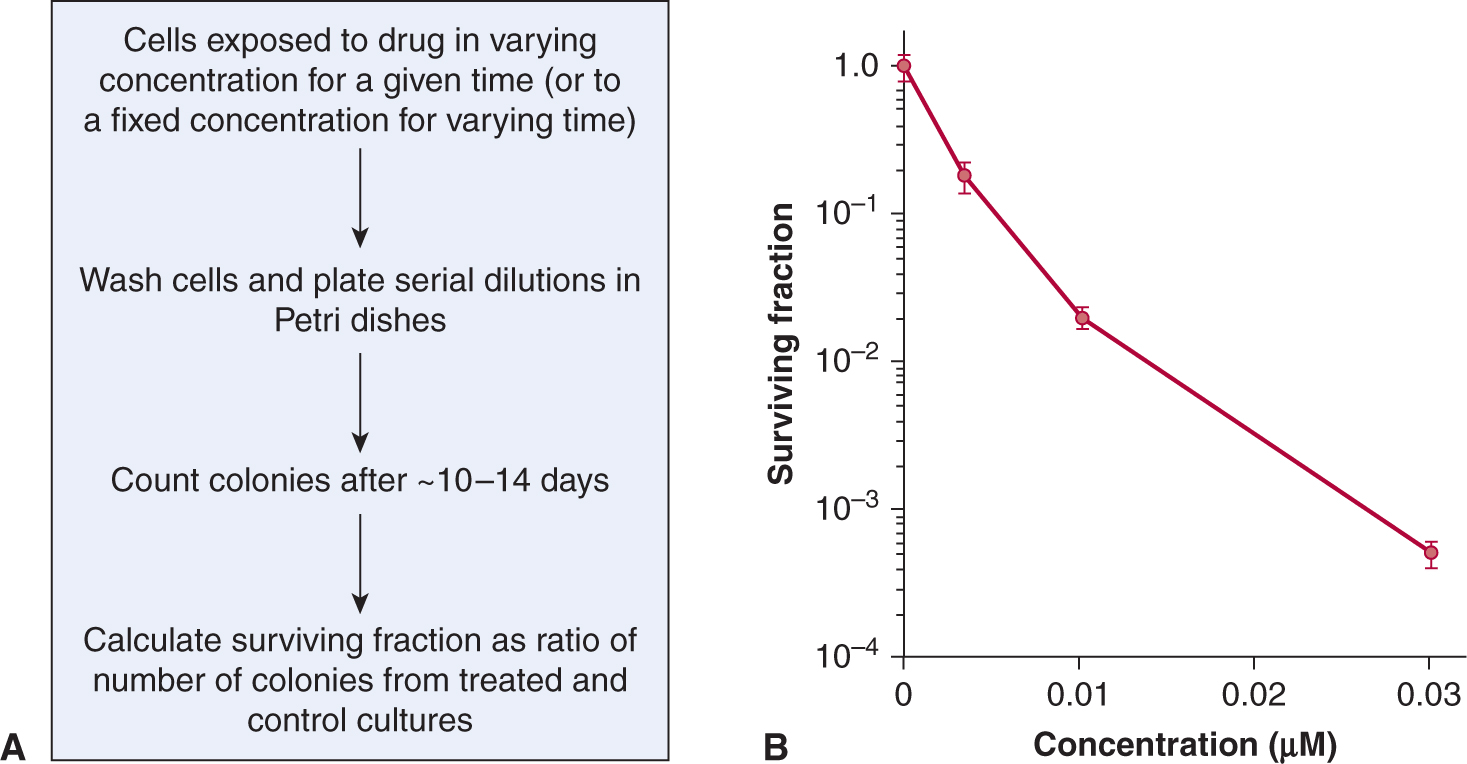
FIGURE 17–7 Cell survival curves. A) Cells are exposed either in monolayer or in suspension culture to different doses of a drug for a given time, or to a single dose of the drug for a variable time. The cells are then washed in fresh medium and serial dilutions are plated in Petri dishes; colonies formed from surviving cells are counted, usually 10 to 14 days later. B) Experimentally determined cell survival curve for murine EMT6 cells exposed to the drug mitoxantrone. Note that surviving fraction is plotted using a logarithmic scale against dose on a linear scale.
Drug activity may depend critically on cell–cell interactions, and such interactions are lost when potential drugs are evaluated against dispersed cells in culture. One model that preserves some of these interactions uses coculture of myeloma plasma cells and stromal bone marrow cells from myeloma patients (Mitsiades et al, 2008). Assays of antimyeloma drugs that determine their ability to preserve activity in this environment are informative about the potentially protective action of stromal components on drug-induced cell death, and about relative effects of the treatment to kill myeloma cells in their microenvironment. Other models that include in vitro-generated blood vessels, or coincubation with immune cells, may offer clues as to antiangiogenic or immunomodulatory properties of the compound of interest. The tumor microenvironment is particularly important for development of drugs with activity against solid tumors. Contact between cells in solid tumors can mediate drug resistance. To be effective drugs must penetrate tissue from blood vessels to reach all of the target tumor cells and achieve a toxic concentration, and should be active under microenvironmental conditions (hypoxia, acidity) that inhibit tumor cell proliferation (see Chap. 12, Sec. 12.2.1 and Chap. 19, Sec. 19.3; Minchinton and Tannock, 2006). Evaluation of drugs in monolayer cell cultures has a limited ability to predict responsiveness in solid tumors, but in vitro models, including tumor spheroids and multilayered cell cultures, can reproduce important properties of solid tumors, such as cellular contact and an extracellular matrix, as well as gradients of nutrient distribution, cell proliferation, and drug access (see Chap. 19, Sec. 19.3.1).
If a potential drug shows activity as assessed by 1 or more of the above assays, it is important to determine its target and mechanism of action. If the agent is designed to target a certain molecule, such as a receptor tyrosine kinase, it is important to show that the agent does, in fact, bind to its target, and that it inhibits downstream signaling from that target at a concentration that might reasonably be expected to be achieved in subsequent in vivo studies. Helpful strategies might include comparing the sensitivity of cells selected for the presence of, and/or dependence on a particular target or pathway, and of genetically modified cells that are not dependent on the target or pathway. Likewise, insights into mechanism of action can be derived by selecting populations of cells resistant to the drug and using genetic approaches, including gene expression profiling or genetic sequencing to identify mechanism of resistance. In the absence of such evaluation, agents might display nonspecific toxicity that is unlikely to lead to selective antitumor effects in vivo.
17.4.2 Evaluation of Potential Anticancer Drugs In Vivo
The in vitro assays described in the preceding sections assess directly the sensitivity of cultured tumor cells to drug treatment, but they do not assess the impact of the drug’s pharmacokinetics or its toxicity to normal cells, both of which are important determinants of the clinical efficacy of a cancer drug. Assays that have been used to assess the effects of drugs on tumors in animals include drug-induced delay in tumor growth and evaluation of colony formation in vitro following treatment of tumors and excision and plating of the tumor cells. Comparison of tumor growth in treated and untreated animals is used most frequently to evaluate drug effects against solid tumors that are transplanted into animals (Fig. 17–8). Tumor shrinkage and delay of regrowth are used to model the clinical assessment of tumor remission, and this approach is relatively humane because animals can be killed painlessly before tumors are sufficiently large to cause discomfort. Drugs that appear promising in in vitro screens are usually evaluated against syngeneic tumors transplanted into inbred mice, and in xenografts where human tumor cell lines are implanted into immune-deficient mice.
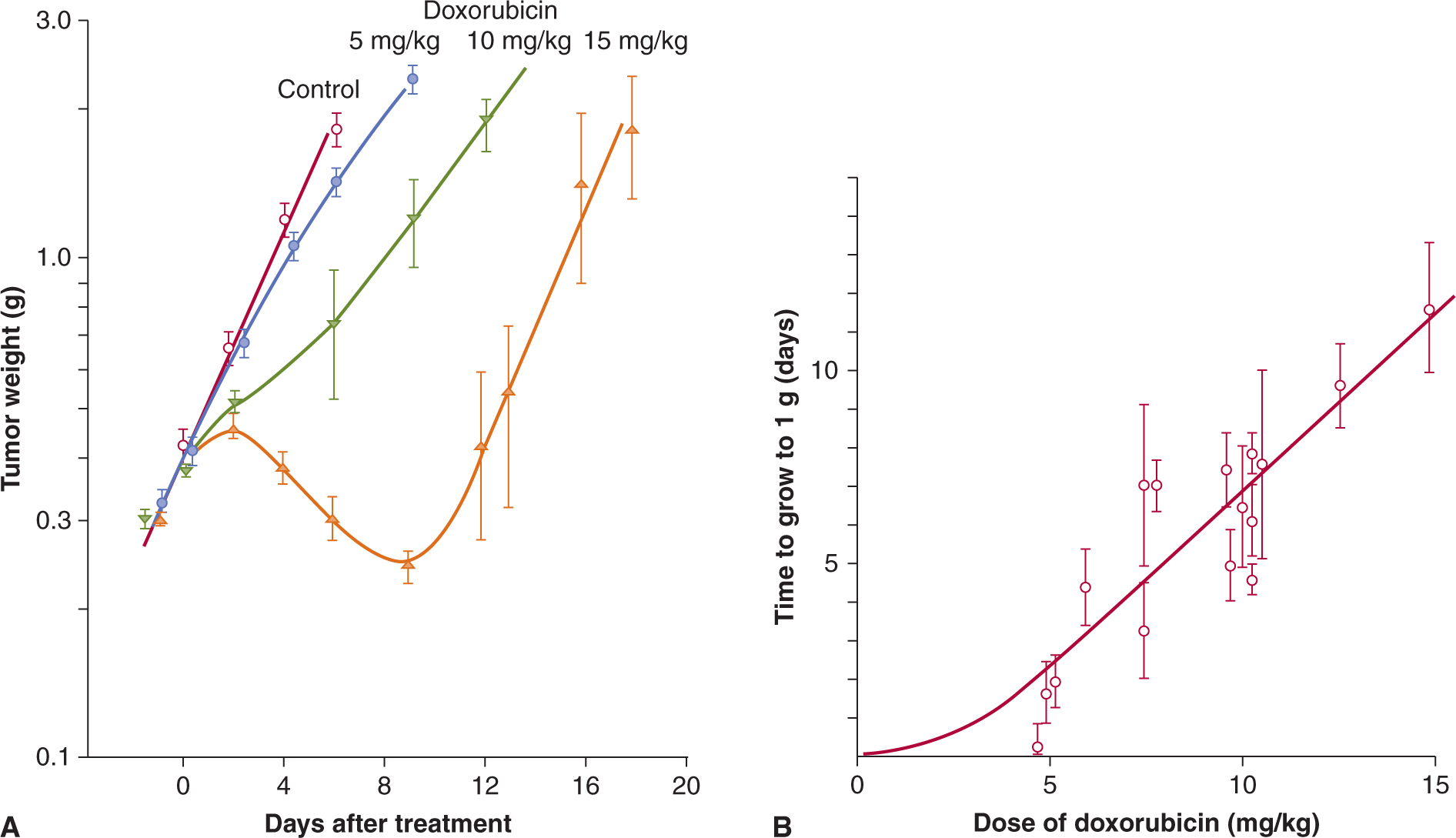
FIGURE 17–8 Tumor growth curve, and dose response relationship. A) Illustration of tumor growth curves for treatment of an experimental tumor with doxorubicin. Tumor weight was estimated by prior calibration with measurements of tumor diameter and is plotted on a logarithmic scale against time on a linear scale. Straight lines then represent exponential growth, and tumor doubling time can be determined from the slope. Note that growth curves after drug treatment are not always parallel to the growth curve for controls and that interanimal variation may lead to large standard errors. B) Dose–response curve relating drug dose to the time for tumors to grow from size at treatment (~0.4 g) to 1 g.
