restore function of these proteins without restoring a fully functional gene to each cancer cell, a significant technical challenge.8, 9 However, there are other approaches that can potentially exploit altered tumor suppressor gene function. The synthetic lethal strategy (identifying a gene that becomes essential for tumor cell survival when a suppressor gene is deleted) has led to the development of polyadenosine diphosphate-ribose polymerase (PARP) inhibitors for BRCA1- and BRCA2-mutated ovarian and breast cancers. In cases where function of a tumor suppressor protein is inhibited (such as by binding of MDM2 to a normal p53 protein) as opposed to being absent due to mutation or deletion, blocking the inhibitor (or unblocking the suppressor) might be able to restore function.10
TABLE 29.1 Types of molecularly targeted compounds | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
TABLE 29.2 Approved molecularly targeted agents | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
optimization through structural studies of the inhibitor and target. This allows more rational design of antineoplastic molecules and, at the same time, efficient screening of a wide range of structures.
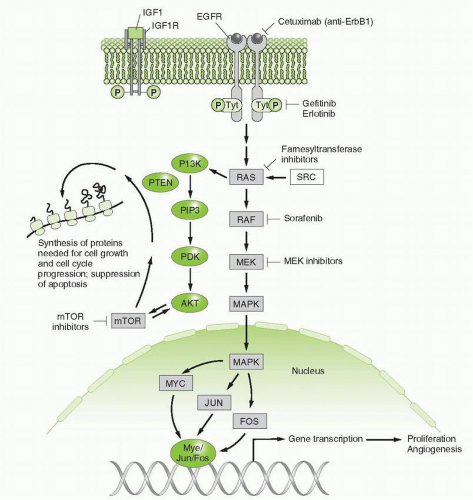 FIGURE 29-1 Two signaling pathways, stimulated by IGF-1 and EGF ligands, which activate downstream signals through PI3 kinase and Ras proteins. Inhibitors of these pathways at the receptor, its intracellular kinase domains, and downstream components are indicated. Cross talk between the pathways occurs at PI3 kinase. Signals in the nucleus stimulate proliferation and angiogenesis, while cytoplasmic signals promote cell growth and survival. |
Mutation, amplification, or overexpression of growth factors or their receptors involved in proliferation and survival of various cancers, such as the EGFR family, including HER2/neu;
Mutation or enhanced activity of intracellular signaling pathways that promote growth, impede cell death, or enhance metastasis, such as activation of the Ras-RAF-MEK or PI3K pathways;
Antiapoptotic mechanisms that antagonize cell death, such as overexpression of bcl-2 or decreased bax expression;
Pathways, such as chaperoning, ubiquination, and the proteosome, that protect mutant or oncogenic proteins or affect their turnover rates;
Epigenetic factors such as methylation or acetylation of histones, or methylation of DNA, which regulate gene expression and differentiation;
MicroRNAs (miRNAs) that promote malignant growth;
Tumor promoting factors in the environment, such as angiogenic pathways (e.g., vascular endothelial growth factor [VEGF] or platelet-derived growth factor [PDGF]);
Are subject gene and its protein found in human tumors, and is there selective expression in tumors versus normal tissues?
Is the subject protein’s function essential to the survival and proliferation, and, indeed, the transformed behavior of the malignant cells?
Does inhibition of the gene product change the phenotype of these cells and lead to the desired result (such as death or growth arrest of malignant cells or a decrease in metastasis) in an appropriate animal model? It is critical to establish that the gene and associated protein product are important for the biology of human tumors and not just an animal model. Short interfering RNAs (SiRNAs) have become invaluable tools for inhibiting the expression of target genes within cells and determining the role of the target.8,27,28 SiRNAs also have potential as therapeutic agents and early evaluation in clinical trials has begun.
Is the protein also expressed in key proliferating normal tissues, such as intestinal epithelium and/or bone marrow progenitors, or even nonproliferating tissues, such as heart, kidney, or brain, and does targeting the protein therefore carry risks for significant toxicity?
The profile of gene expression in normal tissues may provide helpful clues about potential toxicity of an agent directed against that gene, as in the case of trastuzumab, an inhibitor of Her2/neu, a growth factor expressed in myocardium. In this case, gene knockout in animal models had early and fatal consequences for the developing embryo, indicating that inhibition of that gene or its protein product might lead to significant toxicity.
Are there closely related proteins and what is their physiological function? Are they essential for normal tissue function and survival of the host and might be cross-targeted by the agent producing unwanted toxicities?
Are there appropriate biomarkers that can identify likely “responders” and can these be developed as a guides for drug development in early trials?
Aid in selecting appropriate patients likely to respond to a specific agent (e.g., HER2 positivity for trastuzumab, EGFR mutations for erlotinib, and BRAF mutations for BRAF inhibitors);
Help validate that the target is being modulated in vivo (e.g., decreased pERK phosphorylation with BRAF inhibitor);
Aid in optimizing dose and schedule;
Serve as surrogates for response (e.g., HER2 positivity in breast cancer patients treated with trastuzumab and early decline in PET avidity in GIST patients treated with imatinib).
