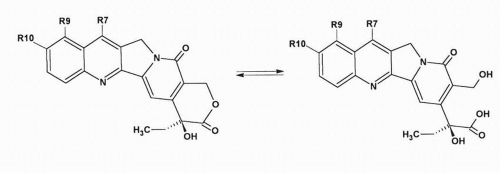
Topotecan is a water-soluble camptothecin derivative containing a stable basic side chain at position 9 of the A ring of 10-hydroxycamptothecin (
Fig. 17-1). Clinical trials of topotecan were initiated in 1989; it was approved for use as second-line chemotherapy in patients with advanced ovarian cancer in 1996, and for the treatment of small cell lung cancer (SCLC) after failure of initial or subsequent chemotherapy in 1998. Key features of topotecan are listed in
Table 17-1.
Clinical Pharmacology
The most common dose and schedule of topotecan administration is a 30-minute IV infusion of 1.5 mg/m
2 daily for 5 days every 3 weeks.
6 This regimen has undergone the most widespread clinical testing, and it is currently the dosage of topotecan approved for treating ovarian and lung cancer. Five-day continuous infusions of topotecan at 2.0 mg/m
2/d have been tested in patients with hematologic malignancies, although in these studies gastrointestinal toxicities such as mucositis and diarrhea became more problematic. Prolonged 21-day infusion schedules at 0.5 to 0.6 mg/m
2/d have been disappointing in phase II studies. Other schedules tested in phase I or phase II studies include a single 30-minute infusion, 24-hour infusions, and 3-, 5-, and 14-day continuous infusions. Oral administration has also been tested clinically and has been compared with IV administration in randomized studies.
7,
8,
9,
10,
11 Furthermore, IP, intrathecal, and individual adaptive, pharmacokinetically guided topotecan studies have been completed.
General Pharmacokinetics
After IV topotecan administration, the lactone ring undergoes rapid hydrolysis to generate the carboxylate species. Less than 1 hour after the start of an infusion, the majority of the circulating drug in plasma is in the carboxylate form, and this species predominates for the duration of the monitoring period. In most studies, the ratio of the lactone to total topotecan area under the concentration versus time curve (AUC) ranged from 20% to 35%. Interindividual variation in the AUC and the total-body clearance is quite large for both lactone and total topotecan (lactone plus carboxylate). In general, plasma concentrations and AUC levels tended to increase with increasing dose levels, consistent with linear pharmacokinetics, although in some studies, nonlinearity in drug clearance at higher dose levels was seen.
12For topotecan lactone, the terminal half-life ranges from 2.0 to 3.5 hours, which is relatively short compared with that of other camptothecin analogs. Consequently, no accumulation of drug is observed when administered daily for five consecutive days. Population pharmacokinetic studies in patients treated with IV or orally administered topotecan revealed that patient characteristics (i.e., gender, height, weight) and laboratory values (i.e., serum creatinine
concentration) give a moderate ability to predict the clearance of topotecan in an individual patient.
13,
14
Absorption
The most common route for topotecan administration has been IV; however, oral formulations using prolonged administration schedules have undergone preclinical and clinical testing. In humans, the reported oral bioavailability ranged from 30% to 42%
15 and is influenced by multiple factors. First, the relatively high pH in the small bowel leads to conversion to the carboxylate form, which is poorly absorbed by the intestinal walls. Second, the bioavailability is reduced by protein-mediated, outward-directed transport of topotecan by ABCG2.
16 Third, the bioavailability is partly influenced by the binding of topotecan to food, proteins, and intestinal fluids and/or by decomposition in the gastrointestinal fluid. From a clinical point of view, as long as equivalent safety and efficacy can be ensured, the majority of patients prefer oral instead of IV administration of chemotherapy.
17
Metabolism
An
N-desmethyl metabolite of topotecan has been characterized and found to be present at relatively low concentrations in human plasma, urine, and feces after IV administration of topotecan.
18 Although a specific interaction with drug metabolism enzymes has not been established, clinical data suggest enhanced topotecan clearance in pediatric patients simultaneously receiving treatment with agents that induce hepatic cytochrome P-450 (CYP) enzymes, such as dexamethasone, phenobarbital, and phenytoin.
19 These observations are consistent with a potential drug interaction at the level of CYP3A; however, additional studies on the metabolism and excretion of topotecan and its potential for drug interactions are warranted.
Topotecan and its main metabolite can also undergo further metabolism into a UGT-mediated glucuronide product.
20 This is a reversible transformation because
β-glucuronidase, actively found in intestinal bacteria, is able to reform topotecan. Because topotecan is metabolized in the liver only to a minor extent, it is not surprising that the pharmacokinetics in patients with impaired liver function did not significantly differ from those in patients with normal hepatic function.
21 In contrast, patients with moderately impaired renal function had significantly reduced plasma clearance.
22 As a consequence, dose modifications are recommended for patients with impaired renal function and are not required for patients with liver dysfunction.
