Antiestrogens
ANTIESTROGENS
Antiestrogen hormonal therapy is the cornerstone of endocrine treatment of hormone-receptor positive breast cancer.
Current antiestrogen treatment options for hormone-receptor positive breast cancer include selective estrogen-receptor modulators (SERMs), selective estrogen-receptor downregulators (SERDs), and aromatase inhibitors (AIs).
 SELECTIVE ESTROGEN-RECEPTOR MODULATORS
SELECTIVE ESTROGEN-RECEPTOR MODULATORS
The SERMs are chemically diverse compounds that lack the steroid structure of estrogen but possess a tertiary structure that allows them to bind to estrogen receptors. Depending on the specific end-organ, they exert selective agonist and/or antagonist effects (1).
There are three currently approved SERMS: raloxifene, toremifene, and tamoxifen (Figure 11-1). The most widely used SERM for treatment of ER positive breast cancer is tamoxifen.
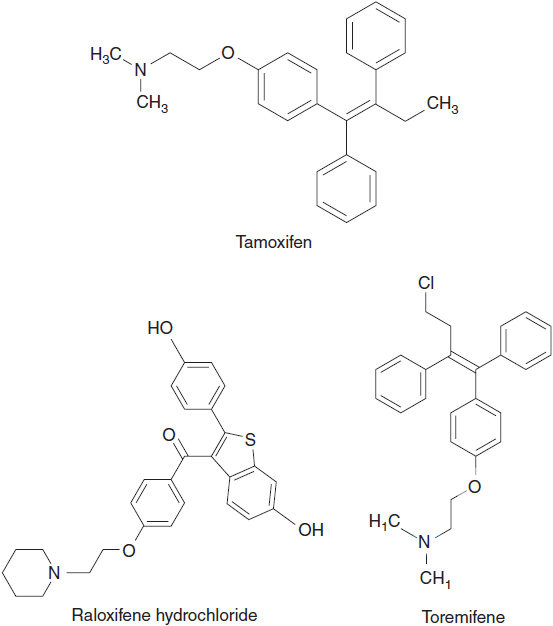
FIGURE 11-1 Chemical structure of selective estrogen receptor modulators: tamoxifen, raloxifene, and toremifene.
Tamoxifen
Mechanism of action. Tamoxifen is a competitive inhibitor of estradiol binding to the ER. In addition to its estrogen antagonist effects on the breast and breast cancer, tamoxifen exerts estrogenic effects on non-breast tissues which influence its overall therapeutic index. Tamoxifen exerts agonist or antagonist effects in part related to ambient estrogen levels. For example on bone metabolism it exerts a partial agonist action in postmenopausal women whereas in premenopausal women its effect on bone is antagonistic (1).
Clinically in women with ER positive disease, 5 years of post-operative adjuvant tamoxifen reduces the annual odds of recurrence of breast cancer by 39% and the annual odds of death by 31%, with comparable effects regardless of age as well as menopausal and nodal status (2).
Absorption, fate, and excretion. Tamoxifen is readily absorbed following oral administration, with peak concentrations measurable after 3–7 h and steady-state levels being reached at 4–6 weeks. It is a prodrug with little affinity for the estrogen receptor and requires metabolization into its active form endoxifen (4-hydroxy N-desmethyltamoxifen) by the sequential action of CYP2D6 and CYP3A4. A second metabolite, N-desmethyltamoxifen, also has strong antiestrogenic activity. Some selective serotonin reuptake inhibitors (SSRIs) like fluoxetine, paroxetine, and sertraline are potent inhibitors of CYP2D6, and may impair tamoxifen’s activation.
It is hypothesized that certain CYP2D6 genotypes and phenotypes are associated with lower endoxifen concentrations and worse breast cancer outcome. However, two published retrospective studies with the largest sample size thus far found no statistically significant association between the presence of poor or intermediate metabolizer phenotype and breast cancer outcome (3, 4). Given the limited and conflicting data, CYP2D6 testing is not recommended as a tool to define the optimal endocrine strategy.
The half-lives of N-desmethyltamoxifen and endoxifen are 14 days or longer. After enterohepatic circulation, glucuronides and other metabolites are excreted in the stool; excretion in the urine is minimal (1).
Therapeutic uses. Tamoxifen citrate (Nolvadex®) is marketed for oral administration. The usual dose prescribed is 20 mg daily.
Tamoxifen is used for (5):
• treatment of ER positive metastatic breast cancer until disease progression.
• adjuvant endocrine treatment of ER positive premenopausal breast cancer alone or in combination with ovarian ablation for 5 years.
• adjuvant endocrine treatment of ER positive postmenopausal breast cancer for 2–3 or 5 years prior to administration of an AI.
• prevention of breast cancer in women at increased risk.
Clinical toxicity. Tamoxifen is generally well tolerated. Side effects are rarely sufficiently severe to require discontinuation of therapy as overall quality of life (QoL) appears not to be impaired (1).
