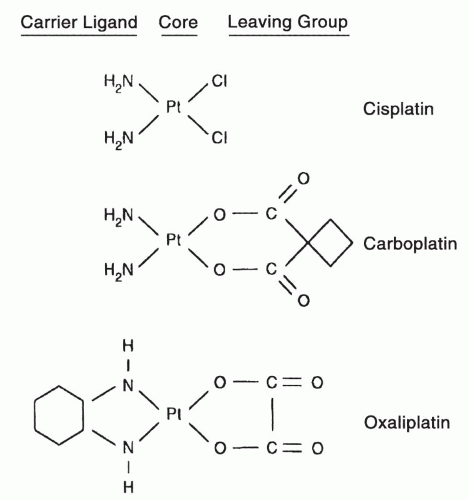
Collectively, cisplatin, carboplatin, and oxaliplatin are major contributors to systemic therapy, for a very broad range of malignancies —with the exception of taxanes, the most active class of anticancer agents. Cisplatin was discovered when Rosenberg and colleagues
1,
2 in a set of experiments involving
Escherichia coli observed the dramatic inhibitory effects of platinum compounds on cellular replication. Following those seminal studies, a rapid series of basic, preclinical, and clinical studies resulted in Food and Drug Administration (FDA) approval for the treatment of testicular cancer. Within 15 years, cisplatin’s effectiveness in testicular, ovarian, lung, head and neck, and bladder cancer was established; new analogues (oxaliplatin) and more recent studies have established the value of this class of drugs in important subset of patients with ovarian cancer, colorectal cancer, breast cancer, lymphoma, childhood malignancies, and numerous rarer cancers.
Because of the particularly troublesome toxicities of renal damage, nausea and vomiting, deafness, and peripheral neuropathy, major efforts were undertaken to identify analogs of cisplatin that have equivalent clinical effectiveness but without the toxicities of the parent compound. Carboplatin, the first such analog to meet achieve widespread clinical use (
Fig. 15-1), proved to be equally effective as cisplatin in ovarian cancer, lung cancer, and several other malignancies but, for unclear reasons, less effective than cisplatin in the treatment of germ cell malignancy. Carboplatin is less neurotoxic, emetogenic, and nephrotoxic than cisplatin but more myelosuppressive.
Among newer platinum analogs, only oxaliplatin (
Fig. 15-1) has received FDA approval. For unclear reasons, oxaliplatin is particularly effective in colorectal cancer (in combination with 5-fluorouracil (
Chapter 9). Colon cancer is a disease for which neither cisplatin nor carboplatin shows meaningful benefit. Understanding the molecular basis for these peculiarities for these three compounds could potentially unlock a treasure trove of new insights as to how cancer cells escape the effects of DNA-damaging agents.
Chemistry
Cisplatin and carboplatin are divalent inorganic complexes and are highly water soluble and readily activated by water displacement of their chloride or carboxylate groups. Oxaliplatin is a divalent oxalate salt, and not entirely cross-resistant with carboplatin and cisplatin in model tumor systems. The more complex leaving groups of carboplatin and oxaliplatin tend to reduce reactivity in aqueous solution, decrease renal toxicity and hearing loss, and the cyclohexyl substitution on oxaliplatin may alter susceptibility to repair of DNA adducts, as will be explained below. The structures of the three FDA-approved analogues are shown in
Figure 15-1, and important aspects of their chemistry are summarized in
Table 15-1 and in previous reviews.
3,
4The most exciting of new developments in the chemistry of platinum analogues is the discovery and characterization of cisplatin and carboplatin nanocapsules.
5,
6,
7,
8 Cisplatin nanocapsules were a product of efforts to develop better methods for enclosing cisplatin in liposomes. For cisplatin, such nanoparticles have approximate bidimensional measurements of 50 to 120 nm. The cisplatin-to-lipid molar ratio is about eleven to one. The solid core of the nanoparticle is 90% the dichloride species of cisplatin, and the outer lipid bilayer is very different from that of conventional liposomes. A slight modification of the cisplatin nanocapsule method was used to develop carboplatin nanocapsules, including modest differences in molar ratios of drug to lipid. Most importantly, when comparing the IC50 values of cisplatin nanocapsules to cisplatin, and that of carboplatin nanocapsules to carboplatin, in both cases, the nanocapsules are two orders of magnitude more cytotoxic than their parent drug against IGROV-1 human ovarian cancer cells.
Thus, platinum compounds may possibly become even more effective through the use of nanotechnology.
Subcellular Pharmacology
The cellular pharmacology of platinum compounds has been extensively studied.
3,
4 Cisplatin is able to cross cellular barriers because of its simple chemistry. However, work by a number of laboratories strongly suggests that simple diffusion of cisplatin across cell membranes does not fully explain transmembrane trafficking of platinum drugs.
9,
10,
11,
12,
13 Specific transmembrane transport proteins may promote influx and efflux of the drug, such as the CTR1, ATP7A, and ATP7B transporters.
9,
10 At physiologic pH of 7.4, dissociation of its chlorides and their replacement by -OH molecules result in cisplatin having a neutral charge (
Fig. 15-2). This makes it possible for ready diffusion across the cellular membrane.
