Identification of solid tumors highly sensitive to etoposide, mainly germ-cell tumors, small cell lung cancers (SCLCs), Ewing’s sarcomas, osteosarcomas, lymphomas, acute leukemias, poorly differentiated adenocarcinomas and poorly differentiated endocrine tumors. Etoposide was approved by the Food and Drug Administration (FDA) for marketing by Bristol Laboratories under the trade name Vepesid in early 1984. Teniposide (VM-26; Vumon) was approved by the FDA in 1992 for refractory childhood leukemia.
Evidence of etoposide-induced secondary leukemias, leading to various attempts to reduce the number of cycles of etoposide when given in a curative intent, especially in testicular cancers.
Molecular biology and genomic analyses revealed the proximity between the topoisomerase II alpha gene (TOP2a) and the HER2 gene, which raised the question of the potential interest of Top2 inhibitors in HER-2-amplified solid tumors.
Molecular pharmacology studies demonstrated that Top2β targeting by etoposide could be responsible for secondary malignancies while Top2α targeting was primarily responsible for the anticancer activity of etoposide. This may justify the search for specific Top2α inhibitors as second-generation etoposide derivatives.
bacterial equivalents of eukaryotic Top2. Quinolones (nalidixic acid, ciprofloxacin, norfloxacin, and derivatives), which are widely used antibiotics, act by inhibiting DNA gyrase and topo IV with little if any, effect on the host human Top2.10,11
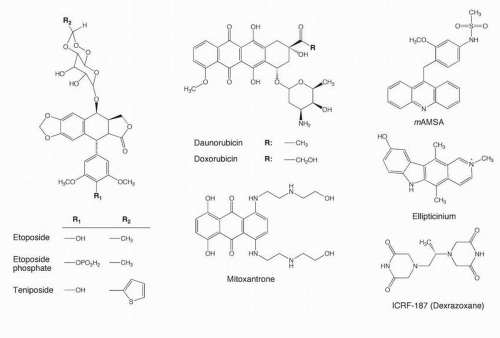 FIGURE 19-1 Chemical structures of the epipodophyllotoxins (VP-16, etoposide phosphate and VM-26), anthracyclines (doxorubicin and daunorubicin), mitoxantrone, acridines (m-AMSA), ellipticines (ellipticinium), and bisdioxopiperazine (ICRF-187). |
TABLE 19.1 Mammalian DNA topoisomerases | ||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||
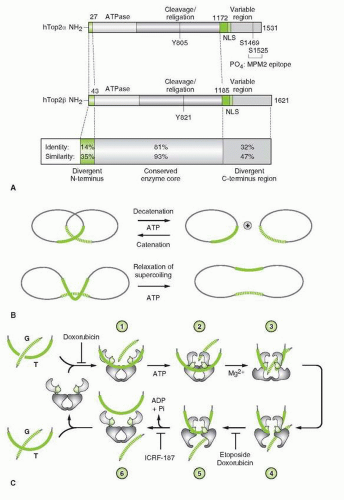 FIGURE 19-2 A. Domain structure of Top2α and β. The three major domains are illustrated, as well as the site of ATP binding (ATPase), the active-site tyrosine (Y), the nuclear localization sequence(s) (NLS), and the sites of phosphorylation (PO4). The N-terminal domain (homologous to the gyrase B subunit) extends from amino acid 1 to about 660. The catalytic core domain (homologous to A subunit of gyrase) extends from about residue 660 to 1,200, and the C-terminal domain (no corresponding homology with gyrase) extends from about residue 1,200 to the C-terminus of the enzyme. Top2 α and β are most divergent in their N- and C-terminus region. B. Schematic representations of two types of topological reactions catalyzed by Top2: catenation-decatenation and relaxation of DNA. C. Model for the catalytic cycle of type II topoisomerases [according to Refs (7,8,12)]. The unliganded enzyme binds to duplex DNA (labeled G) across the TOPRIM domains (step 1). A second duplex DNA strand (labeled T) and ATP bind to the enzyme (step 2). Nucleotide binding promotes dimerization of the ATPase domains and closure of the clamp (step 2). Cleavage of the G-strand (step 3) allows the passage of the T-strand through the cleaved G-strand (step 