The most common side effects (occurring in greater than 30%) are:
• vasomotor symptoms (hot flashes)
• vaginal discharge
• fluid retention
• loss of libido
Less common side effects (occurring in about 10%–30%) are:
• nausea
• menstrual irregularities
• vaginal bleeding
• mood changes
• increased risk of cataracts, retinal deposits, and decreased visual acuity
Rare but serious side effects include:
• two- to threefold increased risk of endometrial cancer, particularly in postmenopausal women over 60 years taking tamoxifen for ≥2 years; monitoring of abnormal vaginal bleeding with prompt gynecological evaluation is recommended.
• doubling of the rate of deep vein thrombosis and pulmonary embolism; it is recommended to discontinue tamoxifen before elective surgery.
 SELECTIVE ESTROGEN-RECEPTOR DOWNREGULATORS
SELECTIVE ESTROGEN-RECEPTOR DOWNREGULATORS
SERDs (also termed “pure antiestrogens”) bind ER with high affinity, without activating any of the normal transcriptional hormonal responses, and are consequently devoid of any estrogen agonist activity. The lead compound of this class currently approved for the treatment of advanced breast cancer is fulvestrant (Figure 11-2).
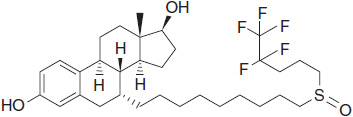
FIGURE 11-2 Chemical structure of fulvestrant.
Fulvestrant
Mechanism of action. Fulvestrant is a steroidal antiestrogen that binds to the ER with an affinity over 100 times that of tamoxifen, inhibits its dimerization, and increases its degradation. In contrast to tamoxifen, which increases the level of ER expression, fulvestrant is associated with a reduction in the number of detectable ER molecules in cells (6).
Absorption, fate, and excretion. Fulvestrant is administered intramuscularly (i.m.) once monthly. Maximum plasma concentrations are reached at about 7 days after i.m. administration and are maintained over a period of 1 month. The plasma half-life is approximately 40 days. Steady state is achieved in 1 month with a loading dose (500 mg on day 0, 250 mg on day 14, 250 mg on day 28 and q4 weeks thereafter) compared to 4–6 months with the approved dose (250 mg q4 weeks).
There is extensive and rapid distribution of the drug, predominantly to the extravascular compartment.
Various pathways similar to those responsible for endogenous steroid metabolism extensively metabolize fulvestrant. The putative metabolites possess no estrogenic activity and only the 17-keto compound demonstrates a level of antiestrogenic activity about one-fifth than that of fulvestrant. Less than 1% is excreted in the urine (6).
Therapeutic uses. Fulvestrant (Faslodex®) is available as a long-acting 50 mg/ml solution. It is typically administered as a 250-mg i.m. injection at monthly intervals, but recent data suggest that a high-dose regimen (500 mg) has greater efficacy compared to the approved 250-mg dose. After many years of clinical trials and development, fulvestrant has been approved at a higher dose of 500 mg by the FDA but is still used at the 250 mg dose in some countries (7).
Fulvestrant is used for (5):
• treatment of postmenopausal women with hormone-receptor positive metastatic breast cancer.
Clinical toxicity. Fulvestrant is generally well tolerated, and QoL outcome measures are maintained over time (8).
Clinical side effects of fulvestrant include:
• nausea
• asthenia
• pain
• vasodilatation (hot flushes)
• headache
• injection site reactions
 AROMATASE INHIBITORS
AROMATASE INHIBITORS
In premenopausal women estrogens are synthesized primarily in the ovaries. Following menopause, estrogen is produced by aromatization of circulating androgens in extra-ovarian peripheral tissues, including liver, muscles, skin fat, and connective tissue, and circulates at low levels. Peripheral aromatization depends on androgenic precursors of adrenal origin to generate estradiol and estrone. Aromatase is the enzyme complex responsible for converting androgens (androstenedione and testosterone) to estrogens (estrone [E1] and estradiol [E2]). In postmenopausal patients, where only baseline levels of aromatase activity are present, aromatase inhibitors (AIs) effectively lower estrogen levels by 90% to nearly undetectable levels.
AIs are not appropriate monotherapy for premenopausal patients, as residual ovarian function can lead to reflex stimulation of FSH, enhanced ovulation, and increased production of estrogen thereby overcoming the effects of the AI.
AIs are classified as type 1 (steroidal aromatase inactivator) or type 2 (nonsteroidal AI) inhibitors according to their structure and mechanism of action (Figure 11-3). Type 1 inhibitors are steroidal analogues of androstenedione and bind to the same site on the aromatase molecule, but unlike androstenedione bind irreversibly because of their conversion to reactive intermediates by aromatase. Thus they are commonly known as aromatase inactivators or suicide inhibitors. Type 2 inhibitors are nonsteroidal and bind reversibly to the heme group of the enzyme by way of a basic nitrogen atom (9).
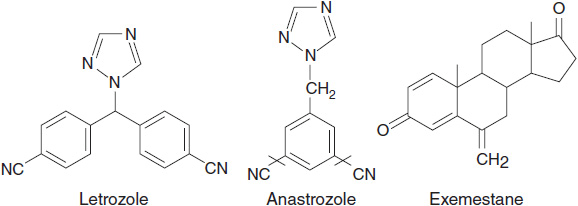
FIGURE 11-3 Chemical structures of aromatase inhibitors.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


