Inhibition of Thymidylate Synthase
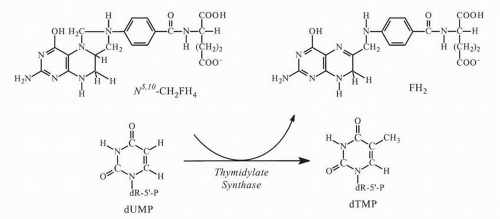
At least two primary mechanisms of action appear capable of causing cell injury: inhibition of TS and incorporation into RNA. FdUMP binds tightly to TS and prevents formation of thymidylate (thymidine 5′-monophosphate, dTMP), the essential precursor of thymidine 5′-triphosphate (dTTP), which is required for DNA synthesis and repair. The functional TS enzyme comprises a dimer of two identical subunits, each of MW ˜30 kDa (bacterial) or ˜36 kDa (human). Each subunit has a nucleotide-binding site and two distinct folate-binding sites, one for 5,10-methylenetetrahydrofolate (5,10-CH
2 FH
4) monoglutamate or polyglutamate, and one for dihydrofolate polyglutamates. FdUMP competes with the natural substrate 2′-deoxyuridine monophosphate (dUMP) for the TS catalytic site.
20,
21 During methylation of dUMP, transfer of the folate methyl group to dUMP occurs by elimination of hydrogen attached to the pyrimidine carbon-5 position (
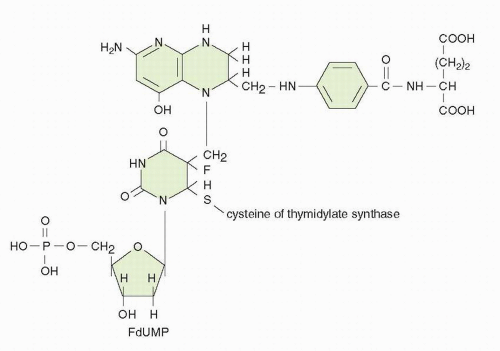
Fig. 9-3). This elimination cannot occur with the more tightly bound fluorine atom of FdUMP, and the enzyme is trapped in a slowly reversible ternary complex with FdUMP and folate (
Fig. 9-4). The “thymineless state” that ensues is toxic to actively dividing cells. Toxicity can be circumvented by salvage of dThd in cells that contain dThd kinase. The circulating concentrations of dThd in humans are not thought to be sufficient (approximately 0.1 μmol/L) to afford protection.
22 The plasma levels of dThd are approximately tenfold higher in rodents, which complicates preclinical evaluation of the antitumor activity of various TS inhibitors.
A reduced-folate cofactor is required for tight binding of the inhibitor to TS. The natural cofactor for the TS reaction, 5,10-CH
2 FH
4, in its monoglutamate and polyglutamate forms, binds through its methylene group to the carbon-5 position of FdUMP. The polyglutamates of 5,10-CH
2 FH
4 are much more effective in stabilizing the FdUMP-TS-folate ternary complex.
23 Other naturally occurring folates promote FdUMP binding to the enzyme but form a more readily dissociable complex. Polyglutamated forms of dihydrofolic acid (FH
2) promote extremely tight binding of FdUMP to the enzyme.
24 FH
2 accumulates in cells exposed to methotrexate (MTX). Although MTX is a relatively weak inhibitor of TS in cell-free experiments, MTX polyglutamates are more potent inhibitors.
24 MTX polyglutamates decrease the rate of ternary complex formation among FdUMP, folate cofactor, and TS. The ability of MTX polyglutamates to inhibit ternary-complex formation is influenced by the glutamation state of the reduced-folate cofactor and is substantially reduced in the presence of 5,10-CH
2 FH
4 pentaglutamate.
24 Similarly, in tissue culture, MTX-induced depletion of intracellular reduced folates causes a marked reduction in the rate of formation of ternary complex.
25FdUMP binds avidly to the mammalian enzyme, with a dissociation half-life (
t1/2) of 6.2 hours.
26 Elucidation of the crystal structure of TS has permitted a complex kinetic and thermodynamic description of ternary complex formation.
27,
28 The interaction proceeds by an ordered mechanism with initial nucleotide binding followed by 5,10-CH
2 FH
4 binding to form a rapidly reversible noncovalent ternary complex (
Fig. 9-4). Enzyme-catalyzed conversions result in the formation of a covalent bond between carbon-5 of FdUMP and the one-carbon unit of the cofactor. The overall dissociation constant of 5,10-CH
2 FH
4 from the covalent complex is approximately 1 × 10
−11 mol/L.
Despite the high specificity and potency of TS inhibition by FdUMP and the well-established lethality of dTMP and dTTP depletion, inhibition of TS may not be the sole cause of 5-FU toxicity. If 5-FU toxicity results from dTTP depletion, then dThd should reverse the toxic effects. Examples of complete protection from 5-FU cytotoxicity by dThd have been reported, but dThd shows variable effectiveness in rescuing cells exposed to 5-FU.
