The anthracycline antibiotics doxorubicin and daunorubicin, initially discovered over 45 years ago,
1,
2 are used widely in current oncologic practice; their antineoplastic spectrum of action compares favorably with the alkylating agents and the taxanes. Doxorubicin and daunorubicin are especially active against the hematopoietic malignancies such as acute lymphocytic leukemia (ALL) and acute myelogenous leukemia (AML), Hodgkin’s and non-Hodgkin’s lymphoma, multiple myeloma, carcinomas of the breast, ovary, stomach, and thyroid; sarcomas of bone and soft-tissue origin; and various childhood malignancies. The key features of the two most commonly used anthracyclines, daunorubicin and doxorubicin, are summarized in
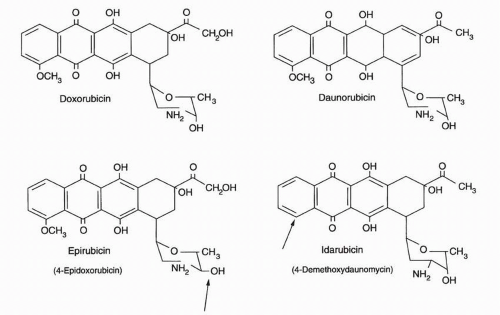
Table 18-1, and their structures are shown in
Figure 18-1. Doxorubicin is currently used principally for the treatment of solid tumors, especially breast cancer and lymphoma, while daunorubicin is routinely included as part of chemotherapeutic induction programs for AML and ALL. The doxorubicin analog epirubicin is similar to the parent compound with respect to its acute toxicity profile and spectrum of antitumor efficacy but is significantly less potent and only slightly less cardiotoxic. The modestly decreased cardiac toxicity of epirubicin is only a marginal advantage since other means are currently available to lessen the risk of anthracycline-induced heart damage. Idarubicin, a daunorubicin analogue, has significant activity in the treatment of AML but is less active against solid tumors and, thus, is an appropriate alternate anthracycline only in the setting of acute leukemia.
In the clinic, the anthracyclines doxorubicin and daunorubicin can be combined with alkylating agents, antimetabolites, and with most other natural products, although enhanced cardiac toxicity has resulted from doxorubicin combinations with paclitaxel and with trastuzumab. The enhanced cardiac toxicity with paclitaxel has been attributed to allosteric effects of the taxane on carbonyl reductase 3, increasing the production of the more cardiotoxic metabolite, doxorubicinol.
As potent radiation sensitizers, the anthracyclines cannot be administered simultaneously with therapeutic irradiation. The anthracyclines are active over a wide range of doses and in a variety of administration schedules; essentially equivalent antitumor activity is observed whether the anthracycline is given as a single large bolus dose once a month, as a weekly intravenous bolus, or as a prolonged infusion.
3,
4 However, changes in drug scheduling do change the pattern of normal tissue injury. The combination of broad antitumor activity, lack of significant drug interactions or toxicity with most other antitumor agents, and flexibility in dose and schedule makes doxorubicin and daunorubicin valuable participants in curative drug combinations for leukemia and lymphomas. Anthracycline-containing combination chemotherapy protocols have become standard therapy for many solid tumors as well, including breast cancer; bone and soft-tissue sarcomas; and many childhood solid tumors. Although the acute toxicities associated with anthracycline administration, such as myelosuppression, mucositis, and alopecia, are important in clinical practice, the toxic reaction that causes the greatest concern is the unique, cumulative cardiac injury produced by these drugs. Elucidation of the biochemical mechanisms of this cardiac toxicity resulted in the identification of an iron-chelating agent with its own modest antineoplastic activity, dexrazoxane (ICRF-187), which can block the cardiac toxicity of the anthracycline antibiotics in a wide range of animal models. Prospective, randomized clinical trials have shown that this agent is highly effective in reducing the cardiac toxicity of doxorubicin. This development, as well as the demonstration of a steep dose-response curve for doxorubicin in the treatment of solid tumors,
5 and the feasibility of utilizing colony-stimulating factors with or without peripheral blood progenitor support to ameliorate the bone marrow toxicity of the anthracyclines have permitted a significant increase in the dose density and duration of anthracycline therapy, further increasing the clinical utility of this family of drugs.
6,
7 The gene encoding topoisomerase IIA, the molecular target for anthracyclines, resides on chromosome 17 and may be co-amplified with the her 2/neu gene in breast cancers. There is evidence that amplification of the region defined by the chromosome 17 centromere enumeration probe, may predict a positive response to this class of agent in breast cancer therapy.
8
Topoisomerase II Interactions
An important advance in our understanding of anthracycline-DNA interactions occurred with the demonstration that anthracyclines cause protein-associated DNA breaks (measured by filter elution), the number of which correlate with cytotoxicity in some cell lines.
51,
52,
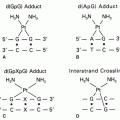
53 Protein-associated DNA breaks result from the formation of a ternary drug-DNA-enzyme “cleavable complex” involving the anthracycline antibiotic and DNA topoisomerase II (top II) (see
Fig. 19-2), an enzyme associated with the nuclear matrix, which plays a critical role in relieving torsional strain in DNA and chromosome condensation.
