Topoisomerase Inhibitors: Camptothecins, Anthracyclines, and Etoposide
Topoisomerases carry out the important function of unwinding DNA by creating temporary breaks in DNA, promoting passage of single strands of DNA through breaks, and then resealing the breaks. This function is critical in allowing access of repair and replication complexes to linear strands of DNA. Similar enzymes are found throughout the eukaryotic and prokaryotic world, a testament to their essential function. Likewise, they are one of the most common targets of naturally occurring poisons. Two classes of topoisomerases (topos) are found in human cells: topo I does not require ATP for strand breakage and resealing, and creates single strand breaks, while the several isoforms of topo II require ATP and create double strand breaks.
TOPOISOMERASE I INHIBITORS: CAMPTOTHECINS
The camptothecins are inhibitors of topo 1 with broad activity against epithelial cancers. Camptothecin was isolated from the Chinese tree Camptotheca acuminata in 1966 and had potent antitumor effects in animal systems. Topotecan (Hycamptin) and irinotecan (Camptosar), two synthetic agents in this class, have been subsequently approved for clinical use in the United States. Topotecan is currently used as second-line chemotherapy for ovarian cancer and small cell lung cancer (SCLC). Irinotecan is indicated for the treatment of metastatic colon cancer, both in first-line and salvage combination therapy, and has been incorporated into regimens to treat small cell lung cancer, gynecologic, and upper gastrointestinal malignancies.
 STRUCTURE
STRUCTURE
The camptothecins consist of a five-ring structure in which a quinolone moiety is joined to a terminal α-hydroxy-δ-lactone ring (Figure 4-1). The electrophilic center of the lactone subunit is responsible for the camptothecins’ biological activity. At the same time, the lactone is also vulnerable to reversible hydrolysis to a less active carboxylate species at neutral and alkaline pH. Substitutions on the C-9 and C-10 positions on the quinolone ring stabilize the lactone and enhance antitumor activity by preventing its conversion to the inactive carboxylate form in human blood and tissues (1).
 MECHANISM OF ACTION
MECHANISM OF ACTION
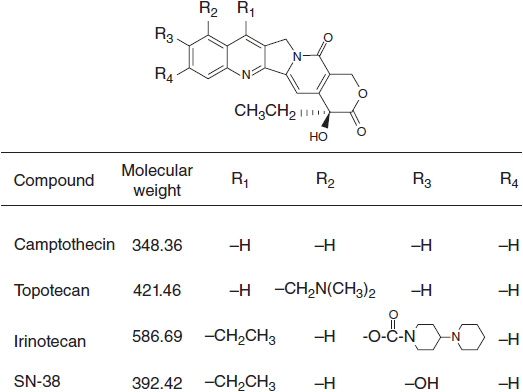
Camptothecin and its analogs exert their antitumor activity by inhibiting the enzyme DNA topo I, a nuclear enzyme that relieves torsional strain in supercoiled DNA during replication, repair, and transcription (Figure 4-2). The enzyme forms a transient, intermediate complex with single-stranded DNA that opens the DNA strand and allows passage of an intact single strand through the nick. The strand break also allows for rotation about the intact strand. Camptothecins bind to and stabilize the otherwise transient DNA-enzyme complex and prevent resealing of the broken strand. The single strand break, when it encounters a replication complex, leads to a double-stranded break, and an accumulation of double strand breaks leads to apoptosis. Active synthesis of DNA is a prerequisite for this interaction (1). In addition to strand breakage, irinotecan causes a downregulation of HIF1-alpha, a transcription factor critical to cell survival in the presence of hypoxia; angiogenesis is inhibited as a result (2). The mechanism of drug effects on HIF1-alpha is not known.
 MECHANISMS OF RESISTANCE
MECHANISMS OF RESISTANCE
In preclinical studies, resistance to the camptothecins may result from multiple mechanisms: increased expression of the multidrug resistance (MDR) efflux pump, p-glycoprotein, and a related transporter, ABCG2, the breast cancer resistance transporter; downregulation of topo I expression; mutations in the catalytic or DNA binding sites of topoisomerase I; upregulation of topoisomerase II; upregulation of NFκB; and inhibition of apoptosis. The clinical significance of these mechanisms of resistance is unproven.
