through a poorly described endocytotic system that may include discrete plasma membrane proteins of 250 kD.7,8 Most investigators believe that the Cu-bleomycin is a prodrug and that cleavagecompetent bleomycin is Fe(II) speciated. Considerable chemical evidence regarding DNA damage produced by Fe(II)·bleomycin exists to support this hypothesis.5 The primary model was first outlined by Umezawa’s group6 and is schematically represented in Figure 16-2. Cu(II) associated with intracellular Cu(II)·bleomycin is reduced, possibly by intracellular cysteine-rich proteins, to Cu(I), which is released, and the apobleomycin quickly complexes with the more abundant Fe(II).9 Nuclear translocation of the Fe(II)·bleomycin complex proceeds with subsequent chromatin damage. The metal coordination chemistry of bleomycin has been the subject of considerable attention, and primarily on the basis of studies using electron paramagnetic resonance (EPR),10 crystallography,11 and Fe(II) surrogate metals such as Cu(II), a square-pyramidal complex, as indicated in Figure 16-1, is most favored.5 Six distinct moieties are required for this metal coordination complex, and the N-1 of the pyrimidine, the N of the imidazole, and the secondary amine are undisputed participants.5 Debate still exists about the arrangement of the remaining ligands, and this presumably will be resolved with further structural analyses. Although many aspects of the cellular scheme found in Figure 16-2 are biologically and chemically appealing, several questions remain unanswered, including the identity of the sulfhydryl-rich reductant for Cu(II), the recipient and fate of the Cu(I), the intracellular source of Fe(II), and the mode by which the metal-bound bleomycin translocates the plasma membrane and the nucleus. Therefore, others12,13 have developed a rival hypothesis to explain the antitumor activity and fate of bleomycin. Even though Cu(II)·bleomycin is not highly cytotoxic,14 persuasive arguments for a potential functional role of Cu(I)·bleomycin in the biologic actions of this antineoplastic agent have been presented.5,13 Thus the widely embraced concept that Fe(II)-complexed bleomycin is the only biologically relevant species may require revision.
TABLE 16.1 Key features of bleomycin pharmacology | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
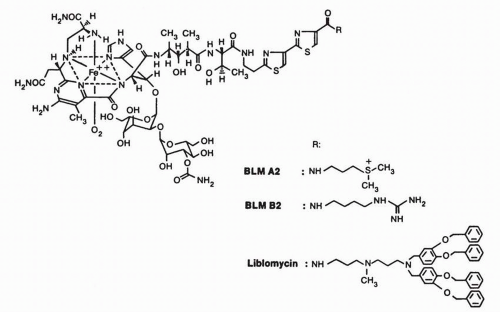 FIGURE 16-1 Structure of bleomycin·Fe(II) complex. The various substitutions on the amino-terminal end of the molecule are shown for bleomycin A2 (BLM A2), for bleomycin B2 (BLM B2; also a component of the clinical preparation), and for one congener, liblomycin. |