54 Anthracyclines inhibit top II by trapping DNA strand passage intermediates and preventing the resealing of the broken ends of DNA. These strand breaks are detected as protein-associated DNA single- and double-strand breaks linked to the enzyme.
55,
56 It had previously been presumed that through intercalation the anthracyclines altered the three-dimensional conformation of DNA, thereby arresting the cycle of top II action at the point of DNA cleavage. However, top II-associated DNA cleavage can be demonstrated at doxorubicin concentrations (10
−8 M) well below the dissociation constant for DNA intercalation
57 and are produced by anthracycline analogs, which do not intercalate into DNA.
58 Thus, it is likely that anthracyclines may stimulate top II-mediated DNA cleavage by a nonintercalative mechanism. Doxorubicinone actively inhibits the purified enzyme, suggesting that the anthracycline sugar is not required for enzyme inhibition. Since the daunosamine sugar plays an important role in DNA binding, it may be possible to dissociate DNA intercalation from top II-mediated strand breakage.
56 The anthracyclines produce topoisomerase-related DNA cleavage in specific regions of the DNA (with an adenine at the 3′-end of one break site); this may provide a clue to gene-specific effects of these drugs and specific sites particularly vulnerable to cleavage by anthracycline analogues.
59,
60 As will be described subsequently, anthracycline resistance may be associated with alteration in expression or mutations of top II, further evidence for a critical role for this enzyme in anthracycline action.
61,
62,
63In human cell lines and in yeast model systems, the role of the alpha isoform of top II plays a central role in the production of protein-associated DNA cleavage after doxorubicin exposure.
64,
65 Overexpression of the antisense construct of top II
α in human U937 monocytic leukemia cells down-regulates top II
α mRNA levels by more than 70% with a concomitant reduction in the cytotoxicity of daunorubicin. Furthermore, doxorubicin-related single-strand cleavage and top II-DNA complex formation are ATP dependent and specific for top II (not I).
66,
67While anthracycline interactions with DNA and top II clearly occur in many mammalian cell lines, it is likely that the formation of cleavable complexes alone is only potentially lethal and is not in itself sufficient for tumor cell killing. Although the initial correlations of protein-associated single-strand cleavage and cytotoxicity in L1210 cells, which were performed at clinically relevant drug
concentrations, suggested a direct relationship between top II-mediated DNA damage and cytotoxicity for doxorubicin,
51,
68 subsequent investigations have demonstrated a dissociation between tumor cell killing by doxorubicin and the kinetics of DNA break formation and resealing.
69 In some cell lines, only DNA double-strand cleavage can be associated with cytotoxicity,
70 and in others, DNA single-strand cleavage is modest and double-strand cleavage essentially undetectable at even supralethal drug concentrations.
71,
72 Furthermore, evidence from several different model systems does not provide uniform support for a causal relationship between the level of top II and the sensitivity of human cell lines to doxorubicin in vitro.
73,
74 Finally, correlations between top II
α content or activity in primary tumors and clinical outcome for breast cancer patients treated with anthracyclines have not been consistent in all studies, although some work indicates a correlation between Top II amplification and clinical benefit in breast cancer.
75,
76In addition to stabilization of the cleavable complex, the anthracyclines and a number of other antineoplastic compounds can inhibit the catalytic activity of the enzyme without trapping the complex.
55,
77 This observation, important for the development of new anthracyclines as well as combination regimens, underlies the recent demonstration that certain anthracycline analogues (not doxorubicin) antagonize the cytotoxicity and DNA cleavage of etoposide through inhibition of cleavable complex formation.
78,
79,
80 Furthermore, other anthracycline analogues inhibit both top I and II, a potential explanation of their nonoverlapping resistance profiles and altered spectrum of action.
81,
82,
83 Doxorubicin exhibits more cytotoxicity than expected per DNA break. This might mean that either doxorubicin-associated breaks are qualitatively different from those produced by other top II-active drugs or that other mechanisms of action might be operating in parallel. Thus, questions remain regarding the precise role of cleavable complex formation in the cytotoxicity of anthracyclines.
Other Effects on DNA
In addition to the physicochemical effects of the anthracyclines on DNA and their interactions with top II, doxorubicin produces other effects on DNA. Doxorubicin and daunorubicin form DNAanthracycline complexes that significantly modify the ability of a specific class of nuclear enzymes, the helicases, to dissociate duplex DNA into DNA single strands in an ATP-dependent fashion; the entire process of strand separation is thus hindered, limiting replication.
84 This effect occurs at clinically relevant drug concentrations (<1 μM) and parallels, at least in part, the cytotoxic spectrum of several anthracycline analogues.
85 The mechanism of helicase inhibition involves the formation of an irreversible ternary complex between glycosidic anthracyclines, DNA, and the helicase.