29 Experimental evidence from in vitro and in vivo studies supports the concept that, depending on the target cell, drug concentration, and modulating factors, 5-FU toxicity may be partially independent of its effect on TS. Coadministration of 5-FU and dThd prevents the early inhibition of DNA synthesis but markedly increases 5-FU toxicity to healthy tissues in the whole animal, increases the antitumor effect of 5-FU against various animal tumors, and increases [
3H]FUrd incorporation into RNA.
30 Pharmacologic measures that increase FUTP formation and its RNA incorporation also increase its toxicity.
RNA-Directed Effects
5-FU is extensively incorporated into nuclear and cytoplasmic RNA fractions, which may result in alterations in RNA processing
and function, such as inhibiting the processing of initial pre-rRNA transcripts to the cytoplasmic rRNA species in a dose- and time-dependent manner.
31,
32,
33,
34Net RNA synthesis may be inhibited during and after fluoropyrimidine exposure in a concentration- and time-dependent fashion. In some cancer cell lines, a highly significant relationship exists between 5-FU incorporation into total cellular RNA and the loss of clonogenic survival.
35 5-FU is incorporated into all species of RNA; substantial amounts of [
3H] 5-FU accumulate in low-MW (4S) RNA at lethal drug concentrations.
33 Although the analog replaces only a small percentage of uracil residues in RNA, the incorporated 5-FU residues appear to be stable and to persist in RNA for many days after drug administration.
365-FU exposure affects mRNA processing and translation. Polyadenylation of mRNA and methylation of tRNA are inhibited at relatively low concentrations of 5-FU,
37,
38 and altered metabolism of specific proteins such as dihydrofolate reductase (DHFR) precursor mRNA has been reported.
39 Incorporation of 5-FU into RNA may affect quantitative and qualitative aspects of protein synthesis.
40,
41In vitro-transcribed TS mRNA with 100% substitution of 5-FU has an altered secondary structure but no differences in the translational efficiency.
40 Hundred percent substitution of uracil residues in human-TS complementary DNA (cDNA) with either FUTP or 5-bromouridine 5′-triphosphate (BrUTP) only inhibited the translational rate in the presence of BrUTP-substituted cDNA.
41 The stability of the transcribed mRNA in a cell-free system is
increased by threefold and tenfold with FUTP and BrUTP, respectively.
Changes in the structure, levels, and function of small nuclear RNAs (snRNA) and small nuclear ribonuclear proteins (snRNP) result from 5-FU treatment.
42,
65 The substitution of FUTP for uridine triphosphate (UTP) in a cell-free system (84% replacement of uracil residues by 5-FU) leads to pH-dependent missplicing of [
32P]-labeled human
β-globin precursor mRNA; pH values favoring 5-FU ionization promote missplicing.
43Further, 5-FU substitution greatly increases the pH and temperature sensitivity of the process. Partial ionization of 5-FU residues at physiologic pH (pK 5-FU = 7.8 versus pK uracil = 10.1) may therefore destabilize the active conformation of RNA.
44Another potential locus of 5-FU action is inhibition of enzymes involved in posttranscriptional modification of RNA particularly the formation of methylated uracil bases that have profound effects on splicing.
45a,
45b,
46,
47 Although 5-FU-associated cytotoxicity in cancer cells exposed in the presence of sufficient concentrations of dThd to circumvent TS inhibition is presumed to result from RNA-directed effects of 5-FU, it is paradoxical that significant incorporation of 5-FU into RNA may occur in some cancer cell lines in the absence of toxicity. The factors that influence whether 5-FU-RNA incorporation results in cytotoxicity are not clear. The rate of RNA incorporation and the species into which the fluoropyrimidine is incorporated may be more important determinants of cytotoxicity than the total amount incorporated. 5-FU and FUrd may be channeled into different ribonucleotide compartments and, ultimately, into distinct classes of RNA.
48In summary, the changes that result in altered pre-RNA processing and mRNA metabolism are not uniform for all RNA species after 5-FU exposure. Effects on precursor and mature rRNA, precursor and mature mRNA, tRNA, and snRNA species suggest inhibition of processing; incorporated 5-FU residues also inhibit enzymes involved in posttranscriptional modification of uracil. Many of the RNA-directed effects of 5-FU undoubtedly occur as a consequence of its fraudulent incorporation into various RNA species. The changes in certain key mRNAs resulting from 5-FU exposure may be relevant to its cytotoxicity. 5-FU-mediated interference with the production of enzymes involved in DNA repair may have cytotoxic consequences, such as 5-FU-mediated inhibition of ERCC-1 mRNA expression in cisplatin-resistant cancer cells.