4). Following G-strand religation (step 5), the T-strand is released through the dimer interface in the C-terminus region (step 5). ATP hydrolysis reopens the ATPase domain and completes the enzyme catalytic cycle. Sites of drug action are indicated: DNA intercalators (doxorubicin at relatively high concentrations) prevent Top2 from binding DNA; Etoposide and lower concentrations of doxorubicin prevent religation of the G-strand (steps 4 and 5), and ICRF-187 traps the G-strand inside the enzyme (steps 5 and 6). |
single-strand and double-strand breaks.2 Hence, the higher cytotoxicity of anthracyclines compared with amsacrine or VP-16 may be due to the higher frequency of DNA double-strand breaks that may be more cytotoxic than single-strand breaks.15 Finally, the DNA sequence and genomic localization of Top2 cleavage complexes vary among drugs.26 Drugs, which are chemically and structurally related, frequently produce closely related patterns of Top2 cleavage, while compounds structurally and electronically unrelated produce different patterns both in purified DNA and in drug-treated cells.15,26,27
TABLE 19.2 Topoisomerase II (Top2) inhibitors | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||


a theory consistent with the concerted action of both enzyme monomers during catalysis.28,38 This type of inhibition, which we refer to as “interfacial inhibition” is one of natures’s paradigm for noncompetitive protein inhibition.39 Top2-mediated DNA cleavage tends to be more pronounced at the DNA site of drug binding,40,41 and reactions triggered by cooperative effects with the other monomer appear less stable than those at sites of preferred binding.38 Recent studies show the DNA interface to include not only each of the DNA breaks generated by Top2 but also intercalation sites within the 4 base pair stagger between the two concerted Top1 cleavage sites.41 The drug-binding site on Top2 is not well defined. Analyses of drug-resistant mutant enzymes suggest that both the A′ and B′ regions of the enzyme reversibly bind to the drugs. Recent studies35,38 indicate that the CAP homology domain determines DNA sequence recognition on the G segment of DNA and drug effects, which is consistent with drug binding at or near the DNA cleavage site and formation of a ternary complex: drug-enzyme-DNA.
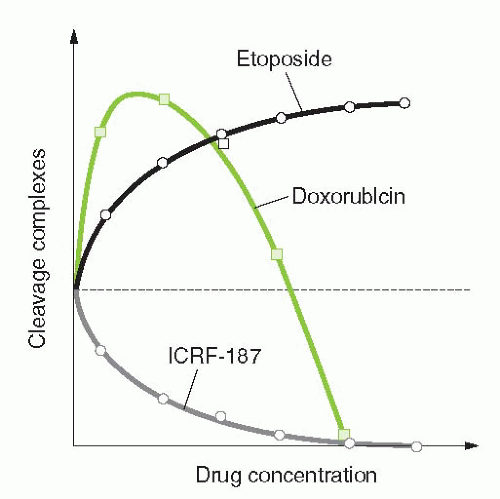 FIGURE 19-3 Different modes of drug inhibition of Top2. Top2 poisons such as the epipodophyllotoxins (VP-16 or VM-26) only trap the Top2 cleavage complexes with increasing efficiency as their concentration increases. Top2 suppressors such as the bis-dioxopiperazines (open circles) are pure catalytic inhibitors that only inhibit the formation of cleavage complexes. Gray squares correspond to biphasic inhibitors such as DNA intercalators (anthracyclines, ellipticines, acridines; see Table 19-2), which enhance Top2 cleavage complexes at low concentrations and suppress cleavage complexes at higher concentrations. |
TABLE 19.3 Differences among Topoisomerase II Inhibitors | ||||||||
|---|---|---|---|---|---|---|---|---|
| ||||||||
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