86 Given the diversity of human DNA helicase, differential effects of the anthracyclines against various tumors could be related to their interactions with members of this class of nuclear enzymes. Additionally, in tumor cells that conditionally express top II
α, doxorubicin can clearly produce DNA double-strand breaks and apoptosis independent of its interaction with top II.
87 Even in the absence of significant DNA double-strand cleavage, doxorubicin interferes with DNA unwinding and produces nonoligosomal fragmentation of nascent DNA during continuous exposure to very low drug concentrations.
72,
88 This DNA effect is associated with tumor cell differentiation and suppression of
c-myc oncogene expression by both doxorubicin and certain of its analogues and suggests yet another potential growth inhibitory pathway.
89,
90As will be reviewed subsequently, doxorubicin can undergo cycles of reduction and oxidation in essentially all intracellular compartments including the nucleus and the mitochondrion, leading to the formation of reactive oxygen species,
91 and oxidized DNA bases in human chromatin and in intact tumor cells, a finding that may provide a cytotoxic mechanism unrelated to strand cleavage.
92,
93,
94 Mitochondrial DNA is also susceptible to oxidative stress. In rats, doxorubicin treatment produces 8-hydroxyguanosine, a by-product hydroxyl radical attack on DNA, in cardiac and liver mitochondria.
95,
96,
97Evidence of DNA base oxidation is also found in patients treated with anthracycline antibiotics. Urinary hydroxymethyluracil (an oxidative by-product of thymine) was identified within 24 hours of drug treatment in patients receiving combination chemotherapy that included an anthracycline.
98,
99 Following bolus therapy with the anthracycline analogue epirubicin, oxidized DNA bases can be found in chromatin isolated from patient lymphocytes.
100 In patients receiving a 96-hour infusion of doxorubicin, producing steady-state drug levels of 0.1 μM, a two- to fivefold increase in 13 different oxidized DNA bases, including thymine glycol, 5-OH-hydantoin, 5-OH-uracil, 4,6-diamino-5-formamindo-pyrimidine, and 5,6-di-OH-uracil, was detected by GC/MS in peripheral blood mononuclear cells, beginning 72 hours after the initiation of treatment.
101 These data are important because this spectrum of DNA base damage is the same as that produced by ionizing radiation and indicates the generation of hydroxyl radicals.
102,
103 Thus, these studies provide unequivocal evidence of the oxidative damage to DNA produced by doxorubicin and its analogues under clinical conditions.
As will be discussed in
Chapter 42, doxorubicin and daunorubicin are mutagens and carcinogens;
104 only recently have investigators begun to map the base substitutions and deletions produced by doxorubicin, which occur adjacent to preferential doxorubicin DNA-binding sequences. Therapy with doxorubicin (or epirubicin) when used in combination with cyclophosphamide is associated with an increased risk of a second malignancy, specifically AML.
105,
106,
107,
108Because of the inhibitory effects of oxidized DNA bases on the action of DNA polymerases and other DNA repair mechanisms, as well as their own intrinsic mutagenic properties, the detection of oxidized DNA bases in hematopoietic cells after treatment with doxorubicin provides a potential mechanism for anthracycline-related mutagenicity and carcinogenicity, as well as insights into the mechanism of tumor cell killing by the anthracyclines. Their potentially adverse consequences for the synthesis of genes comprising the mitochondrial respiratory chain are also raised by these findings.
109,
110,
111,
112,
113In addition to the production of oxidized DNA bases, the intracellular reactive oxygen metabolism of the anthracyclines leads to the iron-dependent production of formaldehyde from a variety of intracellular carbon sources. Formaldehyde can subsequently react to produce a drug-formaldehyde conjugate containing two anthracycline molecules.
114,
115,
116 These conjugates have a unique ability to form novel covalent DNA crosslinks that markedly enhance the therapeutic activity of the
anthracycline and represent targeted anthracycline prodrugs that possess a unique spectrum of anticancer activity,
117,
118 and they are significantly more cytotoxic and more efficient in producing apoptosis than the interaction of doxorubicin with top II.
119Finally, recent microarray-based investigations have revealed the broad range of changes in gene expression that follow doxorubicin exposure,
120,
121,
122 including alterations in multiple biochemical pathways such as DNA repair, RNA metabolism, reactive oxygen metabolism, chromatin remodeling, cell cycle and apoptotic programs, mitochondrial metabolism, and vacuolar function.
Drug Activation by One- and Two-Electron Reduction
During DNA intercalation and binding to top II, the anthracyclines act as chemically inert compounds that owe their activity to their ability to bind to key macromolecules and distort the three-dimensional geometry of these targets. However, the anthracyclines are also chemically reactive and capable of an extraordinarily rich chemistry that even now is not fully documented.
123,
124
One-Electron Reduction
The one-electron reduction of the anthracyclines was initially described in hepatic microsomal systems
125,
126,
127 but was later shown to play a central role in the cardiac toxicity of this class of drugs
128,
129,
130 and may be involved in antitumor activity as well.