Excretion
Elimination of topotecan lactone is thought to result mainly from the rapid hydrolysis to the carboxylate species followed by renal excretion of the open-ring metabolite. Overall, 25% to 49% of the dose administered is excreted in the urine over a 24-hour period,
23 a few pediatric reporting recovery of over 90% of the administered drug during more prolonged periods of urine collection.
24
General Pharmacodynamics
Pharmacodynamic correlations between parameters of systemic drug exposure and topotecan drug effects have been observed inconsistently.
25 In a pediatric study, the topotecan total (lactone plus carboxylate) AUC and lactone plasma AUC correlated with the percentage change in the platelet count and the granulocyte count after a 72-hour drug infusion.
26 Similar correlations between leukocyte or granulocyte counts and total topotecan AUC or topotecan dose have been reported after a 30-minute infusion of topotecan given daily for 5 days, after 20-minute infusions every 3 weeks, and after 24-hour continuous infusions. In each case, measurement of the total topotecan plasma concentration was as informative about drug effects as the lactone drug levels. Thus, the need to measure lactone drug concentrations has been questioned by some investigators. Finally, although interpatient variability in topotecan plasma concentrations is high, the total variability in hematologic toxicity in patients is relatively low when the drug is administered according to standard dosing guidelines. Thus, therapeutic drug monitoring is not useful for this agent.
Adverse Reactions
Dose-related, reversible, and noncumulative myelosuppression is the most important side effect of topotecan.
27 Neutropenia occurs more frequently and is often more severe than thrombocytopenia, and it is more severe in heavily pretreated patients than in minimally pretreated patients. Besides myelosuppression, stomatitis (24% to 28% of patients) and late-onset diarrhea (40%) were noted at higher doses. Other nonhematological toxicities reported include alopecia, nausea, vomiting, fatigue, and asthenia.
Hematologic toxicity is more pronounced with the shorter oral regimens but is still mostly mild and noncumulative, whereas diarrhea is a severe and intractable side effect of more prolonged daily administration.
28 An analysis of the pharmacokinetic-pharmacodynamic relationships revealed that the total area under the curve per course did not differ between the various regimens and that the daily times five schedule provided the best systemic exposure and toxicity profile.
29In acute leukemia, the maximum tolerated dose (MTD) of a daily 30-minute IV infusion for five consecutive days every 3 weeks was 4.5 mg/m
2/d.
30 The dose-limiting toxicity (DLT) at higher dose levels was a complex of symptoms, consisting of high fever, rigors, precipitous anemia, and hyperbilirubinemia, which are possibly related to an acute hemolytic reaction.
Antitumor Activity
The antitumor activity of topotecan, given as a single agent using various schedules of administration, was established in a variety of phase II studies, including ovarian cancer, SCLC, NSCLC, breast cancer, myelodysplastic syndrome, and chronic myelomonocytic leukemia. Marginal activity was seen in head and neck cancer, prostate cancer, pancreatic cancer, gastric cancer, esophageal carcinoma, hepatocellular carcinoma, and recurrent malignant glioma, as well as when topotecan was used as consolidation treatment after first-line standard chemotherapy for ovarian cancer.
31In a phase III study, the daily times five IV topotecan was compared with paclitaxel (3-hour infusion of 175 mg/m
2/d every 3 weeks) in ovarian cancer. In this disease, topotecan and paclitaxel were equally effective with regard to response rates, progression-free survival, and overall survival.
32 In an open-label, multicenter study comparing the activity and tolerability of oral versus IV topotecan in patients with relapsed epithelial ovarian cancer after failure of one platinum-based regimen,
7 oral doses of topotecan were administered as 2.3 mg/m
2/d and IV doses as 1.5 mg/m
2/d for five consecutive days every 3 weeks. No difference in response rates between the two treatment arms was reported. Although a small, statistically significant difference in survival favored the IV formulation, in the context of second-line palliative treatment for ovarian cancer, this difference in outcome has only limited clinical significance.
7 For this reason, oral topotecan could be an alternative treatment modality in this setting because of its convenience and good tolerability.
9,
10,
11,
33Another phase III study compared single-agent topotecan with combination chemotherapy consisting of cyclophosphamide, doxorubicin, and vincristine (CAV) in patients with SCLC relapsing after first-line platinum-based chemotherapy.
34 Although the response rate, time to disease progression, and overall survival were similar, the palliation of disease-related symptoms was better with topotecan. In a randomized trial performed by the Eastern Cooperative Oncology Group (ECOG), topotecan was compared with best support of care in patients with extensive SCLC. In this trial, topotecan was administered as consolidation therapy after response induction with cisplatin and etoposide.
35 Although topotecan produced a moderate increase in the time to disease progression, it did not improve survival. Finally, similar to the study of ovarian cancer, oral topotecan was compared to IV administration of topotecan in patients with relapsed and chemosensitive SCLC. The oral formulation was similar in efficacy, resulted in less severe neutropenia, and was more convenient.
8 Although topotecan has shown some activity against hematological malignancies and advanced cervical cancer,
36,
37 its use for these specific indications is not clearly established.