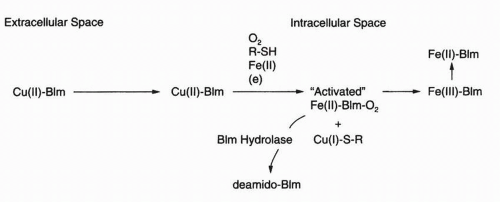 FIGURE 16-2 Schematic representation of bleomycin (Blm) transformation as it moves from the extracellular to the intracellular space. The Cu(II)·Blm complex in the extracellular space is converted to a cytotoxic Fe(II)·Blm·O2 complex in the cell. Inactivation of Blm by Blm hydrolase is also shown. |
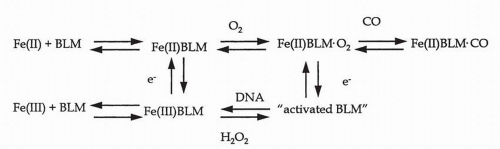 FIGURE 16-3 Model for the activation of cleavage-competent bleomycin (BLM). |
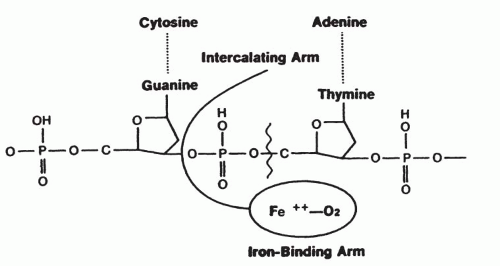 FIGURE 16-4 Intercalation of the bithiazole groups between DNA base pairs, at least one of which contains the GpT of GpC sequence. Also shown is the apposition of the Fe(II)-binding portion of bleomycin to the deoxyribose group, which is cleaved via hydrogen abstraction at the C-4′ of the deoxyribose. |
measurement of oxygen consumption, with a maximum velocity of 27 mol oxygen consumed per minute per mole of bleomycin.26
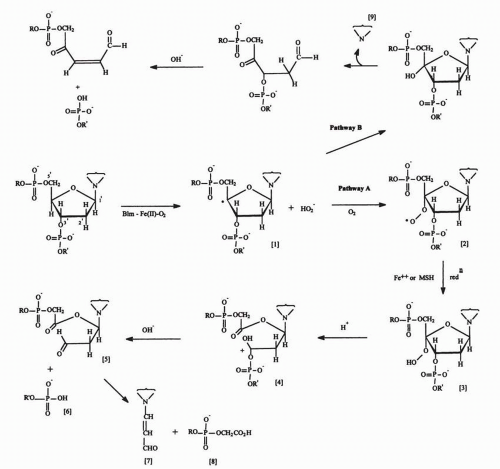 FIGURE 16-5 Scheme for the cleavage of the 3′-4′ deoxyribose bond by the activated bleomycin·Fe(II)·O2 complex. The activated drug complex initially abstracts a hydrogen radical from the 4′ position to produce the unstable intermediate [1] that decomposes in the presence of oxygen (pathway A) to produce the free base propenal [7], leaving a 3′-phosphoglycolate ester [8] and a 5′-phosphate [6] at the free ends of the broken DNA strand. Under conditions of limited oxygen (pathway B), a free base [9] is released, and DNA strand scission occurs only in the presence of alkali (pH 12). |
groups remaining at the site of DNA single-strand cleavage may promote access of a second bleomycin molecule to the opposing strand, resulting in a double-strand break.
particularly double-strand breaks.58,59 Early studies60 demonstrated increased rates of bleomycin inactivation in two bleomycin-resistant rat hepatoma cell lines. Morris et al.61 demonstrated an increased level of bleomycin hydrolase in cultured human head and neck carcinoma cells with acquired resistance to bleomycin. Metabolic inactivation of bleomycin also can contribute to intrinsic bleomycin resistance in human colon carcinoma cells.62
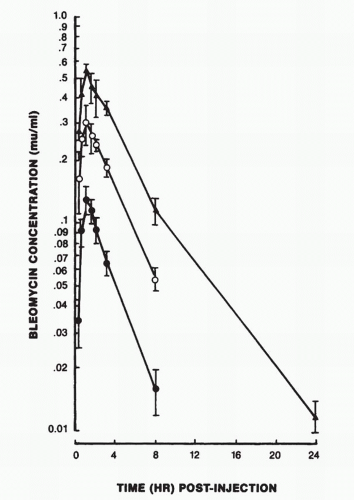 FIGURE 16-6 Pharmacokinetics of bleomycin after intramuscular administration of 2 (•), 5 (m), and 10 (▲) mg of bleomycin per meter square. (From Oken MM, Crooke ST, Elson MK, et al. Pharmacokinetics of bleomycin after IM administration in man. Cancer Treat Rep 1981;65:485.) |
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