131,
132,
133,
134,
135 All of the clinically active anthracyclines are anthraquinones. As is true of quinones in general,
136,
137 the anthracyclines are able to undergo one- and two-electron reduction to reactive compounds that cause widespread damage to intracellular macromolecules including lipid membranes, DNA bases, and thiol-containing transport proteins (
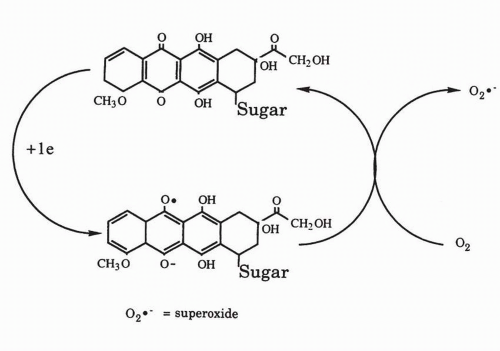
Fig. 18-3).
138,
139,
140,
141 As outlined in
Figure 18-4, the one-electron reduction of doxorubicin or daunorubicin may occur in essentially all intracellular compartments including the nuclear membrane and is catalyzed by flavin-centered dehydrogenases or reductases including cytochrome P-450 reductase, NADH dehydrogenase (complex I of the mitochondrial electron transport chain), xanthine oxidase, and cytochrome
b5 reductase.
142,
143,
144 In addition, all three isoforms of nitric oxide synthase (at their flavoprotein domains) are capable of catalyzing the one-electron reduction of doxorubicin with the subsequent production of superoxide and a decrease in nitric oxide.
145,
146 Furthermore, doxorubicin can directly inhibit nitric oxide synthase activity,
146,
147 which could produce significant alterations in vascular tone both in the heart and in tumors.
148,
149 It can also be metabolized by lactoperoxidase and nitrite.
150 These flavoenzymes are widely distributed in mammalian tissues, and anthracycline-mediated free-radical formation occurs in a wide range of organs and tumor cell lines. In addition to flavoproteins, doxorubicin can be reduced in the heart by oxymyoglobin, producing strong oxidant species.
151One-electron reduction of the anthracyclines leads to the formation of the corresponding semiquinone free radical. In the presence of oxygen, this free radical rapidly donates its electron to oxygen to generate superoxide anion (O
2−). Although not highly toxic itself, the dismutation of superoxide yields hydrogen peroxide (H
2O
2).
152 Furthermore, in the presence of superoxide anion, the nitric oxide system can produce potentially toxic amounts of peroxynitrite.
153 Under biological conditions, the anthracycline semiquinone or reduced metal ions such as iron also reductively cleave hydrogen peroxide to produce the hydroxyl radical (•OH) or a higher oxidation state metal with the chemical characteristics of the hydroxyl radical, one of the most reactive and destructive chemical species known.
154,
155 It is now commonly accepted that reduced metals are critical components in the formation of toxic free-radical intermediates and may well contribute to cell killing by the anthracyclines.
156 However, it is unclear how reduced metal species, including iron, become available for these free radical reactions.
Because oxygen radical formation occurs as a result of normal metabolic processes (including mitochondrial respiration) and is a common mechanism of action for several naturally occurring toxins, most mammalian cells have elaborate defenses against oxygen radical toxicity.
157 Superoxide dismutase, catalase, and glutathione peroxidase act in concert to reduce superoxide, hydrogen peroxide, and lipid hydroperoxides to water or nontoxic lipid alcohols without the formation of the hydroxyl or peroxyl radicals (
Fig. 18-4). Glutathione, a sulfur-containing tripeptide, can react with many radicals. It also functions as a component of the glutathione peroxidase cycle to reduce peroxides to less reactive compounds.
There are also specific DNA repair systems to handle oxidative damage to DNA.
158,
159,
160 However, antioxidant defenses are not equally distributed in various tissues in the body. For example,
glutathione concentration is higher in the liver than in most other tissues and tumors. Catalase activity is lower in the heart than in the liver.
129,
161 Likewise, the activity of several flavoproteins capable of activating the anthracyclines differs from tissue to tissue. These variations in drug activation and antioxidant defense provide ample opportunity for tissue specificity in terms of toxicity and antitumor activity. For example, the unique cardiac toxicity of the anthracyclines may result, in part, from the low level of cardiac catalase coupled with the extraordinary cardiac content of mitochondria and myoglobin, both of which enhance drug activation. An additional contribution to cardiotoxicity may be the sensitivity of cardiac glutathione peroxidase to free radical attack;
129,
162 free radicals destroy this critical enzyme at the same time that anthracyclines stimulate cardiac hydrogen peroxide formation.
163 In contrast, while anthracyclines can easily be activated to reactive intermediates by hepatic enzymes, the liver is infrequently damaged by anthracyclines in clinical chemotherapy, perhaps because it has an active free-radical defense system and is able to efflux anthracyclines and anthracycline
metabolites. The importance of hepatic antioxidant systems is illustrated by the finding that pretreatment of rodents with agents (carmustine or acetaminophen) that significantly diminish the level of reduced glutathione dramatically sensitizes hepatocytes to injury by doxorubicin-induced free radicals.
