Adrenocortical Cancer
BACKGROUND AND PRESENTATION
Adrenocortical carcinoma (ACC) is a rare malignancy, affecting 1.5–2 persons per million each year (1). ACC is slightly more common in women and has a bimodal age distribution, with a higher incidence in children younger than 5 years and in adults in their 4th and 5th decades of life (1). Despite improved methods of diagnosis, ACC usually presents at an advanced stage and as a result 5-year survival ranges from 20% and 45%.
At the time of disease discovery there may be no symptoms (the tumor may be found incidentally on imaging). In other patients there may be symptoms of hormone excess or complaints referable to an abdominal mass. Hormone excess presents clinically as Cushing’s syndrome, virilization, feminization, or, less frequently, hypertension with hypokalemia. Hormone hypersecretion can be found in as many as 73%–79% of ACC patients, although not all patients have symptoms (2). In one study, amongst 45 ACC patients, routine biochemistry documented hormone excess in 33 (73%) with excess glucocorticoid and adrenal androgen in 12, isolated glucocorticoid in 11, isolated adrenal androgen in 7, and 17β-estradiol excess in combination with glucocorticoid and adrenal androgen excess in two and one, respectively. Steroid profiling revealed predominantly immature, early-stage steroid precursors, and their production most likely a consequence of altered expression of steroidogenic enzymes in variably undifferentiated tumors. While the classification of ACCs by hormone profile has limited value, hormone secretion, especially cortisol, may be an independent predictor of poor prognosis (3)—although not all studies agree with this conclusion (4). Older age at diagnosis, stage III (local lymph nodes) and IV (local organ invasion or distant metastases) disease and cortisol hypersecretion are risk factors significantly associated with a shorter survival (3). The poorer prognosis of cortisol-secreting tumors could be attributed to the comorbidity of cortisol hypersecretion, the immunosuppressive effects of excess cortisol possibly favoring development of the tumor and its metastases, or cortisol-mediated metabolic changes that favor the growth of a more aggressive tumor.
GENETICS
Although most ACC lack identifiable risk factors, heredity plays a role in some patients (5). Risk factors include the Li-Fraumeni syndrome (LFS), multiple endocrine neoplasia type 1 (MEN1), familial adenomatous polyposis coli (Gardner syndrome), and the Beckwith-Wiedemann syndrome (BWS) (6). Predisposition is thought to arise from mutations in p53, MEN1, APC, or CDKN1C suppressor genes or dysregulation of the CDKN1C tumor suppressor gene. A unique risk factor has been described in southern and southeastern Brazil. There, a high frequency of an endemic germline p53 R337H mutation is responsible for the highest known incidence of childhood ACC—with a founder germline R337H mutation found in 95% of ACCs reported in young children (7). The frequency of this germline R337H mutation is due to a common ancestor, a conclusion supported by the fact that no case of a de novo R337H mutation has been found (8).
ASSESSMENT/EVALUATION
The initial assessment should determine the extent of disease and whether the tumor is functional. Difficulty in differentiating a benign from a malignant tumor and the risk of seeding tumor argue against a biopsy if the presentation is an isolated adrenal mass without evidence of metastases. Surgical resection can be both diagnostic and therapeutic. Biopsies should be reserved for cases with widespread metastases unlikely to be considered for surgical resection and those in whom there is reason to believe the primary is other than adrenal. Because not all patients present with symptoms of hormonal excess, their hormonal status and the need for steroid replacement should be assessed prior to surgery. Careful assessment may avoid the occurrence of postoperative adrenal insufficiency related to contralateral adrenal involution secondary to tumor hypersecretion of cortisol and suppression of adrenocorticotropic hormone (ACTH) production by the pituitary.