The most widely used host for xenografting of human tumors is the congenitally athymic nude mouse (so called because they lack fur). The nude mouse is not a perfect host because it may produce antibodies and also has large numbers of natural killer cells that may inhibit tumor growth. Alternative hosts include mice with severe combined immune deficiency (SCID) or SCID mice with further mutations to reduce further their ability to reject foreign tissue grafts. These mice may be better recipients for transplanted human tissues and have allowed the establishment of grafts of lymphoid and hemopoietic tissues, as well as of solid tumors. Xenografts possess the advantage that they may have characteristics more similar to those of the human tumors from which they are derived (eg, enzyme activities), as compared to tumors of murine origin. However, xenograft models have several limitations for evaluation of potential new drugs. Firstly, the stromal component of a xenograft is not human, so it is difficult to evaluate the effect of the microenvironment on drug response, or to evaluate drugs such as monoclonal antibodies that may target only human proteins (eg, bevacizumab, which targets human VEGF and does not recognize murine VEGF). Secondly, the rate of growth of xenografts derived from serially transplanted human tumor cell lines is often very rapid (typical doubling times of a few days) compared with that of primary human tumors (typical doubling times of 1 to 3 months), and they are more likely to respond to antiproliferative agents. Thirdly, the animal is immunecompromised so testing of agents with immunomodulatory effects is difficult or impossible. Finally, it may be difficult to select for resistance to drugs using xenograft models, thereby limiting the use of these in vivo models for studying mechanisms of drug resistance.
Transplantation of tumor cells into subcutaneous sites of mice allows easy evaluation of tumor size, but there is evidence that the molecular profile and microenvironment of the tumors is more similar to their human counterpart if they are transplanted orthotopically (eg, mammary tumors are implanted in the mammary fat-pad of female mice, or pancreatic tumors are transplanted into the pancreas of mice) (Nakamura et al, 2007). Humanized mouse models have also been developed; for example, human mammary cells and/or fibroblasts have been transplanted into a cleared mouse mammary gland. Evaluation of tumor growth (or shrinkage after treatment) is more difficult in ortho-topic than subcutaneous sites, but is facilitated by a variety of methods for imaging of small animals (see Chap. 14, Sec. 14.4). Also, a gene encoding a marker such as green fluorescent protein (GFP) or luciferase may be introduced and stably expressed in the malignant cells, allowing them to be detected and quantified by optical imaging in the whole animal. This approach allows estimation not only of tumor volume in the primary site, but also enables the following of metastatic spread and the influence of treatment.
Genetically engineered mouse models (GEMMs), in which the expression of a protein is temporally and/or spatially controlled by genetic manipulation, are being used to better assess transformation events and potential anticancer activity related to their inhibition (Sharpless and Depinho, 2006). These transgenic mouse models (see Chap. 2, Sec. 2.4.5) can simulate some aspects of human cancer by introducing alterations in oncogenes or tumor-suppressor genes in germlines of mice. The GEMM can be used in 2 ways in the evaluation of preclinical agents: the treatment of an established tumor, and their effects on tumor prevention. For example administration of the anti-EGFR/HER2 tyrosine kinase inhibitor lapatinib prevents tumor initiation in MMTV (mouse mammary tumor virus)-erbB2 transgenic mice (Strecker et al, 2009), while the mammalian target of rapamycin (mTOR) inhibitor everolimus prevents the formation of colonic adenomas in the Apc(Delta716) heterozygous mouse, which is a model for human familial adenomatous polyposis (Fujishita et al, 2008). Evidence of activity of a drug to prevent or delay tumor formation in the GEMM can give important insight into mechanisms of activity, and may indicate molecular properties of a tumor (ie, biomarkers; see Fig. 17–6) that may correlate with increased likelihood of activity. However, activity of a drug in the GEMM does not necessarily imply that the drug will also be active in treating tumors with similar types of genetic change.
The benefits of the GEMM for preclinical evaluation of potential anticancer drugs include: (a) that the tumor has developed in an immune-competent animal, and (b) genetic alterations can be modified in a time- and tissue-specific manner. For example, regulatory elements in the inserted transgenic DNA that are sensitive to tetracycline or tamoxifen can be incorporated in these mouse models, so that administration of these well-tolerated drugs to the mice can turn on or off specific molecular pathways (Kistner et al, 1996). However, GEMMs are imperfect models for human tumors, where there are often multiple genetic abnormalities, because the initial molecular alterations in the mice are limited to the ones that have been introduced. However, further mutations in GEMMs may restore activity of a defective molecular pathway. Also the stroma and other normal tissue elements are derived from the rodent, and usually have the same genetic alteration as the tumor. To avoid these potential caveats, some investigators have transplanted the tumors generated in the transgenic mice subsequently into wild-type mice. Transplantation also offers the possibility of shortcutting the long latency for appearance of spontaneous tumors and the possibility of treating genetically similar transplants with different drugs. Thus far, GEMMs have been used more often for the study of the transformation process than for evaluation of novel drugs.
A limitation of most of the above in vitro and in vivo assays of drug activity is that they evaluate relatively short-term effects against cells that have been selected to grow in culture or in mice. However, there is evidence, described in Chapter 13, that tumors may contain a small population of cells with indefinite proliferation, or tumor stem cells, and if so, it is these cells that are the appropriate targets of therapy that may lead to long-term remission or cure. Recently, there have been attempts to screen drugs for activity against tumor cells bearing cell-surface markers that indicate an enriched stem cell population, and compounds with selective activity against such cells were identified (Hassane et al, 2008; Gupta et al, 2009). One approach to evaluating the ability of a drug to target the stem cell fraction is to treat the cells in culture or in immunodeficient mice with the drug or buffer control and then inject the treated tumor cells into further immunodeficient mice. The primary transplants, along with secondary transplants where cells from the primary mice are injected into secondary recipients, can be used to evaluate the effects of the drug on the stem cell fraction. When the stem cells are targeted and killed, the treated tumor cells do not engraft in the secondary mice (Jin et al, 2006).
17.4.3 Relationship Between Tumor Remission and Cure
For most solid tumors the limit of clinical and/or radiological detection is about 1 g of tissue (109 cells). If therapy can reduce the number of malignant cells below this limit of detection, the patient will be described as being in complete clinical remission. Surgical biopsy of sites that were known to be involved with tumor previously may lower the limit of detection, especially if immunohistochemistry is used to identify specific markers on tumor cells, but a pathologist is unlikely to detect sporadic tumor cells present at a frequency of less than 1 in 10,000 normal cells. Therefore, even a surgically confirmed complete remission may be compatible with the remaining presence of a large number of tumor cells (up to 105/g tissue). Tumor cure requires eradication of all tumor cells that have the capacity for tumor regeneration. The proportion of such stem cells among those of the tumor population is unknown, but clinical and even surgically confirmed complete remissions are compatible with the presence of a substantial residual population of surviving tumor stem cells.
High doses of radiation therapy may lead to local tumor control (or to cure in the absence of metastasis) and small groups of mice can be treated with different doses of radiation to their tumors and assayed for tumor remission and long-term recurrence; this approach allows identification of the dose of radiation that will control 50% of the tumors (tumor control dose 50% [TCD50], see Chap. 16, Sec. 16.3.1). Only rarely can tolerated doses of systemic agents cure tumors in mice, and multiple treatments are usually necessary, but when that is possible related experiments can be formulated to define the dose and schedule that leads to cure in 50% of the animals. An advantage of such assays is that they evaluate drug effects against tumor stem cells.