164,
165The role of oxygen radical formation in tumor cell killing.
166,
167,
168,
169,
170 Several lines of evidence support this hypothesis. First, doxorubicin resistance in tumor cell lines can frequently be reversed by agents which decrease glutathione concentration.
170,
171,
172,
173,
174 Second, anthracycline-enhanced free radical formation has been detected in many tumor cell lines.
131,
152,
175,
176 The best studied are human breast cancer cell lines, in which both intracellular and extracellular reactive oxygen species were demonstrated.
132,
177 Extracellular as well as intracellular antioxidants have also been demonstrated to decrease the cytotoxicity of the anthracyclines in normal and malignant cell lines.
132,
133,
156,
178,
180,
181,
182 In addition, some anthracycline-resistant cancer cell lines exhibit increases in various aspects of the oxygen free radical defense system, including increases in glutathione and the selenoprotein glutathione peroxidase.
169,
174,
183,
184,
185,
186 Manipulation of glutathione peroxidase activity produced by changes in selenium status significantly affects tumor cell killing by doxorubicin.
187,
188 Transfection of the human cytosolic glutathione peroxidase in the sense orientation produces doxorubicin resistance
189 while antisense expression sensitizes cells to doxorubicin cytotoxicity.
190 Finally, fourfold overexpression of the manganese superoxide dismutase in CHO cells produces 2.5-fold resistance to doxorubicin,
191 while inhibition of the copper-zinc superoxide dismutase with 1,25-dihydroxyvitamin D
3 significantly enhances doxorubicin cytotoxicity.
192Reservations about the role of oxygen radical formation in tumor cell killing arise from conflicting experimental evidence, although the balance suggests active redox cycling under conditions of tumor therapy. First, many studies demonstrating anthracycline-enhanced hydroxyl radical formation have utilized clinically irrelevant drug concentrations. This limitation is in part technical in that it was not possible in the early experiments to detect hydroxyl radicals at drug concentrations much below 10
−7 M. However, fluorescent probes for hydrogen peroxide production by flow cytometry successfully demonstrated peroxide formation in colon cancer cells after treatment with 0.4 μM doxorubicin;
176 human breast cancer cells exposed to 0.05 μM doxorubicin exhibited enhanced oxidative respiration and reactive oxygen production as measured by chemiluminescent probes.
193 Second, many tumors for which doxorubicin has great clinical utility, such as breast cancer, are clearly hypoxic, and thus, the applicability of the redox chemistry outlined in
Figures 18-3 and
18-4 might be questioned. However, under low partial pressures of oxygen, iron-mediated lipid peroxidation and DNA damage from the doxorubicin semiquinone are actually enhanced.
194 Third, glutathione depletion does not sensitize
all tumor cell lines to the cytotoxic effect of doxorubicin,
195 and many doxorubicin-resistant cells have no alteration in antioxidant defense enzymes.
196 Fourth, a common presumption has been that oxygen radical-mediated effects on DNA are unlikely because intercalated drug could not be reduced. However, doxorubicin covalently bound to oligonucleotides can still be activated by cytochrome P-450 reductase,
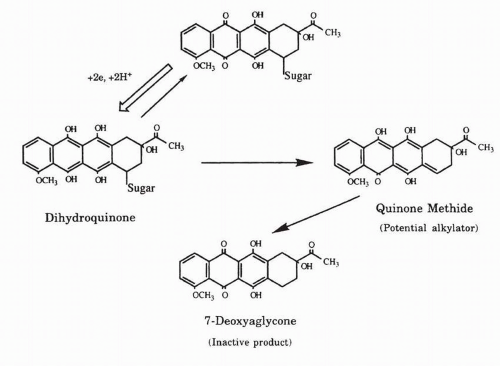
197 and daunorubicin intercalated into calf thymus DNA can be reduced by a superoxide-generating system; under these circumstances the semiquinone is accessible to hydrogen peroxide for reaction, and the disproportionation of the semiquinone to the 7-deoxyaglycone may occur by intramolecular electron transfer with migration of electrons over several base pairs.
198,
199,
200 Finally, as outlined previously, oxidized DNA bases, by-products of anthracycline redox cycling, are present in both the urine
99 and peripheral blood mononuclear cells of patients receiving anthracycline therapy.
100,
201 These reports provide evidence demonstrating the products of redox cycling in human tissues after anthracycline administration according to standard treatment schedules. Whether it is instrumental in cell killing of some or all human tumors is unclear.
Role of Iron
Although free radical formation was originally proposed as the basis for anthracycline cardiac toxicity in 1977 and various animal model studies suggested that hydroxyl radical scavengers could blunt doxorubicin-related heart damage, free radical scavengers were initially unsuccessful cardioprotective agents in humans.