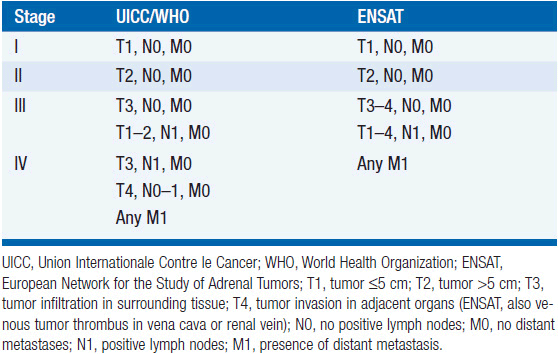
While in the pre-computed tomography (CT) era symptoms of hormone excess most often brought patients to medical attention, increasingly diagnoses are made radiographically, often in asymptomatic patients undergoing evaluation for an unrelated problem. A thin-collimation CT of the chest and abdomen is recommended as the initial imaging technique, with magnetic resonance imaging (MRI) reserved for selected patients. Both CT and MRI can help discriminate benign adenomas from malignant lesions, an important distinction that should be made before any operative intervention. On CT scans, ACCs usually have higher density values (i.e., lower lipid content) and are typically inhomogeneous; on MRI, they are usually isointense with liver on T1 images, with intermediate to high intensity on T2 images (9). MRI is superior in assessing vascular invasion and should be obtained before a surgical resection if there is concern regarding inferior vena cava involvement with right adrenal tumors. The value of [18F] fluorodeoxyglucose (FDG)-positron emission tomography (PET) is not established in routine evaluation or follow-up (10). While imaging might help in discriminating a benign adenoma from a malignant tumor, it is not definitive and cannot differentiate ACC from other tumors that may have similar metabolic activities. Once all information is available, staging is undertaken using the UICC/WHO TNM classification or a revised classification proposed by European Network for the Study of Adrenal Tumors (ENSAT) (11) (Table 66-1). In the latter, stage III includes tumors that infiltrate into surrounding tissue or tumor thrombus in vena cava/renal vein or those that have positive lymph nodes, whereas stage IV is defined by the presence of distant metastasis. The prognostic value of ENSAT staging has been confirmed in an independent cohort from the United States (12).
TABLE 66-1 CLASSIFICATION OF ADRENOCORTICAL CARCINOMA, WORLD HEALTH ORGANIZATION (WHO) AND EUROPEAN NETWORK FOR THE STUDY OF ADRENAL TUMORS (ENSAT)
PATHOLOGY
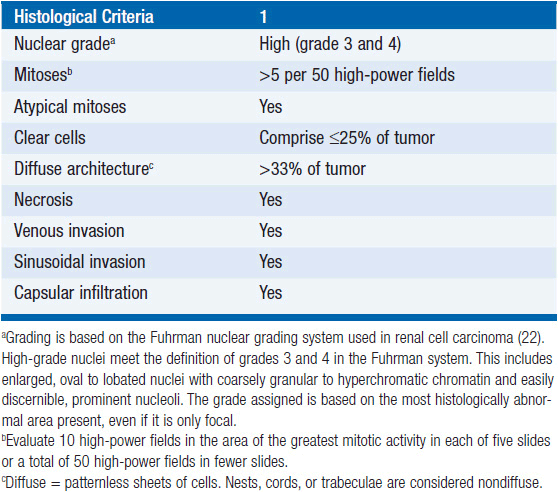
In the absence of distant metastases or local spread, it may be difficult to distinguish small (less than 6 cm) ACC’s from a benign adenoma. Several multi-parametric approaches have been proposed for establishing malignancy. Among these, the Weiss system, first proposed in 1984, is the standard (13). It is based on the presence of nine histopathologic properties associated clinically with metastatic potential or local recurrence. The criteria are related to tumor structure: (1) 25% or less clear cells, (2) a “patternless” diffuse architecture, and (3) necrosis; cytological features: (4) atypical mitoses, (5) a mitotic rate greater than 5 per 50 high-power fields, and (6) nuclear grade 3 or 4; and invasive properties: (7) venous, (8) sinusoidal, and (9) capsular invasion. The individual criterion in the Weiss system are simply given a score of 1 if present and 0 if absent, yielding an overall score from 0 to 9 (Table 66-2). In the initial report 18/19 patients with adreno-cortical tumors with a score of 4 or more had a recurrence or metastatic disease, whereas all 24 tumors with scores of 2 or less had a benign clinical course. Subsequently the threshold was lowered from 4 to 3 criteria based on one patient with a recurrent adrenal tumor that exhibited only 3 adverse histologic features. It is unclear whether higher scores above 4 are associated with an increasingly poor outcome. The latter may not be surprising, since as has been shown, many of the Weiss components including high mitotic rate, tumor necrosis, atypical mitosis, capsular, venous, and adjacent organ invasion, as well as nuclear grade are adverse predictors of tumor-related mortality in a univariate analysis (14).
TABLE 66-2 THE WEISS CRITERIA (THE PRESENCE OF THREE OR MORE CRITERIA HIGHLY CORRELATES WITH MALIGNANCY)
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