17.5 TOXICITY OF ANTICANCER DRUGS
17.5.1 The Concept of Therapeutic Index
In addition to their antitumor effects, all anticancer drugs are toxic at some level to normal tissues, and it is this toxicity that limits the dose of drugs that can be administered to patients. The relationship between the probability of a biological effect of a drug and the administered dose is usually described by a sigmoid curve (Fig. 17–9), although it is possible that for targeted agents there is an optimal dose where the target is completely inhibited and using higher doses only augments the toxicity. If the drug is to be useful, the curve describing the probability of antitumor effect (eg, complete clinical remission) must be displaced toward lower doses as compared with the curve describing the probability of major toxicity to normal tissues (eg, myelosuppression leading to infection). The therapeutic index (or therapeutic ratio) may be defined from such curves as the ratio of the dose required to produce a given probability of toxicity and the dose required to give a defined effect against the tumor. The therapeutic index in Figure 17–9 might be represented by the ratio of the drug dose required for a 5% level of probability of severe toxicity (sometimes referred to as toxic dose 05 [TD05]) to that required for 50% probability of antitumor effect (ie, effective dose 50 [ED50]). Any stated levels of probability might be used. The appropriate end points of tumor response and toxicity will depend on the limiting toxicity of the drug and the intent of treatment (ie, cure versus palliation). Improvement in the therapeutic index is the goal of systemic treatment. However, although dose–response curves similar to those of Figure 17–9 have been defined in animals, they have rarely been obtained for drug effects in humans. They emphasize the important concept that any modification in treatment that leads to increased killing of tumor cells in tissue culture or animals must be assessed for its effects on critical normal tissues prior to therapeutic trials.
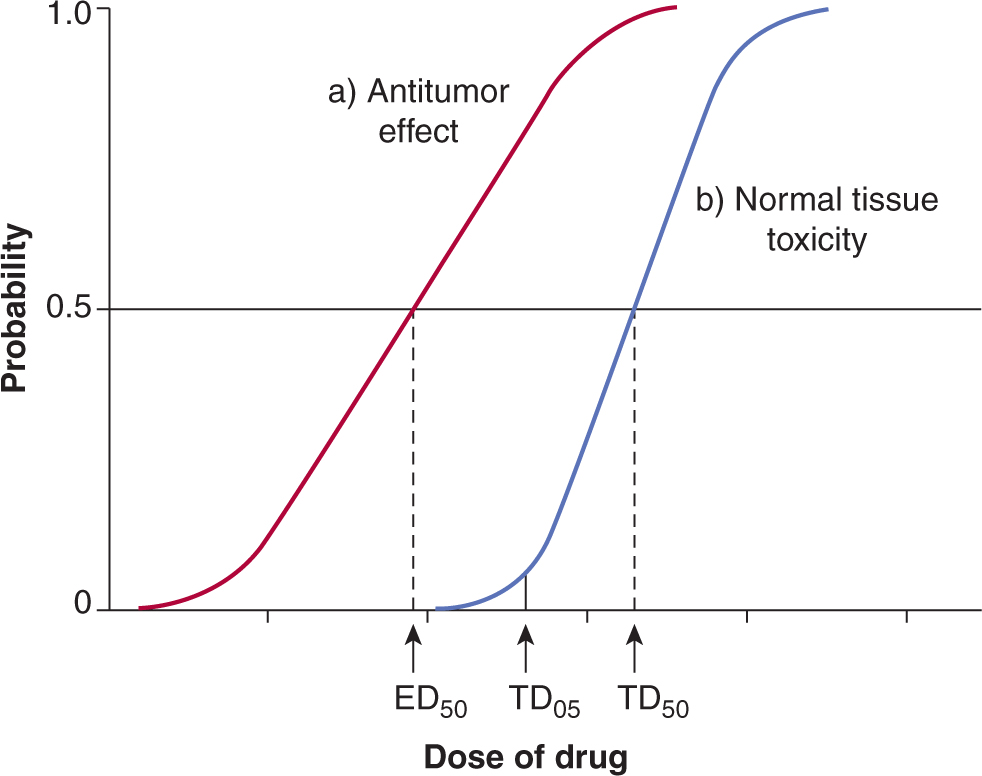
FIGURE 17–9 Concept of Therapeutic Index. Schematic relationship between dose of a drug and the probability of a given measure of antitumor effect (curve a), and the probability of a given measure of normal-tissue toxicity (curve b). Heterogeneity among tumors often leads to a shallower slope of the line representing tumors, as shown in red. The therapeutic index might be defined as the ratio of doses to give 50% probabilities of normal-tissue damage and antitumor effects. However, if the end point for toxicity is severe (eg, sepsis as a result of bone marrow suppression), it would be more appropriate to define the therapeutic index at a lower probability of toxicity (eg, toxic dose [TD05]/effective dose [ED50]).
Toxicity to normal tissues limits both the dose and frequency of drug administration. Many drugs cause toxicity because of their preferential activity against rapidly proliferating cells, and this is especially true for chemotherapy, but also for some targeted agents—normal adult tissues that maintain a high rate of cellular proliferation include the bone marrow, intestinal mucosa, hair follicles, and gonads (see Chap. 12, Sec. 12.1.3). Nausea, vomiting, fatigue, and carcinogenic effects are also common side effects of many drugs. In addition, several drug-specific toxicities to other tissues of the body may be observed. The biological basis for toxic damage to normal tissues that may occur through a common mechanism is discussed below, whereas toxic effects specific for individual drugs are described in Chapter 18. Targeted agents, in particular, may cause a variety of side effects that are not specific to rapidly proliferating tissues.
17.5.2 Toxicity to Bone Marrow and Scheduling of Treatment
Within the bone marrow there is evidence for a pluripotent stem cell that under normal conditions proliferates slowly to replenish cells in the myelocytic, erythroid, and megakaryocytic lineages (Fig. 17–10A). Lineage-specific precursors proliferate more rapidly than stem cells, whereas the morphologically recognizable but immature precursor cells (eg, myeloblasts) have a very rapid rate of cell proliferation. Beyond a certain stage of maturation, proliferation ceases and the cells mature into circulating blood cells. The relationship between proliferation and maturation in bone marrow precursor cells provides a plausible explanation for the observed fall and recovery of blood granulocytes (and more rarely of platelets) that follows treatment with most chemotherapy drugs (Fig. 17–10B). Proliferation-dependent cytotoxic drugs, including most types of chemotherapy, deplete the rapidly proliferating cells in the earlier part of the maturation series, with minimal effects against the more mature nonproliferating cells and against slowly proliferating stem cells. Blood counts may remain in the normal range while the more mature surviving cells continue to differentiate but will then fall rapidly at a time when the cells depleted earlier would normally have completed maturation. A substantial decrease in the number of mature cells is common for granulocytes because their lifetime is only 1 to 2 days, less common for platelets (lifetime of a few days), and rare for red blood cells (mean lifetime of approximately 120 days), but it may also be influenced by differences in the intrinsic sensitivities of their precursor cells for different drugs. The number of mature granulocytes usually decreases at 8 to 10 days after treatment with drugs such as cyclophosphamide, doxorubicin, or paclitaxel, but may do so earlier for other drugs (eg, vinblastine). The variation in time from treatment to the fall in peripheral blood counts for different drugs probably reflects their different effects on the rate of cell maturation. When the peripheral granulocyte count falls, proliferation of stem cells is mediated by release of growth factors, with subsequent recovery of the entire bone marrow population. Administration of growth factors (eg, granulocyte colony-stimulating factor [G-CSF]) after chemotherapy can accelerate the reappearance of mature cells in the peripheral blood and decrease the possibility of infection that can occur in the absence of mature granulocytes. For many drugs (eg, cyclophosphamide, doxorubicin, taxanes), recovery of peripheral blood counts is complete at approximately 3 weeks after therapy (or at ~2 weeks if growth factors are given), and further treatment may be given with little or no evidence of residual damage to bone marrow.