202 These results suggested that some additional variable was involved. This variable proved to be the interaction of anthracyclines with iron. Because of its reactivity, free iron concentrations in the body are in the range of approximately 10
−13 M, whereas total iron concentrations are between 10
−4 and 10
−5 M. Most iron in tissues is stored bound to ferritin. Doxorubicin releases iron from ferritin in two ways. The drug can slowly abstract iron from the ferritin shell
203; a much more rapid release of iron follows conversion of the anthracycline to its semiquinone, which can release iron under hypoxic conditions or through the reducing power of the superoxide anion in air.
204,
205 Doxorubicin can release nonheme, nonferritin iron from microsomes.
206,
207,
208,
209 Doxorubicin can also increase the uptake of iron by a transferrin receptor-mediated process, enhancing intracellular iron availability.
210 The hydroxyquinone structure of doxorubicin or daunorubicin represents a powerful site for chelation of metal ions, especially ferric iron. Iron anthracycline complexes possess a wide range of interesting biochemical properties in vitro.
211,
212 These complexes can bind DNA by a mechanism distinct from intercalation, cause oxidative destruction of membranes, and oxidize critical sulfhydryl groups. It remains unclear, however, whether these tightly bound iron-anthracycline complexes are stable and produce toxic effects intracellularly,
213,
214 or whether the “delocalization” of catalytic amounts of protein-bound iron by anthracyclinestimulated free radical formation is responsible for hydroxyl radical formation in tissues.
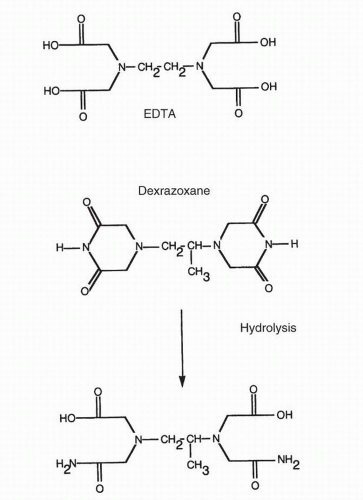
These investigations suggested that the most effective way to interfere with the generation of highly reactive oxidants after anthracycline exposure would be to pretreat with a chelating agent that might withdraw iron from free radical reactions. This hypothesis has been confirmed; an iron chelator, dexrazoxane (ICRF-187), prevents doxorubicin-induced lipid peroxidation
215 and cardiac toxicity in a wide range of animal models.
216,
217 Randomized, controlled clinical trials in humans have confirmed that this agent markedly reduces the cardiac toxicity of doxorubicin.
218,
219 Dexrazoxane is a highly effective iron chelator in vivo; during phase I studies, it caused a tenfold increase in urinary iron clearance.
220,
221 Dexrazoxane is itself a prodrug in that it must undergo hydrolysis to become an effective iron chelator (
Fig. 18-5). This hydrolysis to ICRF-198, the major iron-binding metabolite of dexrazoxane, can occur spontaneously at physiologic pH but is markedly enhanced after uptake into cardiac myocytes with conversion of the parent
drug to ICRF-198 in less than 60 seconds.
222 The parent drug is very lipid soluble and enters cells by passive diffusion. ICRF-198 has been demonstrated to efflux iron from iron-loaded myocytes; these studies suggest that the same chelating ability may remove iron that has been released from cardiac iron-storage proteins.
222Important additional studies have further elucidated the role of iron in free radical generation and other iron-dependent reactions that may contribute to the cardiac toxicity of the anthracy-clines.
223,
224,
225,
226 Down-regulation of cellular iron uptake by overexpression of the wild-type hemochromatosis gene in MCF-7 cells decreases doxorubicin-induced reactive oxygen production and apoptosis.
227 On the other hand, feeding rats a diet that is iron enriched dramatically enhances cardiac apoptosis and mitochondrial injury after treatment with doxorubicin.
228 Further, the alcohol metabolite of doxorubicin, doxorubicinol, which is produced by the two-electron reduction of the C-13 side chain carbonyl group, causes the delocalization of low molecular weight Fe(II) species from the iron-sulfur center of aconitase in a redox-dependent fashion. Further evidence supporting the role of alcohol metabolite in cardiotoxicity comes from a study of polymorphisms of carbonyl reductases. The CBRV244M variant of carbonyl reductase 3 is associated with an 8.5-fold increased risk of cardiotoxicity in children receiving doxorubicin. The recombinant variant had nearly threefold greater activity in synthesizing doxorubicinol, as compared to its more common counterpart.
613 The formation of a doxorubiciniron-complex with aconitase interferes with critical interconversions of cytosolic aconitase with iron regulatory protein-1 (IRP-1); IRP-1 plays an essential role in iron homeostasis and, hence, in the regulation of critical intracellular metabolic processes (such as the action of mitochondrial electron transport proteins, myoglobin, and various cytochromes). These effects of doxorubicin metabolites suggest that iron-dependent reactions occur that may not be directly related to the formation of reactive oxygen species. This could help to explain the utility of dexrazoxane, compared to free radical scavengers that do not chelate iron, in the prevention of both acute and chronic anthracycline cardiotoxicity.