As suggested above, there is good evidence that CTR1, a copper transporter in the cell membrane, is a significant contributor to the active uptake of platinum drugs by cells, including cisplatin, carboplatin, and oxaliplatin.
9,
11 Also, ATP7A and ATP7B, normal copper efflux proteins in the cell membrane, are major contributors to the efflux of platinum drugs in cancer cells.
10 In addition, the human organic cation transporters 1, 2, and 3, have been implicated in
modulating the cellular uptake of platinum compounds, specifically as it may relate to human renal toxicity.
12,
13Once inside the cell, three different fates await the compound.
3,
4 It may be exported from the cell by one of several specific active transport proteins. A second fate is to become reactive by displacement of the chloride groups of cisplatin or carboxylato groups of carboplatin (
Fig. 15-2) and form a covalent complex with sulfhydryl groups, such as glutathione or metallothioneins, or sulfhydryl on proteins. A third fate is for the activated platinum complex to react in a relatively nonspecific way with a number of intracellular nucleophiles, which include electron-rich sites on proteins, RNAs, and DNA (cellular and mitochondrial).
Through this relatively nonspecific interaction of the highly reactive platinum moiety with a range of subcellular molecules, cisplatin damages the cell. The measured affinity for RNA is greater than that for DNA, which, in turn, is greater than that for proteins. Because of time of transit from cellular membrane into the nucleus, the sum total of reactions with DNA is lower than that with intracellular protein on a molar basis.
In most circumstances, the reactions with cellular DNA determine the bulk of cisplatin-related cellular effects. One exceptional study found that in a Burkitt’s lymphoma cell line, adduct formation with protein was correlated with cell death in a time frame that could not be explained by DNA damage.
3Early studies comparing cisplatin with transplatin established that the DNA adduct formation by cisplatin was responsible for its cell-killing effects.
3 Further, it was found necessary for reactive groups to be in the
cis configuration to generate effective cell killing. In subsequent, more detailed studies, the intrastrand N7-d(GpG) and the N7-d(ApG) adducts were most clearly correlated with cell killing (
Fig. 15-3).
4 The relative contribution of these and other adducts to cell killing, as well as other specific DNA lesions such as interstrand cross-links, has never been completely resolved.
The N7-d(GpG) and the N7-d(ApG) intrastrand adducts account for more than 80% of total platinum-DNA damage that forms after an exposure of cisplatin to isolated DNA
14 or to cells in tissue culture
15,
16 or to cells from patients in clinical settings.
17,
18,
19 These two adducts are associated with severe kinking of the DNA double helix.
20 This kinking is recognized and repaired by the nucleotide excision repair pathway, which involves the genes ERCC1, XPA,
and others.
21,
22,
23 Table 15-2 summarizes the relative proportions of the DNA lesions formed after exposure to cisplatin, carboplatin, or oxaliplatin.
Kinking of the DNA is caused by rigid the bond angles within the cisplatin molecule.
20 The DNA double helix thereby bends to conform to the structure of the platinum adduct. In contrast, most bifunctional alkylating agents have bond angles that allow the drug to bend and thereby to accommodate to the structure of the DNA. Carboplatin is similar to cisplatin in most respects and is active as a clinical agent in many of the same malignancies as the parent drug. The major subcellular differences between these two drugs include the need for an esterase activity to release the carboxylato moiety of the carboplatin molecule and thereby to expose the reactive arms for covalent binding to target sites, and a delayed time frame for the formation of the specific DNA lesions such as the N7-d(GpG) and N7-d(GpG) adducts, as compared with cisplatin.
24 Differences in clinical pharmacology and in clinical toxicity are discussed later in this chapter. For almost all matters, the subcellular behaviors of cisplatin and carboplatin appear to be the same.
Oxaliplatin is FDA approved for the treatment of colorectal cancer. The major subcellular differences between cisplatin and oxaliplatin include the carrier ligand effects involving the less reactive leaving groups of the oxaliplatin compound
25,
26 and differences in the rates of formation and repair of oxaliplatin-DNA damage, as compared with cisplatin.
25,
27,
28 Differences in clinical pharmacology and in clinical toxicity are also discussed in this chapter.
Mechanism(s) of Action
The consensus is that cisplatin and its analogs exert their cytotoxic effects by covalently binding to purine DNA bases and disrupting the normal functions of cellular DNA. Platinum analogs that have therapeutic activity form a preponderance of DNA intrastrand adducts as opposed to DNA interstrand cross-links or DNA-platinum-protein cross-links.
3,
4 Cisplatin binding to mitochondrial DNA has been described
29,
30,
31 but is of unclear biologic significance.
Early studies of cisplatin and transplatin compared the relative importance of DNA damage versus protein binding in terms of causing tumor cell kill in tissue culture.
4 Some laboratories have sought to correlate tumor cell kill with one or more of the different intrastrand lesions, the N7-d(GpG) adduct or the N7-d(ApG) adduct or the N7-d(GpXpG) adduct.