CAMPTOTHECINS: CLINICAL PHARMACOLOGY
 TOPOTECAN
TOPOTECAN
Pharmacokinetics
Topotecan is usually given as a 30-min intravenous infusion. Plasma concentrations of the inactive carboxylate species begin to predominate over the active lactone form within 5–10 min after the end of infusion. The plasma half-life of topotecan is 2.4–4.3 h. Topotecan is eliminated primarily by plasma hydrolysis to the inactive carboxylate form, followed by renal excretion. Approximately one-third to one-half of a dose of drug is excreted unchanged in the urine. The clearance of topotecan and the carboxylate metabolite is reduced by 33% in patients with renal dysfunction and a creatinine clearance between 40 and 59 ml/min, and by 75% in patients with a creatinine clearance between 20 and 39 m/min. There is no significant change in elimination or toxicity in patients with liver disease, even in patients with total bilirubin levels up to 10 mg/dl. Administration of cisplatin decreases topotecan clearance, presumably through renal tubular damage. Topotecan pharmacokinetics are not altered when it is combined with anthracyclines, cyclophosphamide, or cytarabine (1).
Dosing and Schedule
The standard dosing schedule for topotecan is 1.5 mg/m2 given as a 30-min intravenous infusion on 5 consecutive days, repeated every 21 days. Continuous infusion regimens up to 21 days in duration have attempted to take advantage of the in vitro observation that prolonged exposures of low concentrations are more efficacious than intermittent exposures to high concentrations. Results have been equivocal.
Toxicity
Neutropenia is the most significant dose-limiting toxicity for all schedules of topotecan administration, with grade 4 neutropenia in up to 81% of patients and febrile neutropenia in 26%. Renal dysfunction requires a dose reduction as indicated above. There is no dose modification required for patients with hepatic dysfunction. Non-hematologic toxicities of topotecan include nausea, vomiting, mucositis, elevated transaminases, fatigue, and rash. These side effects are generally minimal and easily managed (1).
 IRINOTECAN
IRINOTECAN
Pharmacokinetics
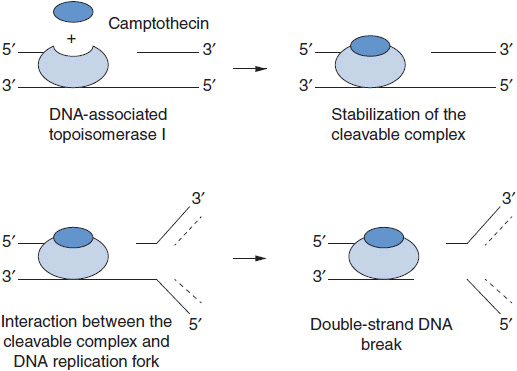
Irinotecan (Camptosar) is a congener of camptothecin specifically designed to facilitate generation of its lactone-stabilized metabolite, the 7-ethyl-10-hydroxy analog, SN-38, which is a 1000-fold more potent inhibitor of topo I than the parent drug. Irinotecan is primarily eliminated through the liver via two clinically relevant mechanisms (Figure 4-3). First, irinotecan is a substrate of the cytochrome p450 system and is metabolized to inactive derivatives by CYP2B6 and CYP3A4. These inactivation pathways are inducible by phenobarbital or phenytoin (1). Second, irinotecan is converted to SN-38 by ubiquitous esterases, and SN-38 is then cleared by glucuronidation and excreted in the biliary system. The half-life of the active lactone form of SN-38 is 11.5 h (1).