209Doxorubicin is a powerful chelator of other metal ions, including Cu2+ and Al3+. The chelation of aluminum by doxorubicin is so avid that a doxorubicin solution left in contact with aluminum foil for only 1 hour will change from the orange-red of doxorubicin to the bright cherry red of the aluminum complex. A similar reaction occurs with ironcontaining alloys; doxorubicin left within a syringe needle for any significant period of time will also change color by virtue of chelation of metal from the needle. For this reason, every effort should be made in the clinic to keep anthracyclines from prolonged contact with any metal surface.
Signal Transduction and Anthracyclines
Communication between the cell surface and the nucleus plays a crucial role in growth control; important signal transduction pathways for mitogenic stimuli are initiated at the plasma membrane.
248,
249,
250,
251,
252 Anthracyclines impact signaling pathways in ways that may be important for cell survival and response to DNA damage.
253Anthracycline action affects specific signal transduction pathways, most importantly the protein kinase C(PKC) system. Anthracyclines inhibit PKC, but only at suprapharmacological concentrations (above 100 μM)
254,
255; the doxorubicin-iron complex inhibits diacylglycerol stimulation of PKC at 10-fold lower concentrations.
256 Even lower concentrations of doxorubicin increase the turnover of phosphoinositide and phosphatidylcholine in Sarcoma 180 cells, which leads to the accumulation of diacylglycerol and inositol phosphates and a twofold increase in cytosolic PKC activity.
257 Furthermore, activation of the PKC pathway by phorbol esters enhances doxorubicin cytotoxicity and drug-related DNA-protein cross-links, whereas down-regulation of PKC reduces cell kill.
258 Since PKC can phosphorylate topoisomerase II,
259 it is possible that the initiation of membrane signaling by doxorubicin could be involved in the regulation of anthracycline-mediated DNA damage.
The importance of the sphingomyelin pathway in signal transduction has become increasingly clear.
248,
260 In addition to participating in PKC-related signal transduction, sphingolipid metabolites transduce signals from cell surface molecules including interferon-
γ, TNF-
α, and Fas/APO-1. Activation of sphingomyelinase by cellular stresses, including exposure to the anthracyclines, leads to the release of the critical signaling intermediate ceramide from membrane sphingomyelin.
261,
262,
263 Intracellular ceramide accumulation can produce profound effects on cell cycle progression as well as on the effector arm of the cell death program.
264,
265,
266 Expression of glucosylceramide synthase (the enzyme that converts ceramide to glucosylceramide) in human MCF-7 cells blocks doxorubicin-induced increases in ceramide after drug exposure, leading to an 11-fold increase in IC
50 concentration.
267Signal transduction pathways involving PKC and ceramide may contribute to the regulation of P170-glycoprotein function and the enhanced export of anthracyclines in drug-resistant cells
268,
269,
270; inhibition of PKC down-regulates P170-glycoprotein function and enhances the sensitivity of myeloid leukemia cells to daunorubicin, providing a novel strategy for overcoming multidrug resistance.
271 Agents that reverse multidrug resistance, such as cyclosporin A and verapamil, may in part act by inhibiting ceramide glycosylation.
272As might be expected from the multiplicity of effects of anthracyclines on signaling pathways, doxorubicin alters activity of the phosphoinositide 3-kinase and AKT survival pathway, both in tumor cells and in the heart.
273,
274,
275,
276 In malignant cells, doxorubicin exposure leads to increased phosphorylation of AKT, which, if blocked, increases tumor cell killing by the anthracycline. In related fashion, overexpression of PTEN, which dephosphorylates AKT, leads to an increase in the apoptotic program. In the heart, doxorubicin treatment decreases both AKT and ERK phosphorylation (critical for cell survival), an inhibition that is blocked by pretreatment with dexrazoxane.
277 Anthracyclines inhibit signaling through TGF
β1 and block activation of HIF-1
α-mediated gene expression.
278,
279,
280 Both of these latter effects may modify tumor-related angiogenesis by the anthracycline antibiotics.
Anthracyclines and Apoptosis
Anthracycline-related alterations in membrane biochemistry, signal transduction, mitochondrial metabolism, DNA damage, and free radical formation all contribute to apoptosis.
281,
282,
283Doxorubicin or daunorubicin exposure produces the morphological changes associated with apoptosis including chromatin condensation, internucleosomal DNA fragmentation, reduced cell volume, and cytoplasmic blebbing in many normal and malignant cell types.
284,
285,
286,
287,
288,
289,
290,
291 In general, the degree of anthracycline-related apoptosis varies considerably between experimental model systems; in cell culture, the full expression of apoptotic morphology is often not observed until 48 to 120 hours after drug treatment. This variability is due, in part, to wide variations in the expression of both proapoptotic and antiapoptotic molecules in cultured tumor cells, including human tumor samples.