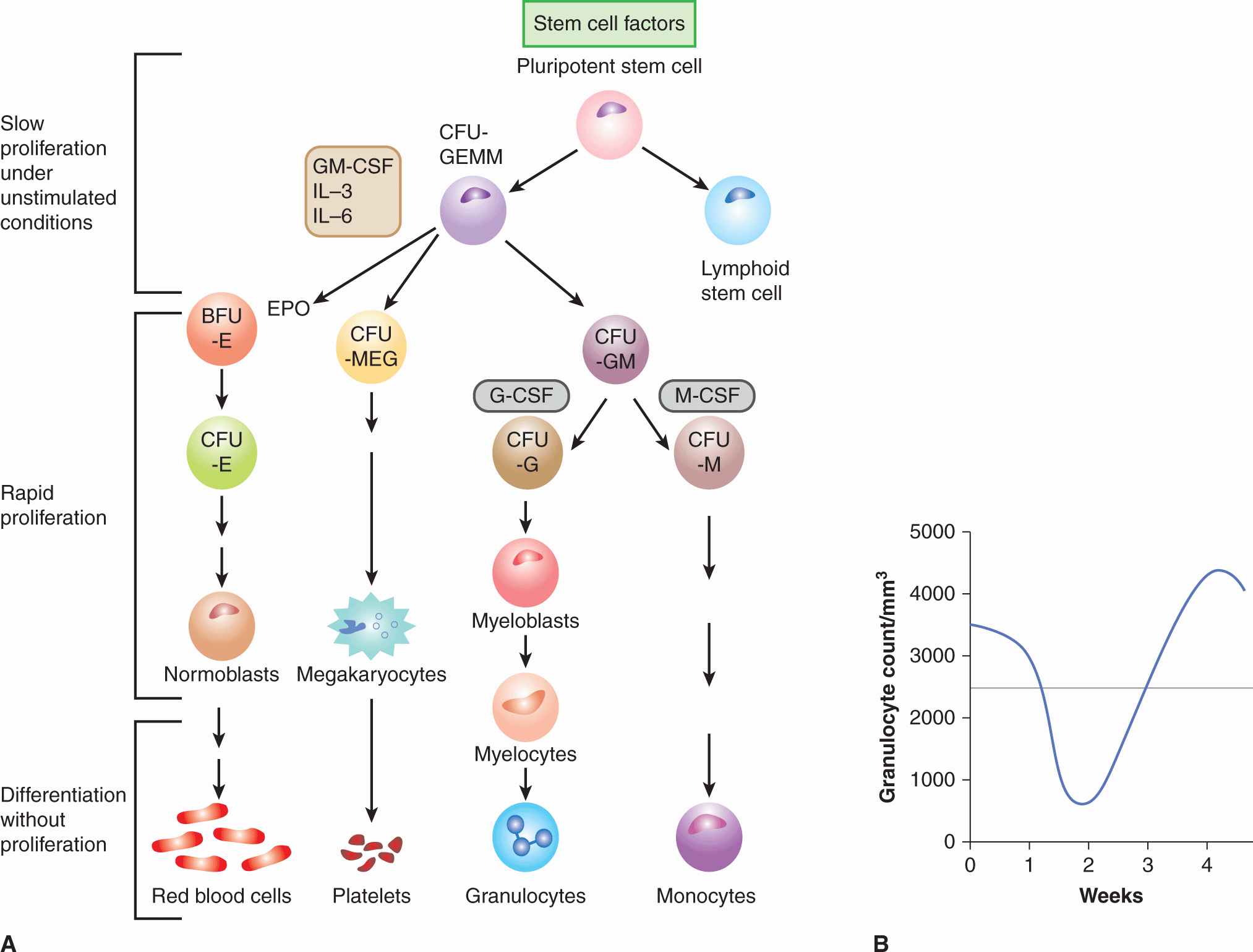
FIGURE 17–10 Pattern of proliferation in bone marrow and explanation of scheduling. A) Schematic of the differentiation of hematopoietic precursor cells in the bone marrow, leading to the production of red blood cells, platelets, granulocytes, and monocytes. CFU, colony-forming unit; BFU, blast-forming unit; E, erythroid; MEG, megakaryocytic; G, granulocytic; M, monocytic. Various cells are stimulated to proliferate and/or differentiate by the growth factors interleukin (IL)-3 and -6, granulocyte-macrophage colony-stimulating factor (GM-CSF), granulocyte colony-stimulating factor (G-CSF), monocyte colony-stimulating factor (M-CSF), erythropoietin (EPO), stem cell factors (eg, Sox2, Klf4, Nanog), and others; only their main target cells are indicated here. Under normal conditions, the early precursor cells proliferate slowly, intermediate precursors proliferate rapidly (in the megakaryocytic series there is nuclear replication without cell division) to expand the population, and later precursors of the functional cells differentiate without further cell division. B) Fall and recovery of the peripheral granulocyte count after chemotherapy. For most drugs the count falls to a nadir at 10 to 14 days after treatment, with complete recovery by 3 to 4 weeks.
Following treatment with some drugs, such as melphalan, or with wide-field radiation to a high proportion of the bone marrow, recovery of mature granulocytes and platelets to normal levels is slower, usually requiring approximately 6 weeks after treatment. Drugs that produce prolonged myelosuppression cause direct damage to slowly or nonproliferating stem cells. Thus recovery is delayed because of repopulation from a smaller number of bone marrow stem cells, and some damage may be permanent because of incomplete repopulation of the stem cell pool.
Recovery of blood counts after treatment with anticancer drugs is the usual determinant of the interval between courses of chemotherapy. If myelosuppressive drugs are given when peripheral blood counts are low, they will not only delay recovery and increase the chance of infection and bleeding, but will also have a higher chance of depleting the stem cell population, because it is likely to be proliferating rapidly. Drug administration can be repeated up to 1 week after initial treatment, before the decrease in mature granulocytes and platelets is observed; this schedule has been incorporated into several drug regimens where anticancer drugs are given on days 1 and 8 of a 21- or 28-day cycle. Some drugs (eg, bleomycin, vincristine, and many of the newer targeted agents) cause only minimal toxicity to bone marrow, probably because of intrinsic resistance of the precursor cells; they can be given when peripheral granulocyte and platelet counts are low following the use of myelosuppressive agents.
Red blood cells have a long lifetime, which usually prevents the rapid development of anemia following initiation of chemotherapy. However, repeated courses of chemotherapy cause repeated interruptions of red blood cell production so that the serum level of hemoglobin tends to decrease slowly, leading to anemia and contributing to fatigue. This effect can occur with all types of drug therapy, but occurs more rapidly following the use of some drugs, such as cisplatin. Injection of erythroid-stimulating agents, which are analogs of the growth factor erythropoietin, can be used to stimulate production of red blood cells, thus minimizing the effects of chemotherapy to cause anemia and reducing the associated fatigue. However, these agents must be used with caution, as some tumor cells may also express the erythropoietin receptor and be stimulated by these agents, and they have also been shown to induce thrombosis in people with near-normal levels of hemoglobin, which may be life-threatening (Hadland and Longmore, 2009).
Chemotherapy is scheduled most often using intermittent large doses, but there is evidence that continuous daily administration of low doses of drugs (known as metronomic therapy) can give superior effects in animal models, perhaps because of superior effects against tumor blood vessels (Kerbel and Kamen, 2004). Although such schedules can have activity (and low toxicity) against some human tumors, there is no evidence as yet that they provide better long-term outcomes.
Many of the molecular-targeted agents described in Section 17.3 require chronic administration to provide sustained inhibition of their target receptor or pathway. Monoclonal antibodies such as trastuzumab or cetuximab have long half-lives in the circulation, and sustained levels can be achieved by dosing at weekly intervals or less often. However, most inhibitors of receptor tyrosine kinases are small molecules that are given orally (eg, everolimus, an inhibitor of mTOR) and these agents are most often given on a continuous daily schedule. Many of these agents inhibit pathways that stimulate proliferation of cancer (and other cells) and although when used alone they do not usually lead to myelosuppression, they can add substantially to this and other toxicities when used in combination with chemotherapy, and a dose reduction of both agents is then generally required.
17.5.3 Toxicity of Drugs to Other Proliferative Tissues
Ulceration of the mucosa in the mouth, throat, esophagus, or intestine may also occur after treatment with antiproliferative drugs, and can lead to soreness, intestinal bleeding, and diarrhea. It is caused by interruption of the production of new cells that normally replace the mature cells continually being sloughed into the intestine (see Chap. 12, Fig. 12–4). Damage to bone marrow is more commonly dose-limiting in humans, but mucosal ulceration may occur after treatment with several drugs, including methotrexate, 5-fluorouracil, bleomycin, and cytosine arabinoside; it may also occur after treatment with several targeted agents, including sunitinib and sorafenib, presumably because they inhibit proliferation and maturation in mucosal epithelium. Mucosal damage usually begins approximately 5 days after treatment, and its duration increases with the severity. Full recovery is usually possible if the patient can be supported through this period; recovery is analogous to that in the bone marrow, with repopulation from slowly proliferating stem cells.
Partial or complete hair loss is common after treatment with many anticancer drugs and is a result of lethal effects of drugs against proliferating cells in hair follicles; this usually begins approximately 2 weeks after treatment. Full recovery usually occurs after cessation of treatment, suggesting the presence of slowly proliferating precursor cells. In some patients, regrowth of hair is observed despite continued treatment with the agent that initially caused its loss. Regrowth of hair might reflect a compensating proliferative process that increases the number of stem cells, or may represent the development of drug resistance in a normal tissue akin to that which occurs in tumors.
Spermatogenesis in men and formation of ovarian follicles in women both involve rapid cellular proliferation and are susceptible to the toxic effects of many anticancer drugs. Men who receive chemotherapy often have decreased production of sperm and consequent infertility. Testicular biopsy usually demonstrates a loss of germinal cells within the seminiferous tubules, presumably because of drug effects against these rapidly proliferating cells. Antispermatogenic effects may be reversible after lower doses of chemotherapy, but some men remain permanently infertile; it is now usual to recommend sperm banking for young men who undergo intensive chemotherapy for potentially curable malignancies such as Hodgkin disease or testicular cancer. Chemotherapy given to premenopausal women often leads to temporary or permanent cessation of menstrual periods and to menopausal symptoms, and is accompanied by a fall in serum levels of estrogen. Reversibility of this effect depends on age, the types of drug used, and the duration and intensity of chemotherapy. Biopsies taken from the ovaries have shown failure of formation of ovarian follicles, sometimes with ovarian fibrosis. The pathological findings are consistent with a primary effect of drugs against the proliferating germinal epithelium.
17.5.4 Nausea, Vomiting, and Other Common Toxicities
Nausea and vomiting are frequent during the first few hours after treatment with many types of chemotherapy, but occur rarely after use of targeted agents. Drug-induced vomiting may occur because of direct stimulation of chemoreceptors in the brainstem, which then emit signals via connecting nerves to the neighboring vomiting center, thus eliciting the vomiting reflex. Major evidence for this mechanism comes from studies in animals, where induction of vomiting by chemotherapy is prevented by removal of the chemoreceptor zone. In addition to a central mechanism, some chemotherapeutic agents exert direct effects on the gastrointestinal tract that may contribute to nausea and vomiting. Several neurotransmitters, such as serotonin (5-HT3) and substance P are involved in transmitting signals involved in producing nausea and vomiting. Medications have been developed that inhibit nausea and vomiting after chemotherapy. The most effective of these are the serotonin antagonists (such as ondansetron, tropisetron, and granisetron), which block 5-HT3 receptors, and the neurokinin 1 (NK1) receptor antagonists (such as aprepitant), which block substance P.
Some drugs can produce diarrhea without directly damaging the intestinal mucosa. For example, irinotecan can produce diarrhea soon after administration as a consequence of a direct cholinergic effect on the cells of the intestinal mucosa. This type of diarrhea may be prevented through the use of anticholinergic drugs. A second form of diarrhea, secretory in nature, may occur several days following administration of irinotecan, and may be a result of damage to the mucosa coupled with cytokine release causing fluid secretion (see Chap. 18, Sec. 18.4.1).