5 There are conflicting reports over which lesion(s) may be more associated with the cytotoxic effects of these drugs and which lesion(s) may be more associated with the mutagenic effects of these drugs. These studies have not been definitive because of the complexity of the mix of DNA adducts after cisplatin exposure, as shown in
Table 15-2. When platinum agents are allowed to react with isolated DNA or cells, or are given to animals, the proportions of the various DNA adducts are relatively constant, as listed in
Table 15-2.
Cells deficient in double-strand break repair, such as BRCA-1 or BRCA-2 mutant cells, and basal-type breast cancer cells (which tend to express low levels of BRCA-1 RNA and are often p53 mutant), are highly sensitive to cisplatin, raising the possibility that accumulation of double-strand breaks, as a result of either single strand or double strand cross-links, is ultimately responsible for cell death. In the presence of cisplatin, these cells are uniquely able to phosphorylate and activate p73, a proapoptotic factor.
32Cell death may occur through apoptotic or nonapoptotic pathways. The apoptotic pathways may be mediated through mismatch repair (MMR) genes:
33,
34,
35,
36 p5337 and/or decreased
bcl2/bax.38,
39 Overwhelming DNA damage is associated with acute, nonapoptotic cell death. Reports of experimental and clinical studies over the past three decades show that cisplatin enhances immune-mediated killing of tumor cells
3,
4 as will be discussed below. The advent of oxaliplatin has raised questions about the effects of different carrier ligands on platinum’s ability to induce cellular damage and evade DNA repair processes. Saris and colleagues
27 found that a ligand bound to the platinum core, when opposite the
cis configuration of the reactive bonds, can exert tremendous influence on subcellular
pharmacology of the drug. The carrier cyclohexyl ligand reduces DNA repair efficiency and increases cell killing.
25,
26,
27,
28,
35Repair of oxaliplatin adducts with DNA seems less dependent on recognition of the adduct by high mobility group (HMG) proteins, which are required for effective repair of cisplatin and carboplatin adducts. These proteins are found in high concentration in testicular cancers. The adducts formed by oxaliplatin are bulkier, create greater distortion of the DNA helix, and cause a different pattern of hydrogen bonding to adjacent regions of DNA.
40Another difference between oxaliplatin and cisplatin or carboplatin is the relative inability of DNA polymerase alpha to perform replicative bypass over an oxaliplatin-DNA lesion as compared with a cisplatin or carboplatin-DNA lesion. DNA polymerase beta is a key protein involved in repair of the 1,2GG adduct formed by oxaliplatin; cells deficient in this enzyme are hypersensitive to oxaliplatin, but not to cisplatin.
106 Thus, cells that depend on replicative bypass as a major mechanism of platinum resistance may be comparatively more sensitive to oxaliplatin than the other platinum compounds. Others have shown that the carrier ligand has substantial effects on the clinical pharmacology of platinum analogs, as will be discussed later.
Platinum agents give additive or synergistic activity with a range of other anticancer agents. Cisplatin is thought to be relatively non-cell cycle-specific in terms of its cell-killing effects, although cross-links form with greatest efficiency during S-phase. It tends to synergize with agents that reduce the intracellular levels of purine and pyrimidine precursors needed for DNA replication or repair, including 5-fluorouracil
41 and gemcitabine.
42 Oxaliplatin downregulates thymidylate synthase, perhaps adding to its beneficial interaction with fluoropyrimidines (see
Chapter 9). Platinums also show additive or supra-additive cell killing with agents that alter mitosis (paclitaxel),
43 and with inhibitors of DNA repair activity (PARP)
44,
99 and with down-regulation or inhibition of ERCC1.
45,
46,
47,
48,
49 Positive interactions with topoisomerase inhibitors have been described,
50 as well as with agents from other drug classes.
3,
4Cisplatin is widely used as a radiation sensitizer for head and neck cancer, locally advanced lung cancers, and cervical cancer. The mechanism of sensitization is poorly understood but likely relates to its generation of DNA strand breaks, in conjunction with the similar action of irradiation, which damages DNA through the production of reactive oxygen species. Both modalities call into play nucleotide-excision repair (NER) pathways and double-strand break pathways for repair of DNA, and their active intermediates sequester and deplete sulfhydryls and other nucleophiles that protect against reactive molecules. Thus, their effects are likely to be additive, if not synergistic. The same synergy is seen experimentally with many other cytotoxic drugs, although the mechanistic basis is poorly understood.
In summary, the primary mechanism of cell killing for this class of compounds is covalent binding to purine bases of cellular DNA. This covalent binding leads to bending of the DNA helix at a fixed angle, with local denaturing of the DNA strand. This DNA damage is detected by components of the repair complex and is converted into a strand break. Adducts are removed and breaks are repaired primarily by the NER and double strand break repair process. When not effectively repaired, cell killing may occur through apoptotic or nonapoptotic pathways. In clinical use, platinum compounds are highly synergistic with irradiation, and with other DNA damaging agents, perhaps as a result of their ability to overwhelm mechanisms that protect and repair DNA. The possible contribution of drug-induced, immune-mediated cell killing, which may occur in the intact host, is discussed next.