Inhibition of EGFR in certain preclinical models leads to death of malignant cells and significant antitumor response. HER2/neu is expressed in a subset of patients with breast and gastric cancers and has been successfully targeted.30, 31, 32, 33 Anti-EGFR mAbs or small molecules have successfully treated lung, colorectal, and head and neck cancers.34,35 Finally, as discussed above, mutant proteins provide the most specific target in cancer. EGFRs are mutated in a subset of lung adenocarcinomas, primarily in nonsmokers or light smokers, and these are very sensitive to inhibition by small molecular inhibitors of the tyrosine kinase activity of the EGFR.36
of messenger RNAs (mRNAs) for a number of proteins involved in DNA synthesis. RB’s function is, in turn, tightly controlled by a complex sequence of protein interactions that regulate its phosphorylation state. For example, two of the responsible kinases, cdk4 and cdk6, are activated by cyclin D and inhibited by p16 and p21. Multiple sites of mutation or other alterations in this pathway can be involved in the neoplastic process; essentially any mutation or modification that eliminates or inactivates RB function (including by phosphorylation) will activate E2F and allow cell-cycle progression. Examples of these alterations in human malignancies include loss of RB itself in patients with retinoblastoma; activation of cdk4 in melanoma; overexpression of cyclin D in many human tumors; and loss of p16 function (such as by mutation), which can occur in a number of malignancies.51 Most human tumors display an alteration of at least one component of this pathway, most frequently p16 deletion or cyclin D overexpression.9 Experimental models of RB loss or inactivation have confirmed the tumorigenic effect of mutations in this pathway.
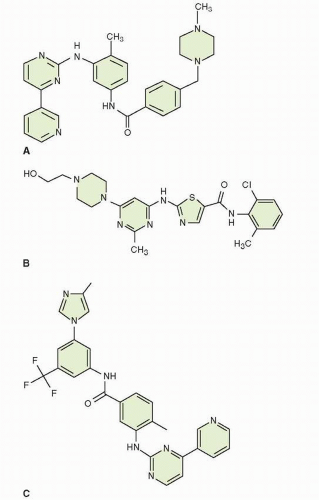 FIGURE 29-2 Structure of the small molecule tyrosine kinase inhibitors of BCR-ABL. A. imatinib; B. dasatinib; and C. nilotinib. |
is important to monitor for toxicity in individual patients and to consider the possibility of underdosing in nonresponsive patients (see below).
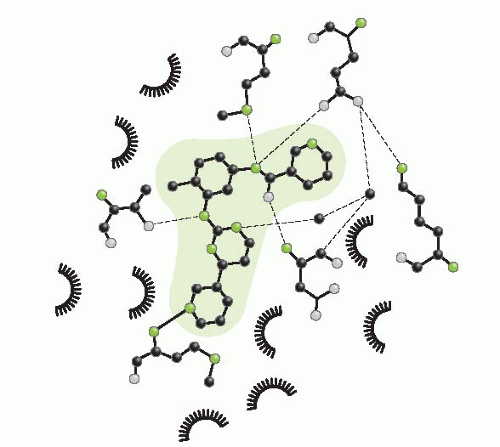 FIGURE 29-3 Detailed diagram of the intermolecular interactions between the drug (shaded in blue) and amino acids in BCR-ABL. Hydrogen bonds are indicated by dashed lines, while van der Waals interactions (indicated by halos around the amino acid name and its position in the protein sequence) are shown for nine amino acids with hydrophobic side chains. |
TABLE 29.3 Features shared by most small molecule tyrosine kinase inhibitors approved to date | ||||||
|---|---|---|---|---|---|---|
|
Neutropenia, thrombocytopenia, and anemia
Hepatotoxicity, with elevated liver enzymes. Toxicity can be decreased by holding or adjusting the dose, but occasionally it can be severe, especially if given with acetaminophen; thus, acetaminophen should be used with caution in patients taking imatinib.
Fluid retention or edema (often periorbital edema, which is usually manageable though occasionally serious, with a rare incidence of ascites, pleural effusions, brain edema, and congestive heart failure)
Musculoskeletal pains and cramps
Abdominal pain
Rash and other skin conditions, including blistering
Nausea and vomiting
GI irritation (imatinib should be taken with food and a large glass of water)
GI bleeding or intratumoral bleeding (especially in patients with GIST), which can be significant
GI perforation
with T315 I substitution. Resistant forms of the enzyme have been isolated from patients prior to therapy, indicating that mutations arise spontaneously, but are selected by the presence of drug.133 The most common mutations conferring resistance occur at Glu 255 and Thr 315, both creating high-level resistance to imatinib and nilotinib. Dasatinib remains effective against Glu 255. Mutations at Met 351 and Glu 355 create low-level resistance to imatinib, but not to the other drugs. Monitoring of peripheral blood BCR-ABL transcripts by polymerase chain reaction (PCR) aids the assessment of levels of response, detects early evidence of the emergence of resistance, and aids in the selection of secondary therapies.134
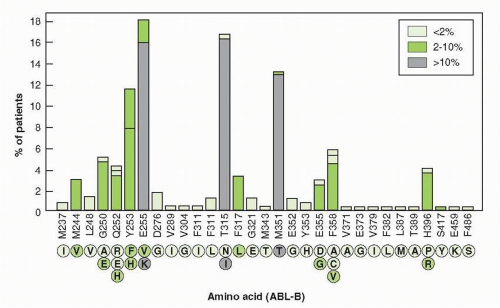 FIGURE 29-4 The amino acid substitutions, found in BCR-ABL kinase domain in 219 patients with CML and in 26 patients with Ph-positive acute lymphoblastic leukemia. Substitutions are shown below each amino acid position. Their color coded (as shown in the box) ties to their frequency, as depicted above each amino acid position. The T315I substitution creates resistance to all approved kinase inhibitors. (Figure from Hughes T, Deininger M, Hochhaus A, et al. Monitoring CML patients responding to treatment with tyrosine kinase inhibitors: review and recommendations for harmonizing current methodology for detecting BCR-ABL transcripts and kinase domain mutations and for expressing results. Blood 2006;108:28-37, with permission.) |
have been largely unresponsive to standard chemotherapeutic agents.
TABLE 29.4 c-KIT mutations and response to imatinib | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