Fatigue is a common side effect of both the presence of cancer and its treatment, and several types of chemotherapy (eg, taxanes) and targeted agents that inhibit multiple receptors (eg, sunitinib, sorafenib, and pazopanib) can cause profound fatigue. Unfortunately there are no pharmacological treatments that have been found to relieve fatigue, and the only strategy of proven benefit is exercise, possible only for select patients with metastatic cancer.
The hand-foot syndrome (or palmar-plantar erythrodysesthesia) may occur during treatment with several types of chemotherapy (eg, capecitabine) and with targeted agents such as sunitinib and sorafenib (Lipworth et al, 2009). Patients have redness and pain on the palms of the hands and soles of the feet, sometimes with blistering and desquamation, which can be quite disabling. The condition responds to reduction of dose of the anticancer drug. The cause of this condition is uncertain, although it might relate in part to sensitivity of proliferating cells in the basal layer of the skin in these sites.
Subtle cognitive dysfunction has also been identified as a side effect of chemotherapy, especially in women who are receiving adjuvant chemotherapy for breast cancer (Ahles et al, 2007; Vardy et al, 2007). The mechanisms underlying these effects are unknown; they may be mediated in part by changes in the levels of sex hormones and induction of menopausal symptoms, but are probably also a result of direct effects of anticancer drugs on the brain.
17.5.5 Drugs as Carcinogens
Many anticancer drugs cause toxic damage through effects on DNA; they can also cause mutations and chromosomal damage. These properties are shared with known carcinogens (see Chap. 4), and patients who are long-term survivors of such chemotherapy may be at an increased risk for developing a second malignancy. This effect has become apparent only under conditions where chemotherapy has resulted in long-term survival for some patients with drug-sensitive diseases (eg, Hodgkin disease, other lymphomas, testicular cancer) or where it is used as an adjuvant to decrease the probability of recurrence of disease following local treatment (eg, breast cancer). Many of the second malignancies are acute leukemias, and their most common time of presentation is 2 to 6 years after initiation of chemotherapy. Increased incidence of solid tumors may also be observed after longer periods of follow up. Alkylating agents are the drugs most commonly implicated as the cause of second malignancy, and there is increased risk if patients also receive radiation. It is often difficult to separate an increase in the probability of second malignancy that may be associated with the primary neoplasm (eg, in a patient with lymphoma) or with a shared etiological factor, either environmental or as a result of genetic predisposition, from that associated with treatment.
Comparisons of the incidence of leukemia and other malignancies in clinical trials that randomize patients to receive adjuvant chemotherapy or no chemotherapy after primary treatment have given conclusive evidence of the carcinogenic potential of some drugs. The relative risk of leukemia in drug-treated, as compared with control patients, was increased in women receiving adjuvant therapy for breast cancer that included an alkylating agent (especially when melphalan was used) but for modern regimens that include conventional dose cyclophosphamide there is no significant increase in relative risk (Curtis et al, 1992). There is a 2- to 3-fold increased relative risk of endometrial cancer following use of tamoxifen, but the absolute risk is below 1% and most are curable by surgery (Matesich and Shapiro, 2003). Drugs that target topoisomerase II (eg, doxorubicin, epirubicin, mitoxantrone, and etoposide; see Chap. 18, Sec. 18.4) have also been identified as causes of treatment-related leukemia, with a relative risk of approximately 1.5 compared to those not receiving this treatment (Patt et al, 2007). Leukemias that occur following treatment with these drugs have a limited number of characteristic chromosomal translocations that distinguishes them from those that occur following alkylating agents, and they tend to occur after a shorter latent period of 1 to 3 years after treatment of their primary cancer (Mistry et al, 2005).
The risk of second solid tumors following treatment with chemotherapy is far lower than that of leukemia. Nonetheless, a 4.5-fold increase in the risk of transitional cell carcinoma of the urothelium has been demonstrated in patients who had received cyclophosphamide for the treatment of non-Hodgkin lymphoma (Travis et al, 1995), and there is an increased risk of breast and other cancers in patients who are treated for Hodgkin lymphoma with radiotherapy or chemotherapy, and especially in those receiving both treatments. The absolute risk of second malignancy is small compared with the potential benefits in treating curable cancers but care is needed in using carcinogenic drugs as adjuvant chemotherapy for malignancies where benefit is minimal.
17.5.6 Determinants of Normal-Tissue Toxicity
When chemotherapy is given to a patient, a drug dose is selected on the basis of early phase clinical trials that have determined the average dose (usually per unit of body surface area) that gives some toxicity, but at an acceptable level. At this dose, there may be a small proportion of patients, who experience severe, potentially lethal toxicity. Multiple factors influence the distribution of drugs to tissues in the body (see Chap. 18, Sec. 18.1) and the response of normal cells to these drugs. Some patients have genetically determined traits that influence drug metabolism or excretion, and the study of genetically determined factors that influence the probability of drug toxicity is known as pharmacogenetics (see Chap. 3, Sec. 3.6.2 and Chap. 18, Sec. 18.3.1). For example, patients who lack the enzyme dihydropyrimidine dehydrogenase (DPD), which catabolizes 5-fluorouracil show extreme sensitivity to this drug (see Chap. 18, Sec. 18.3.2; Milano et al, 1999). Genetic abnormalities that give rise to the DPD-deficient phenotype have been identified, and screening tests can identify susceptible individuals. Changes in the activity of enzymes that metabolize other drugs, either genetically determined or induced by concomitant medications, may also have a profound effect on drug-induced toxicity.
Because lethal damage caused by chemotherapy results most often from interaction of drugs with DNA, patients with deficiencies in DNA repair (see Chap. 5, Sec. 5.5) are very sensitive to anticancer drugs, as they are to radiation. People who are heterozygous for such gene mutations (eg, xeroderma pigmentosum or ataxia telangiectasia) may also be at high risk for severe toxicity if treated by chemotherapy. Predictive assays, based on assessing chromosomal damage in lymphocytes, are being developed that could allow identification of individuals who may exhibit extreme radio- (and possibly chemo-) sensitivity (Pfuhler et al, 2011). However, the clinical utility of such tests will need to be evaluated carefully, given the low prevalence of the abnormalities being tested, although such individuals may be overrepresented among cancer patients (see also Chap. 5, Table 5–1).
17.6 TREATMENT WITH MULTIPLE AGENTS
17.6.1 Influence on Therapeutic Index
Patients are treated frequently with drug combinations or with drugs and radiation therapy. When 2 or more agents are combined to give an improvement in the therapeutic index, this implies that the increase in toxicity to critical normal tissues is less than the increase in damage to tumor cells. Because the dose-limiting toxicity to normal tissues may vary for different drugs and for radiation, 2 agents are often combined with only minimal reduction in doses as compared with those that would be used if either agent were given alone. Additive effects against a tumor with less than additive toxicity for normal tissue may then lead to a therapeutic advantage. Mechanisms by which different agents may give therapeutic benefit when used in combination have been classified by Steel and Peckham (1979) as follows: (a) independent toxicity, which may, for example, allow combined use of anticancer drugs at full dosage; (b) spatial cooperation, whereby disease that is missed by one agent (eg, local radiotherapy) may be treated by another (eg, chemotherapy); (c) protection of normal tissues; and (d) enhancement of tumor response, where there is selective sensitization of effects of one agent against tumor cells by another.
The above mechanisms suggest guidelines for choosing drugs that might be given in combination. Most drugs exert dose-limiting toxicity for the bone marrow, but this is not the case for vincristine (dose-limiting neurotoxicity), cisplatin (nephrotoxicity), or bleomycin (mucositis and lung toxicity). Some (but not all) of these drugs can be combined with myelosuppressive agents at close to full dosage and have contributed to the therapeutic success of drug combinations used to treat lymphoma and testicular cancer. Most trials of combined chemotherapy and targeted agents have led to increased toxicity and a requirement for dose reduction, even though the targets of the drugs are different. Most such combinations have employed concurrent treatment, which is conceptually counterintuitive: targeted agents often act initially to inhibit proliferation of target cells, which might then protect them from cycle-active chemotherapy. A more logical schedule may be to use chemotherapy and targeted agents in sequence, and might have the added advantage of inhibiting tumor cell repopulation in the intervals between chemotherapy (see Chap. 19, Sec. 19.3.4; Kim and Tannock, 2005).
17.6.2 Synergy and Additivity
Claims are made frequently that 2 agents are synergistic, implying that the 2 agents given together are more effective than would be expected from their individual activities. Confusion has arisen because of disagreement as to what constitutes an expected level of effect when 2 noninteracting agents are combined. The use of multiple agents may lead to an increase in the therapeutic index, but it is rare that a claim for synergy of effects against a single population of mammalian cells can be substantiated (Ocana et al, 2012). Two methods have been used to evaluate possible synergy, additivity, or antagonism between 2 agents: isobologram analysis as proposed by Steel and Peckham (1979) and calculation of an “interaction index,” based on the median effect principle (Chou and Talalay, 1984; Lee et al, 2007).
The above concepts require consideration of the dose–response relationship following treatment of a single population of cells either in a tumor or in a normal tissue, by either agent alone, or of a combination of the two agents. Thus, suppose a given dose of agent A gives a surviving fraction of cells (SA), that a surviving fraction (SB) follows treatment with a given dose of agent B, and a combination of the agents gives a surviving fraction (SAB; Fig. 17–11). Claims for synergy are often made if SAB is less than the product SA × SB. This conclusion is correct only if cell survival is exponentially related to dose for both agents. If the survival curves have an initial shoulder, as in Figure 17–11, then combined treatment will be expected to lead to a lower level of survival if, after treatment with the first agent, A, the survival falls exponentially with dose (in the absence of a shoulder effect) for the second agent, B (Fig. 17–11). The fallacy of defining this lower level of survival as a synergistic effect can be illustrated by replacing agent B with a second, equivalent dose of agent A given immediately after the first dose: the combined survival curve then follows that for agent A (Fig. 17–11A). If agent A has a survival curve with an initial shoulder, one would then conclude erroneously that agent A was synergistic with itself.