49
DNA-Directed Mechanisms of Potential Toxicity
The biochemical consequences of TS inhibition and the potential effects on DNA integrity have been extensively studied, but their relationship to cytotoxicity is incompletely understood. Inhibition of TS results in depletion of dTMP and dTTP, thus leading to inhibition of DNA synthesis and interference with DNA repair. Accumulation of dUMP occurs behind the blockade of TS, and further metabolism to the deoxyuridine triphosphate (dUTP) level may occur.
50 Inhibition of TS is accompanied by elevated concentrations of deoxyuridine in the extracellular media in cell culture models and in plasma of rodents; monitoring changes in plasma deoxyuridine levels may, therefore, serve as an indirect reflection of TS inhibition.
FdUTP and dUTP are substrates for DNA polymerase, and their incorporation into DNA is a possible mechanism of cytotoxicity.
51,
52,
53,
54 5-FU cytotoxicity in some models correlates with the level of 5-FU-DNA.
52,
53 Two mechanisms prevent incorporation of FdUTP and dUTP into DNA. The enzyme dUTP pyrophosphatase or dUTP hydrolase catalyses the hydrolysis of FdUTP to FdUMP and inorganic pyrophosphate.
55,
56 The DNA repair enzyme uracil-DNA-glycosylase hydrolyzes the 5-FU-deoxyribose glycosyl bond of the FdUMP residues in DNA, thereby creating an apyrimidinic site.
57 The base deoxyribose 5′-monophosphate is subsequently removed from the DNA backbone by an AP (apurinic/apyrimidinic) endonuclease, creating a single-strand break, which is subsequently repaired. With dThd triphosphate depletion, however, the efficiency of the repair process is substantially weakened. Uracil-DNA-glycosylase is a cell cycle-dependent enzyme with maximal levels of activity at the G1 and S interface, such that excision of the fraudulent bases occurs before DNA replication. The activity of uracil-DNA-glycosylase inversely correlates with the level of FdUrd incorporation into DNA in human lymphoblastic cells. Because the affinity of human uracil-DNA-glycosylase is much lower for 5-FU than for uracil, 5-FU is removed more slowly from DNA by this mechanism.
57 FdUTP itself inhibits the activity of uracil-DNA-glycosylase.
58 Accumulation of deoxyadenosine triphosphate (dATP) accompanies TS inhibition.
59 The combined effects of deoxyribonucleotide imbalance (high dATP, low dTTP, high dUTP) and misincorporation of FdUTP into DNA may have deleterious consequences affecting DNA synthesis, the integrity of nascent DNA, and induction of apoptosis.
A variety of DNA-directed effects have been described.
60,
61,
62,
63,
64 5-FU treatment inhibits DNA elongation and decreases the average DNA chain length.
60 DNA strand breaks accumulate in 5-FU-treated cells and correlate with excision of [
3H]5-FU from DNA. 5-FU and FdUrd result in single- and double-stranded DNA breaks in HCT-8 cells in a concentration- and time-dependent fashion, a process that is enhanced by LV and limited by dThd.
61 FdUrd
exposure may result in the formation of large (1 to 5 Mb) DNA fragments as a result of double-strand DNA breaks; the time course and extent of DNA megabase fragmentation correlate with loss of clonogenicity in HT29 cells.
62 The pattern of DNA fragmentation is distinct from that associated with
γ radiation, which produces random breaks. The pattern of high-MW DNA damage differs in SW620 cells, which are equally sensitive as HT29 cells to FdUrdinduced inhibition of TS, but require higher drug concentrations and longer exposures to achieve a comparable degree of DNA fragmentation and cytotoxicity. Resistance to 5-FU appears to be related to the higher activity of dUTPase and failure to accumulate dUTP of SW620 cells.
63 Simple dThd starvation of a TS-deficient murine cell line produces much smaller DNA fragments, 50 to 200 kb in length.
64Inhibition of protein synthesis by cycloheximide within 8 hours of FdUrd exposure dramatically reduces DNA double-strand breakage and lethality in murine FM3A cells, suggesting that FdUrd exposure triggers the synthesis of an endonuclease capable of inducing DNA strand breaks.
60 Factors that regulate recognition of DNA damage and apoptosis contribute to 5-FU lethality. The oncogene
p53 plays a pivotal role in the regulation of cell-cycle progression and apoptosis and influences the sensitivity of murine embryonic fibroblasts to 5-FU.
65 Transfection and expression of the
bcl-2 oncogene in a human-lymphoma cell line render it resistant to FdUrd. TS inhibition, dTTP depletion, and induction of single-strand breaks in nascent DNA are similar in vector control cells and
bcl-2-expressing cells.
66 In vector control cells, induction of double-stranded DNA fragmentation in parental DNA coincides with onset of apoptosis. The contribution of DNA damage to cell lethality varies among different malignant lines, and DNA fragmentation does not appear to contribute to 5-FU-mediated cytotoxicity in some cancer cell lines.