36,
292,
293,
294Anthracycline-related apoptosis may involve biochemical interactions that produce several different initiating signals that produce apoptosis and/or necrotic cell kill. In selected cell lines, anthracyclines stimulate the interaction of the CD95 (APO-1/Fas) surface receptor with its natural ligand CD95L to form a signaling complex. This complex in turn activates proteases of the caspase family, resulting in apoptosis. The fas pathway, which plays a critical role in the regulation of lymphoid cell growth, is active in some solid tumors and leukemias. In some cell lines, doxorubicin produces apoptosis by inducing CD95L and CD95 receptor formation. CEM cells, Jurkat T cells, and neuroblastoma cells resistant to anti-CD95 antibody were resistant to doxorubicin-induced apoptosis.
295,
296,
297 Doxorubicin clearly appears to up-regulate CD95L expression after doxorubicin exposure in HeLa cells transfected with a CD95L reporter construct.
298 However, acquired resistance to CD95 by clonal selection or by treatment with other anti-CD95 antibodies that inhibit fas-mediated apoptosis does not concomitantly engender resistance to doxorubicin-related apoptosis.
299,
300 Further, CD95 and CD95L are frequently not up-regulated following doxorubicin exposure,
301 and anthracycline-mediated activation of caspase 8, a component of the fas pathway, also occurs in the absence of signaling through the fas pathway.
302 These experiments suggest that doxorubicin and CD95 utilize common downstream effectors of apoptosis but that the CD95 pathway is not required for initiating death stimulus in all cell lines or tumors.
In other experimental systems, apoptosis due to the anthracyclines has also been clearly related to an alternative mediator of cell death, ceramide generation,
261,
303 which links the plasma membrane biochemistry of doxorubicin with the induction of the cell death cascade. In these studies, ceramide generation following sphingomyelinase activation leads to increased mitochondrial permeability, the activation of proapoptotic caspases, and serine-threonine protein phosphatases.
248,
264,
304 It remains to be determined, however, whether ceramide production after anthracycline exposure is associated principally with the activation of cell death signals to the mitochondria or with the effector phase of apoptosis.
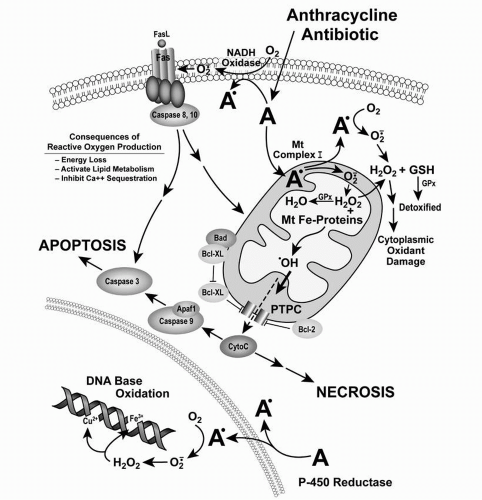
305One of the critical steps in translating a wide variety of apoptotic stimuli into either apoptotic or necrotic cell death is the induction of cytochrome c release from the space between the inner and outer mitochondrial membranes.
281,
306,
307 Cytochrome c release can lead to caspase activation or to altered mitochondrial electron transport with subsequent apoptosis or necrosis. Anthracyclines are fully capable of inducing cytochrome c release independent of DNA damage.
308,
309,
310 In light of the previously described extensive binding and metabolism of the anthracyclines by complex I of the mitochondrial electron transport chain,
143 it is likely that reactive oxygen species, produced by anthracycline-treated mitochondria, can damage mitochondrial membrane integrity and may play an important initiating role in doxorubicin-related apoptosis. Various free radical scavengers inhibit programmed cell death following anthracycline exposure.
93,
168,
180,
181,
311,
312 Furthermore, cytokine-mediated induction of ceramide production is redox-sensitive, and overexpression of antioxidant genes in human tumor cells prevents ceramide production and partially blocks apoptosis.
313,
314 These observations link many of the known but pleiotropic biochemical effects of the anthracyclines; further studies are likely to define the order of anthracycline-related death signals at the molecular level and the relationship of these signals to intracellular anthracycline metabolism.
Two major endogenous modulators of programmed cell death, Bcl-2 and p53, as well as their associated downstream effectors, play critical roles in regulating anthracycline-related apoptosis. The Bcl-2 protein functions in the outer membranes of mitochondria, nuclei, and the endoplasmic reticulum as an inhibitor of cell death; it blocks apoptosis following anthracycline exposure in many experimental systems.
315,
316 Furthermore, in acute myelogenous leukemic blasts and HL-60 cells, the cytotoxicity of daunorubicin increases in cells treated with
bcl-2 antisense oligonucleotides.
317 Bcl-2 suppresses the p53-mediated transcriptional activation of several genes involved in the apoptotic process after doxorubicin exposure in MCF-7 cells.
318 The biochemical mechanisms through which Bcl-2 inhibits cell death continue to be elucidated; however, it is currently appreciated that Bcl-2 represses cytochrome c release from mitochondria, interferes with caspase activation, blocks the apoptotic effects of reactive oxygen species, and binds to transcription factors involved in doxorubicin-mediated apoptosis, such as NF-kappa B.
318,
319,
320,
321,
322,
323