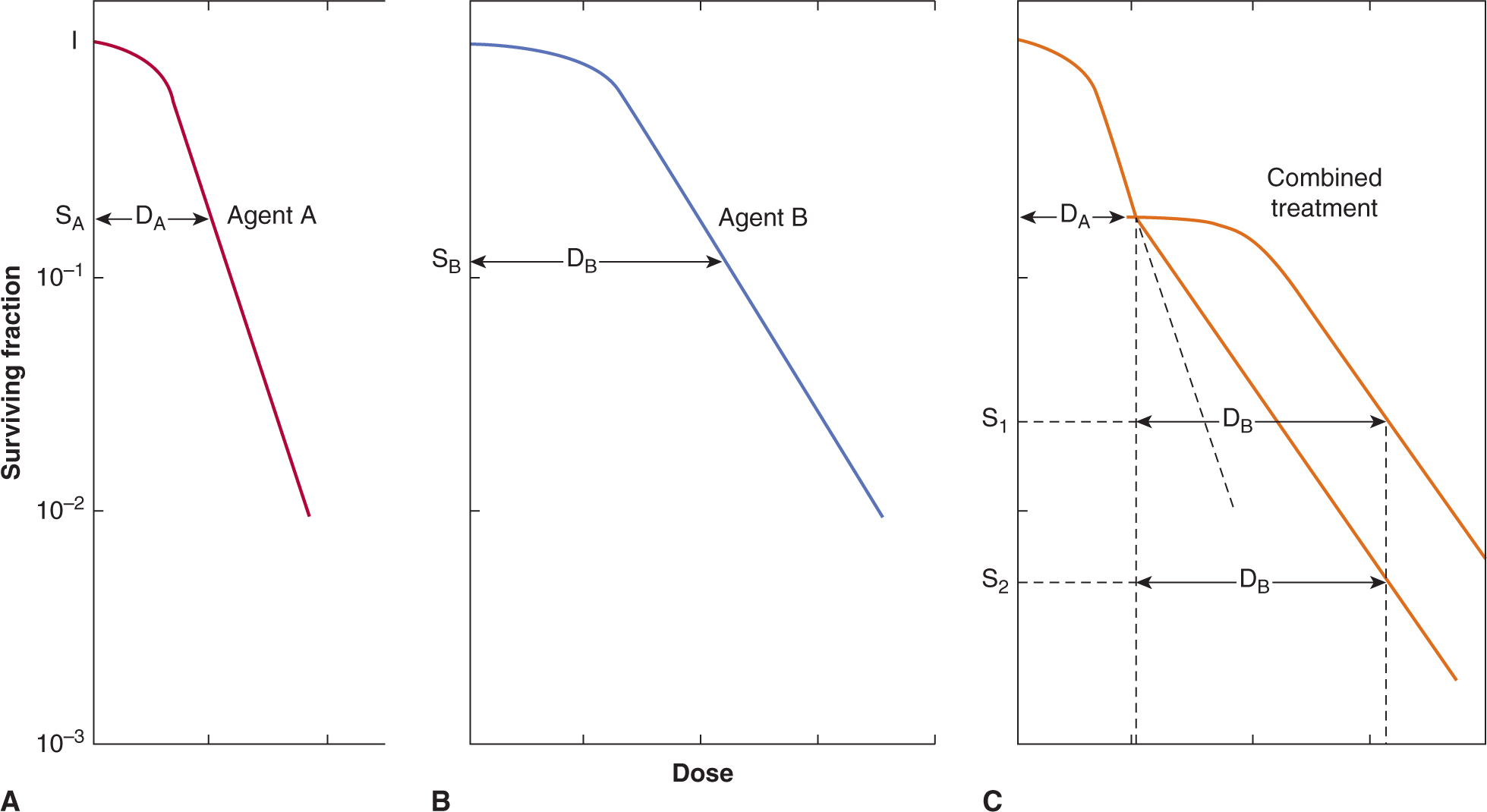
FIGURE 17–11 Interaction of 2 agents A and B. A, B) Cell survival (SA or SB) is indicated following treatment with either of 2 agents, A and B, each of which has a survival curve characterized by an initial shoulder followed by an exponential fall with increasing dose. C) Survival (SAB) after combined use of dose DA of agent A and dose DB of agent B will be equal to S1 = (SA × SB) if there is no overlap of damage, and the “shoulder” representing accumulation of sublethal damage is retained for the second agent. Survival after combined treatment (SAB) will be equal to S2 if cells have accumulated maximum sublethal damage from the first agent A, and the “shoulder” of the curve is lost for the second agent, B.
The above discussion implies that there is a range over which 2 agents can produce additive effects. Isobologram analysis provides a method for defining this range of additivity (Steel and Peckham, 1979). Dose–response curves are first generated for each agent used alone. These Dose–response curves are then used to generate isoeffect plots (known as isobolograms). These curves relate the dose of agent A to the dose of agent B that would be predicted, when used in combination, to give a constant level of biological effect (eg, cell survival) for the assumptions of (a) independent damage and (b) overlapping damage (Fig. 17–12). These curves define an envelope of additivity. If, when the 2 agents are given together, the doses required to give the same level of biological effect lie within the envelope, the interaction is said to be additive. If they lie between the lower isobologram and the axes (ie, the combined effect is caused by lower doses of the 2 agents than predicted) the interaction is supraadditive or synergistic. If the required doses of the 2 agents in combination lie above the envelope of additivity (ie, the effect is caused by higher doses than predicted), the interaction is subadditive or antagonistic (Fig. 17–12).
The median effect principle represents an alternative method of evaluating additivity or synergy between agents and a simplified explanation of the principle has been provided (Chou, 2010). The method also depends on the availability of a dose–effect relationship for each of the agents used alone, and for both agents in combination, and relies on the calculation of a “combination index” (CI); a computer program is available to facilitate this calculation. A CI of 1 represents additivity, a CI greater than 1 represents synergy, and a CI less than 1 represents antagonism. The method can also be used to plot a normalized isobologram, and although the mathematical formulation is more complex, it is in conceptual agreement with the representation described in Figures 17–11 and 17–12.
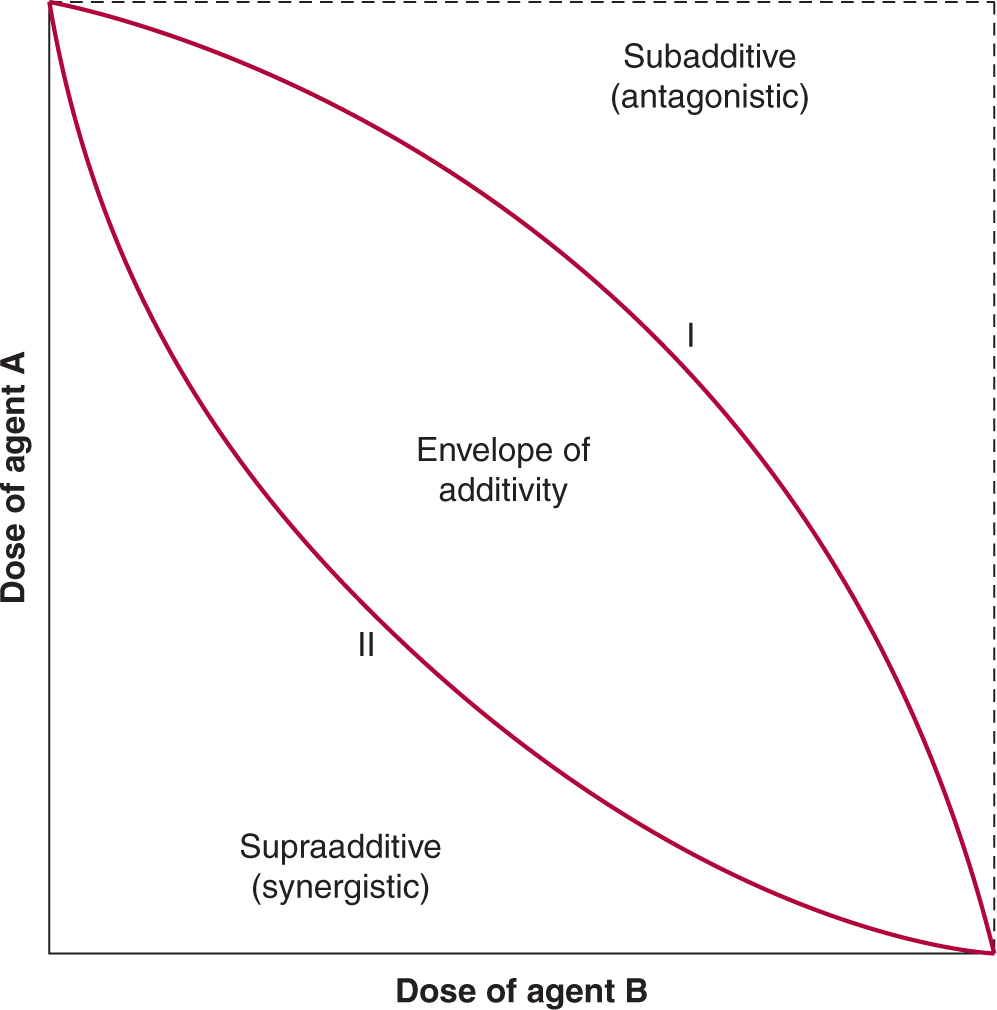
FIGURE 17–12 Isobologram analysis. Isobologram relating the doses of 2 agents that would be expected to give a constant level of biological effect when used together. It was generated from dose–response curves for each agent separately. Assumptions about overlap or non-overlap of damage (see Fig. 17–11) lead to the generation of 2 isobologram curves (I and II) that describe an envelope of additive interaction. Experimental data falling outside this envelope may indicate synergistic or antagonistic interactions, as shown. (Adapted from Steel and Peckham, 1979.)