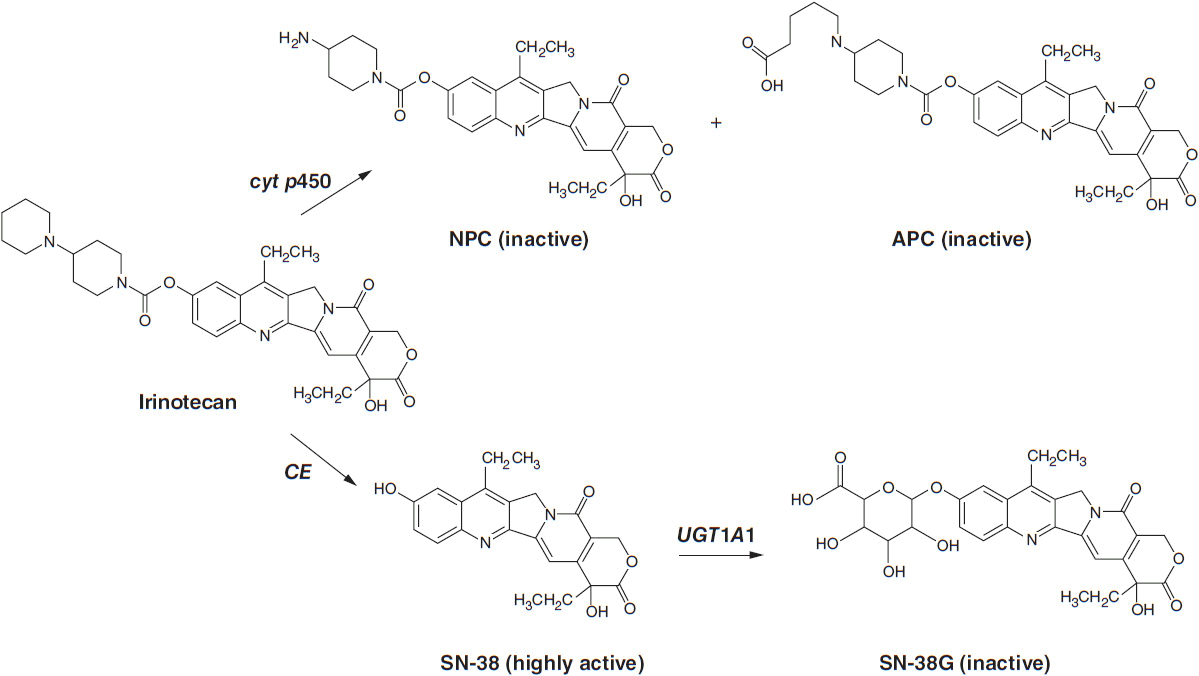
FIGURE 4-3 Metabolic pathway for irinotecan showing the conversion to inactive metabolites NPC and APC via cytochrome p450 enzymes and the conversion by liver carboyxylesterase (CE) to the active form, SN-38, and Its subsequent inactlvation to SN-38G by the enzyme UGT1A1.
SN-38 undergoes glucuronidation by the polymorphic enzyme uridine diphosphoglucuronosyl-transferase (UGT1A1), which is also responsible for bilirubin glucuronidation. The activity of this enzyme is significantly reduced in patients homozygous for the allele UGT1A1*28, the same defect seen in subjects with Gilbert’s syndrome. This homozygous deficiency is found in approximately 10% of patients. When treated with standard doses of irinotecan, such patients have slower clearance of the parent drug and higher levels of plasma SN-38 and encounter higher rates of toxicity, particularly neutropenia (3, 4). In patients with Gilbert’s syndrome, or with an unexplained elevated indirect bilirubin level, lower starting doses of irinotecan should be used. A commercially available test for the UGT1A1*28 polymorphism (Invader UGT1A1*28 Molecular Assay) can be used, although it has not been widely accepted in clinical practice. Common polymorphisms in ABC cassette drug transporters, which export the parent drug and SN-38 from intestinal epithelium, may account for the severe gastrointestinal toxicity that affects 20%–30% of patients (5).
Dosing and Schedule
Irinotecan, as a single agent, can be given at a dose of 125 mg/m2 over a 90-min intravenous infusion every week for 4 of 6 weeks or at a dose 350 mg/m2 over a 90-min intravenous infusion every 3 weeks. The weekly schedule appears to be equally effective, although there are lower rates of diarrhea with the every 3-week regimen (6).
Toxicity
The most common adverse effects of irinotecan are diarrhea, which can be life-threatening in some instances, myelosuppression, and an acute cholinergic syndrome of nausea, vomiting, mucositis, diarrhea, and flushing. The latter responds to atropine. Interstitial pneumonitis has been reported in Japanese patients receiving irinotecan. Grade 3–4 diarrhea was observed in up to 35% of patients in early clinical studies. Irinotecan, when given at 125 mg/m2 on a weekly basis for 4 of 6 weeks in combination with 5-FU and leucovorin (7), led to toxic deaths due to severe diarrhea and neutropenia. An alternative and better tolerated schedule of biweekly irinotecan at 180 mg/m2, whether as single agent or combined with 5-FU and leucovorin (FOLFIRI), has been widely adopted. Loperamide starting at 4 mg should be given at the first sign of diarrhea, and repeat doses of 2 mg every 2 h may be given until resolution of the diarrhea.