67In summary, TS inhibition, as seen in “pure” form with FdUrd treatment in the absence of dThd salvage, and 5-FU incorporation into RNA are capable of producing lethal effects on cells. DNA damage also contributes to cytotoxicity and can occur in the absence of detectable FdUTP incorporation into DNA. The combined effects of deoxyribonucleotide imbalance (high dATP, low dTTP, high dUTP) and misincorporation of FdUTP and dUTP into DNA result in deleterious consequences affecting DNA synthesis and the integrity of nascent DNA. The pattern and extent of DNA damage induced by fluoropyrimidines in human colorectal cancer cells vary and may be affected by the activity of enzymes involved in DNA repair and by downstream pathways that are required to implement cellular destruction.
It is now recognized that the genotoxic stress resulting from TS inhibition activates programmed cell-death pathways, resulting in induction of parental DNA fragmentation. Depending on the cell line in question, two different patterns of parental DNA damage may be noted: internucleosomal DNA laddering, the hallmark of classical apoptosis, and high-MW DNA fragmentation with segments ranging from approximately 50 kb to 1 to 3 Mb. Differences in the type and activity of endonucleases and DNA-degradative enzymes triggered in a given cell line most likely explain these disparate patterns of parental DNA fragmentation. In “apoptosis-competent” cancer cell lines, such as HL60 promyelocytic leukemia cells, genotoxic stress results in rapid (within hours) induction of programmed cell death, with classic DNA laddering. In contrast, many cancer cell lines derived from epithelial tumors, including colon cancer, appear to undergo delayed programmed cell death. This phenomenon may reflect a “postmitotic” cell death, in which one or more rounds of mitosis are needed before cell death occurs.
68 In such cell lines, the duration of the genotoxic insult may determine whether induction of cytostasis or programmed cell death occurs. One possible explanation for delayed apoptosis is that originally sublethal damage to genes, essential for cell survival, may ultimately lead to cell death with subsequent rounds of DNA replication.
Factors operating downstream from TS clearly influence the cellular response to genotoxic stress, such as overexpression of the cellular oncoproteins
bcl-2 and mutant
p53. Disruption of the signal pathways that sense genotoxic stress or lead to induction of programmed cell death, or both, may render a cancer cell inherently resistant to 5-FU. In some cancer cell lines, thymineless death may be mediated by Fas and Fas-ligand interactions.
69 Cancer cell lines insensitive to Fas-mediated apoptosis are insensitive to 5-FU, suggesting that modulation of their expression may influence sensitivity to 5-FU.
70 As previously mentioned, base excision repair (BER) plays an essential role in removing incorporated 5-FU and uracil residues from DNA, resulting in single-strand DNA breaks. BER recognizes the mispairing of 5-FU with guanine in DNA and excises the fluoropyrimidine. Because BER involves multiple proteins, deficiencies in one of the components, such as uracil DNA glycosylase, XRCC1 or DNA polymerase-
β, may reduce the toxic effects of TS inhibitors.
71 Abrogation of uracil DNA glycosylase activity affords protection from cytotoxicity in selected cell lines
72,
73 but not others.
73Microsatellite instability (MSI) is a manifestation of genomic instability in human cancers that have a decreased overall ability to faithfully replicate DNA and is a surrogate phenotypic marker of underlying functional inactivation of the human DNA mismatch repair (MMR) genes.
74 In vitro studies suggest that MMR recognizes DNA breaks resulting from 5-FU incorporated into DNA and signals cell cycle (G2M) arrest. Functional loss of a MMR gene results from inactivation of both alleles via some combination of coding region mutations, loss of heterozygosity, and/or promoter methylation, which leads to gene silencing. In vitro studies suggest that MMR-proficient cells are more sensitive to 5-FU or FdUrd than MMR-deficient cells.
75,
76 The loss of MMR proficiency leads to fluoropyrimidine resistance in some cell lines, but not others,
77 perhaps because of differences in cell lines and conditions of drug concentration and duration of exposure in vitro. The MSI phenotype has been associated with a better prognosis in stage-for-stage matched tumors in primary colorectal cancer,
78 but data are conflicting as to whether MSI status influences benefit from 5-FU-based adjuvant therapy. The MSI phenotype has been associated with a better prognosis in stage for stage-matched tumors in primary colorectal cancer. While there is conflicting evidence for the predictive effect of the MSI phenotype in stage 3 and 4 patients, there is an emerging consensus that stage 2 patients with an MSI high phenotype do not gain any benefit from adjuvant 5-FU therapy and may actually be harmed by 5-FU treatment through undetermined mechanisms.
392 An ongoing prospective trial in stage 2 colon cancer patients will help clarify this issue.