Demonstration that 2 or more agents have a supraadditive or synergistic interaction has been used as a rationale for their inclusion in clinical protocols (Ocana et al, 2012). This rationale is valid only if the interaction leads to a greater effect against the tumor as compared with that against limiting normal tissues (ie, if it leads to an improvement in therapeutic index; see Sec. 17.5.1). It is theoretically possible that antagonistic agents (subadditive interaction) could improve therapeutic index provided that there was greater antagonism of toxic effects for normal tissues as compared to toxicity for the tumor, or they have non-overlapping toxicities.
17.6.3 Modifiers of Drug Activity or Toxicity
Some drugs with little or no toxicity for tumor cells may modify the action of anticancer drugs to produce increased antitumor effect or may protect normal tissue. Examples of interactions that might lead to therapeutic benefit through increased antitumor effects include (a) the use of doxorubicin with agents such as verapamil, cyclosporine, or their analogs (which inhibit multidrug resistance; see Chap. 19, Sec. 19.2.3.4), and (b) use of folinic acid with 5-fluorouracil (5-FU), which may provide a necessary cofactor for inhibition of the target enzyme thymidylate synthase (see Chap. 18, Sec. 18.3.2). Alternatively, reduction of the toxic effects of chemotherapy against bone marrow may be achieved by coadministration of growth factors such as G-CSF, which can stimulate earlier recovery of mature granulocytes after bone marrow suppression by chemotherapy, or after stem cell transplantation (see Sec. 17.5.2). G-CSF is used commonly in situations where reduction in dosage of chemotherapy might lead to a decrease in the probability of cure or long-term survival of patients. Growth factors are also being developed to protect against the effects of chemotherapy (and radiotherapy) on other body systems. For example, recombinant human thrombopoietin, and other agents that stimulate platelet production, have shown activity in clinical trials (Vadhan-Raj, 2010). Also palifermin (recombinant keratinocyte growth factor), can stimulate proliferation of the oral and gastrointestinal mucosa, and has been used in selected patients to decrease mucosal injury following chemotherapy (Vadhan-Raj et al, 2010).
Two other agents that may protect normal tissues from damage caused by chemotherapy are dexrazoxane and amifostine. Dexrazoxane is a prodrug with an active form that chelates iron. Because complexes between iron and anthracyclines, such as doxorubicin (and the consequent formation of free radicals), appear to mediate cardiac toxicity but not antitumor effects, dexrazoxane may decrease cardiac toxicity of these drugs and increase their therapeutic index (Speyer et al, 1988; Venturini et al, 1996). Amifostine is also a pro-drug that is converted to a sulfhydryl-containing active form. Amifostine is localized selectively in normal tissues, probably because of increased activity of the activating enzyme alkaline phosphatase on the membranes of normal cells. Therefore, it may offer selective protection against a variety of drugs (and radiation) that damage cells by producing reactive intermediates that bind to sulfhydryl groups (Kemp et al, 1996). There remain concerns, however, that these agents might also provide some protection of tumor cells from drug effects.
The crucial test for all modifiers is the demonstration in well-designed clinical trials that higher doses of chemotherapy given with the modifier improve therapeutic index (for example, by increasing the probability or duration of tumor response with no increase in toxicity) as compared to lower doses of chemotherapy used alone.
17.6.4 Drugs and Radiation
Many patients receive treatment with both drugs and radiation, and there is evidence that concurrent treatment with radiation and drugs such as cisplatin leads to improvement in therapeutic index in a variety of cancer sites such as the head and neck and uterine cervix. Here, radiation therapy is the primary treatment and local failure is a major problem that can be decreased with use of concurrent chemotherapy. Mechanisms of interaction between drugs and radiation at the cellular level may be evaluated from cell survival curves for radiation obtained in the presence or absence of the drug (Fig. 17–13). Drugs may influence the survival curve in at least 3 ways: (a) the curve may be displaced downward by the amount of cell kill caused by the drug alone; (b) the shoulder on the survival curve may be lost, suggesting an inability to repair radiation damage in the presence of the drug; and (c) the slope of the exponential part of the survival curve may be changed, indicating sensitization or protection by the drug. Most drugs influence survival curves according to the first 2 patterns; this effect corresponds to the limits of additivity defined in Section 17.6.2, where sublethal damage may be independent or overlapping. The third pattern, leading to a change in slope of the dose response curve, defines agents that are radiation sensitizers or protectors (see Chap. 16, Secs. 16.4.4.2 and 16.5.7). Sensitization of this type has been reported inconsistently for cisplatin and for prolonged exposure to 5-FU after radiation.
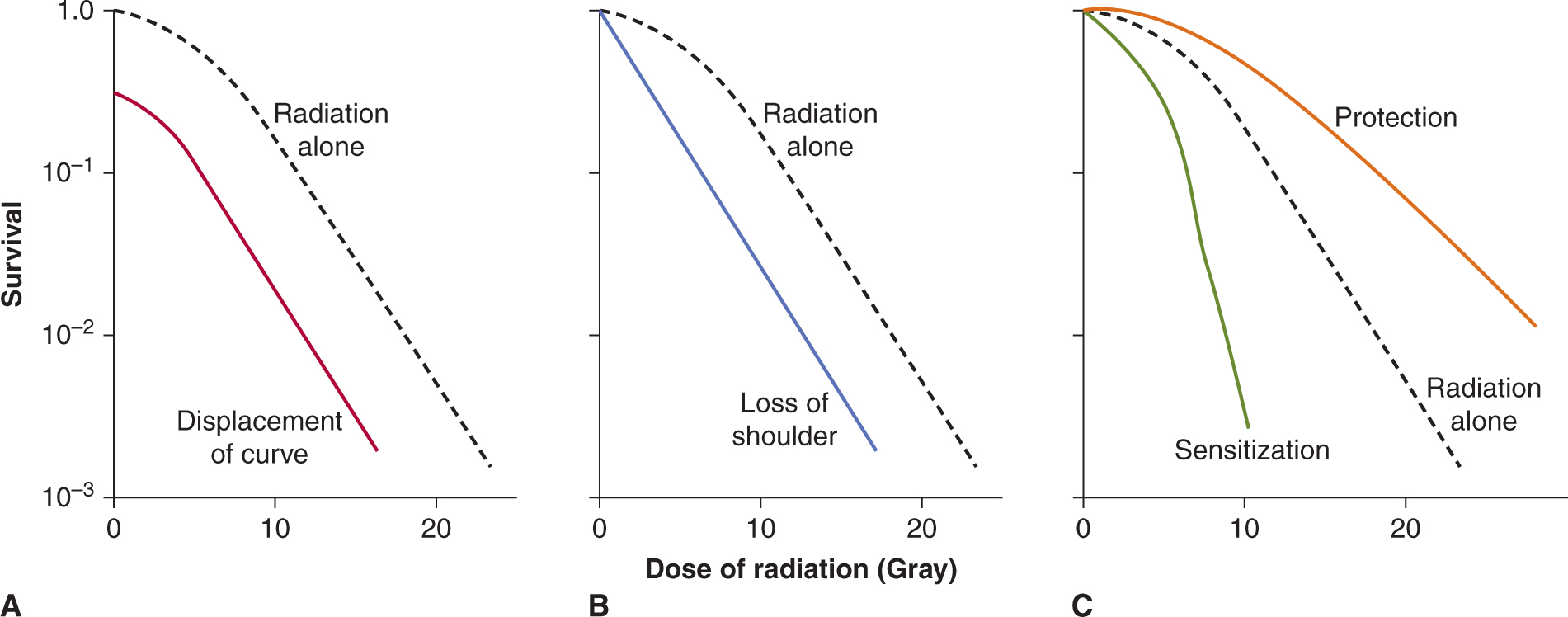
FIGURE 17–13 Drugs and radiation. Possible influences of drug treatment on the relationship between radiation dose and cell survival: (A) displacement of curve; (B) loss of shoulder, indicating effects of drug on the repair of sublethal radiation damage; (C) change in the slope of the curve, indicating sensitization or protection.
Improvement in therapeutic index from use of drugs and radiation requires selective effects to increase damage to tumor cells as compared to those in normal tissues. One mechanism by which combined treatment with radiation and drugs leads to therapeutic advantage arises when radiation is used to provide effective treatment for sites of bulky disease (usually the primary tumor) and drugs are used to treat metastatic sites containing smaller numbers of cells. This spatial cooperation (see Sec. 17.6.1) requires no interaction of the 2 modalities, but involves different dose-limiting toxicities. There are also mechanisms whereby the combined use of radiation and drugs might be used to obtain therapeutic advantage for treatment of a primary tumor. Table 17–2 lists some properties of cells that might be exploited to give therapeutic advantage for the combined use of radiation and drugs.