Myelosuppression is common with irinotecan; grade 3–4 neutropenia occurs in 14%–47% of patients.
Clinical Indications
Irinotecan is most commonly used in advanced colorectal cancer. As first-line therapy in metastatic disease, it is combined with 5-FU and leucovorin, with or without bevacizumab. It can also be given in the second-line therapy as a single agent, or in patients with k-ras wild-type tumors, it may be combined with cetuximab (8).
TOPOISOMERASE II INHIBITORS: ANTHRACYCLINES
Drugs of this class, derived from the fungal culture broths of Streptomyces peucetius, have become critical components of treatment for acute leukemias, lymphomas, breast cancer, and sarcomas. The two original members of this family, daunorubicin (DN) and doxorubicin (DX), remain in active clinical practice: DN for acute myelogenous leukemia (AML), and DX for lymphomas and solid tumor chemotherapy. Two semisynthetic derivatives, idarubicin (IDA) and epirubicin (EPI), have made inroads as valuable agents for leukemia and breast cancer, respectively. Doxil, a liposomal formulation of DX has found limited use, primarily for ovarian cancer, and a similar liposomal DN preparation is indicated for Kaposi’s sarcoma.
 MECHANISM OF ACTION
MECHANISM OF ACTION
The anthracyclines share a rigid planar four-ring structure complemented by a glycosidic substitution on the D ring and variable side groups on the A and D rings (Figure 4-4). The planar configuration allows anthracyclines to intercalate between strands of DNA, and this action was originally thought to be responsible for its inhibition of DNA synthesis. However, the anthracyclines possess other important features. The quinone on ring C readily undergoes oxidation/reduction cycling in the presence of Fe++, producing free radicals from oxygen and/or lipids (9). These free radicals are responsible for the cardiac toxicity inherent in this class of agents. The anthracyclines bind to and inhibit topo II, an enzyme that promotes DNA strand unwinding essential for DNA synthesis and repair. In its normal function, topo II binds to DNA and creates a double strand break that allows strand passage. The enzyme then reseals the break. Anthracyclines bind to and stabilize the DNA-topo II complex, preventing the resealing of the strand break. An accumulation of strand breaks signals the p53 system to halt cell cycle progression, and to initiate DNA repair. If the breaks are sufficiently numerous, the cell undergoes apoptosis. High levels of topo II expression correlate positively with response to DX in patients with breast cancer (10, 11). Cells become resistant to anthracyclines through diminished expression of topo II activity or through topo II mutations that decrease binding affinity of the enzyme for the drugs of this class. An interesting correlation has been observed between amplification of the isoenzyme topo IIa and the HER2/neu receptor. The topo IIa gene is located on chromosome 17 adjacent to the gene coding for the HER2/neu. HER2/neu amplification is found in one quarter of breast cancers. In one-third of patients with amplified HER2, topo IIa is co-amplified, and this subset of breast cancer patients has a higher response rate to DX.
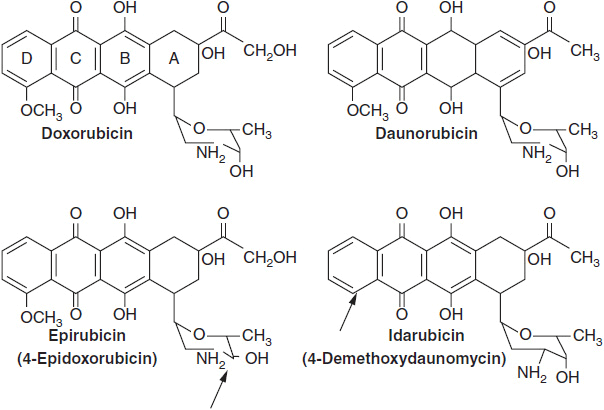
FIGURE 4-4 Anthracyclines in current clinical use. For epirubicin and idarubicin, arrows point to the sites where these new drugs differ from doxorubicin and daunomycin, respectively.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