Genetic instability in tumors often leads to the presence of sub-clones, which coexist in the tumor with different levels of sensitivity to drugs and to radiation (see Chap. 5, Sec. 5.2). When therapy is applied, any resistant cells that are present will have a selective survival advantage and will determine tumor response; thus, heterogeneity in therapeutic response may tend to make tumors more resistant to treatment than normal tissues. Combined treatment with radiation and drugs might then lead to improved therapeutic index if radiation can eradicate small populations of drug-resistant cells, or if drugs can eliminate populations that are relatively resistant to radiation therapy. This cooperative effect requires that mechanisms of resistance to the 2 therapeutic agents are independent. Mechanisms (other than hypoxia) that convey clinical resistance to radiotherapy remain poorly understood, but probably include enhanced ability to repair damage to DNA, increased levels of sulfhydryl compounds such as glutathione (or of associated glutathione S-transferase enzymes) that scavenge free radicals (especially in hypoxic cells), and decreased ability to undergo apoptosis. These mechanisms may also convey resistance to some anticancer drugs, whereas many other mechanisms of drug resistance (see Chap. 18) are unlikely to cause resistance to radiation. Resistance to any given drug may be caused by multiple mechanisms so that a radiation–drug combination that provides therapeutic advantage for one tumor may not do so for another if different mechanisms of drug resistance are dominant. Effective use of combined treatment would be facilitated by rapid pre-treatment assays that give insight into mechanisms of resistance prior to initiation of therapy.
Proliferation of surviving cells during a course of fractionated radiation (ie, repopulation; see Chap. 19, Sec. 19.3.4) acts to increase the total number of cells that must be killed. Anticancer drugs given during the course of fractionated radiation might be expected to inhibit repopulation (Fig 17–14). Combined treatment may then convey therapeutic advantage if the rate of repopulation is greater for the tumor cells than it is for normal tissues within the radiation field. Greater specificity would be expected for agents that inhibit specifically the proliferation of tumor cells; this might be achieved through use of hormonal agents (tamoxifen, antiandrogens) used concurrently with radiation for treatment of breast or prostate cancer (see Chap. 20, Sec. 20.4), or through use of molecular-targeted agents. Improved survival of patients with head and neck cancer treated with radiotherapy and concurrent cetuximab, an inhibitor of EGFR, as compared to radiotherapy alone (Bonner et al, 2010), is most likely a result of inhibition of repopulation of tumor cells during the course of radiotherapy, although it is not clear that this targeted agent provides better therapeutic outcome than concurrent cisplatin.
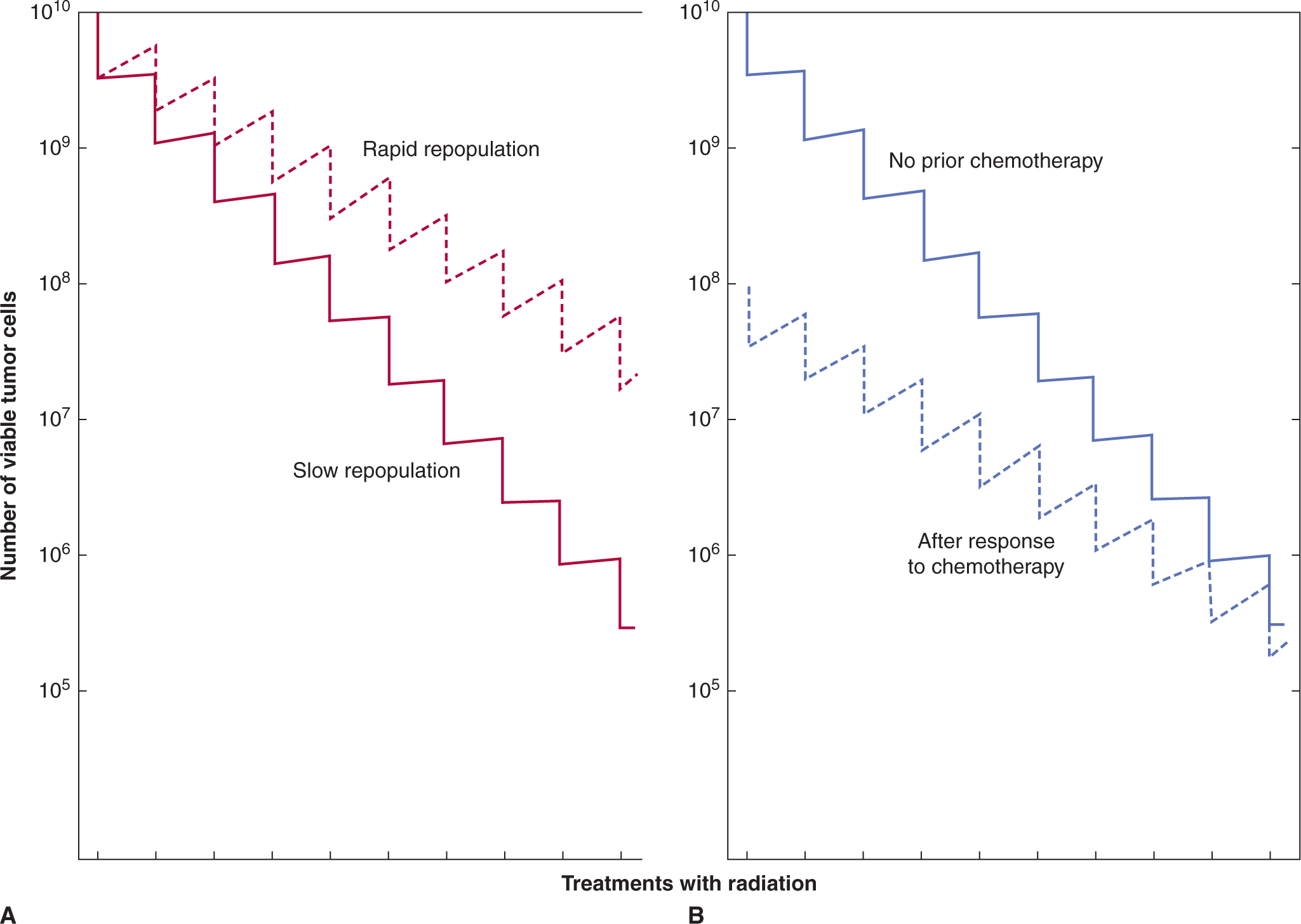
FIGURE 17–14 Schematic illustrating the effect of repopulation in a tumor during a course of fractionated irradiation. Each radiation fraction is assumed to kill the same fraction of tumor cells. In (A), the effect of different rates of repopulation is illustrated. In (B), it is assumed that prior chemotherapy kills 99% of the cells but induces “accelerated” repopulation by the survivors. Response to radiation treatment alone (solid line) or radiation treatment following the chemotherapy (dashed line) is illustrated. The accelerated repopulation induced by the prior drug treatment may rapidly negate the extra cell kill achieved by the drug treatment.
Repopulation during fractionated radiation therapy might also be influenced by prior treatment with neoadjuvant chemotherapy. Such chemotherapy may cause tumor shrinkage, followed by improved nutrition of surviving cells, with consequent stimulation of cell proliferation (Withers et al, 1988). If there is increased repopulation of surviving cells during the subsequent course of fractionated radiation therapy, any advantage from initial shrinkage of the tumor caused by chemotherapy may be lost or reversed because of the decreased net effectiveness of subsequent radiation treatment (Fig. 17–14).
Regrowth of tumors following radiotherapy depends on maintenance of a vascular supply to provide nutrients to surviving cells. Clinical courses of radiotherapy are likely to lead to killing of endothelial and other cells that constitute the preexisting vasculature and it has been proposed that tumor regeneration depends on recruitment of circulating myeloid precursors that form new blood vessels in irradiated tumors (Ahn and Brown, 2009). These circulating precursors might be killed or suppressed by chemotherapy given concurrently with radiation, or by agents that target them more specifically (Ahn et al, 2010).
A fourth mechanism that has potential for exploitation through combined use of radiation and drugs depends on the presence of a hypoxic microenvironment within solid tumors (see Chap. 12, Sec. 12.2). An hypoxic environment conveys resistance to radiation because cell killing is dependent in part on the presence of oxygen (see Chap. 16, Sec. 16.4). Several drugs have been developed that require bioreduction under hypoxic conditions for activity, and therefore have selective toxicity for hypoxic cells; effective drugs may also diffuse to influence neighboring aerobic regions (Brown, 1999). Such drugs would be expected to have fewer effects against normal tissues, where adequate vasculature usually prevents development of a hypoxic microenvironment, although there is evidence for hypoxic regions in bone marrow and some other normal tissues (Parmar et al, 2007). Hypoxia-selective drugs might augment both the effects of radiation, and of conventional chemotherapy, whereas hypoxic cells and their neighbors might be spared because of limited drug distribution within solid tumors and their low proliferative rate (Minchinton and Tannock, 2006; see Chap. 19, Sec. 19.3.2). The first-generation hypoxia-selective agent, tirapazamine, has been evaluated with cisplatin with and without radiotherapy for patients with lung or head and neck cancer (von Pawel et al, 2000; Rischin et al, 2010). Despite some positive effects especially in tumors selected for hypoxia (Rischin et al, 2006), this drug adds toxicity without long-term benefit but more promising hypoxia-activated pro-drugs, such as TH-302, are being evaluated in clinical trials.
Whenever radiation and drugs are used together or in sequence, there is potential for increased damage to normal tissues in the radiation field. Some of the effects of combined treatment may lead to changes in function that occur months to years after treatment. Both clinical experience and studies in animals have shown that most anticancer drugs can increase the incidence of toxicity from radiation, sometimes in organs (eg, the kidney) where the drugs alone rarely cause overt toxicity (von der Maase, 1986). The effect of a drug on radiation toxicity to any organ may be expressed in terms of a dose-enhancement ratio (DER), which is the dose of radiation to produce a given effect when used alone divided by the dose of radiation that gives the same effect when combined with the drug. For acute effects of radiation on normal tissues of mice, typical values of DER range from 1.0 to 1.5, depending on the drug and normal tissue; maximum interaction occurs when drug and radiation are administered within a short time span (von der Maase, 1986). The therapeutic gain factor equals the ratio of DER for the tumor to the DER for the dose-limiting normal tissue in the radiation field. In experimental systems, this ratio may vary widely depending on the drug used, the doses of drug and radiation, and the treatment sequence. Thus, it is difficult to predict the dose schedules that are likely to lead to therapeutic gain in patients.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


