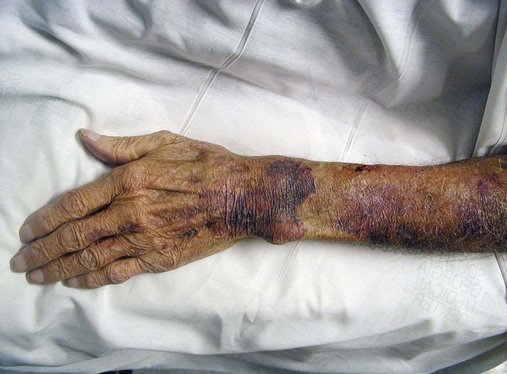
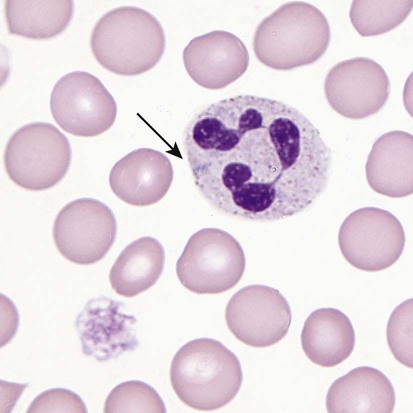
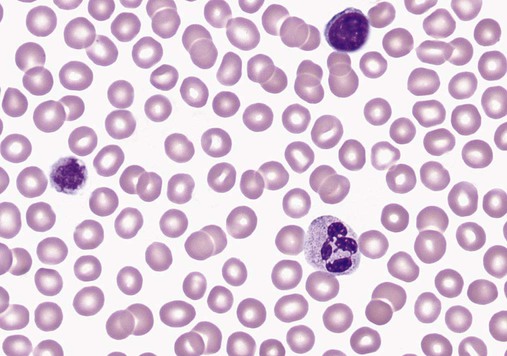
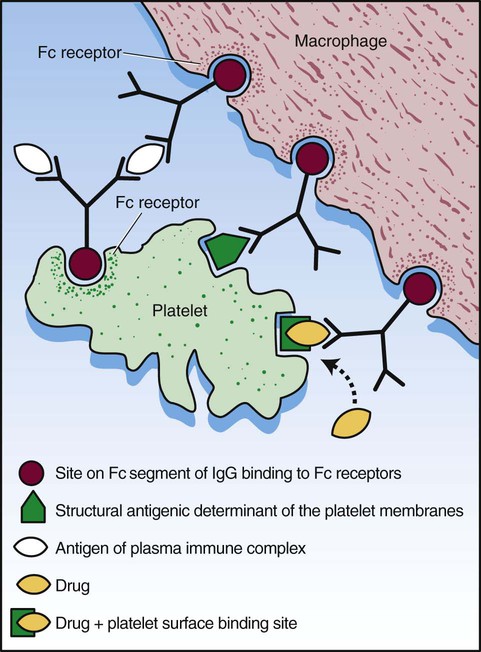
After completion of this chapter, the reader will be able to: 1. Define thrombocytopenia and thrombocytosis, and state their associated platelet counts. 2. Compare and contrast the clinical symptoms of platelet disorders and clotting factor deficiencies. 3. Explain the primary pathophysiologic processes of thrombocytopenia. 4. Name and list the unique diagnostic features of at least four disorders included in congenital hypoplasia of the bone marrow and describe their inheritance patterns. 5. Differentiate between acute and chronic immune thrombocytopenia. 6. Describe the immunologic and nonimmunologic mechanisms by which drugs may induce thrombocytopenia. 7. Differentiate between neonatal isoimmune thrombocytopenia and neonatal autoimmune thrombocytopenia. 8. Explain the laboratory findings and pathophysiology associated with thrombotic thrombocytopenic purpura and hemolytic uremic syndrome. 9. Summarize the pathophysiology of thrombotic complications in heparin-induced thrombocytopenia and describe the sequence of treatment options. 10. Given clinical history and laboratory test results for patients with thrombocytopenia or thrombocytosis, suggest a diagnosis that is consistent with the information provided. Although the reference range for the platelet count varies among laboratories, it is generally considered to be approximately 150,000 to 450,000/mcL (150,000 to 450,000/mm3 or 150 to 450 × 109/L). Thrombocytopenia (platelet count of fewer than 100,000/mcL) is the most common cause of clinically important bleeding. The primary pathophysiologic processes that result in thrombocytopenia are decreased platelet production, accelerated platelet destruction, and abnormal platelet distribution (sequestration) (Box 43-1). Small-vessel bleeding in the skin attributed to thrombocytopenia is manifested by hemorrhages of different sizes (Figure 43-1). Petechiae are small pinpoint hemorrhages about 3 mm in diameter, purpura are about 1 cm in diameter and generally round, and ecchymoses are 3 cm or larger and usually irregular in shape. Ecchymosis corresponds with the lay term bruise. Clinical bleeding varies and often is not closely correlated with the platelet count. It is unusual for clinical bleeding to occur when the platelet count is greater than 50,000/mcL, but the risk of clinical bleeding increases progressively as the platelet count decreases from 50,000/mcL. Patients with platelet counts of 20,000/mcL or sometimes lower may have little or no bleeding symptoms. In general, patients with platelet counts of fewer than 10,000/mcL are considered to be at high risk for a serious hemorrhagic episode. May-Hegglin anomaly is a rare autosomal dominant disorder; the exact frequency is unknown. Large platelets (20 µm in diameter) are present on the peripheral blood film, and Döhle bodies are present in neutrophils (Figure 43-2) and occasionally in monocytes. Other than the increase in size, platelet morphology is normal. Thrombocytopenia is present in about one third to one half of affected patients. Platelet function in response to platelet-activating agents is usually normal. In some patients, megakaryocytes are increased in number and have abnormal ultrastructure. Mutations in the MYH9 gene that encodes for nonmuscle myosin heavy chain (a cytoskeletal protein in platelets) have been reported.1 This mutation may be responsible for the abnormal size of platelets in this disorder. Most patients are asymptomatic unless severe thrombocytopenia is present, but bleeding times may be prolonged in some patients in the absence of bleeding complications. Three other disorders involving mutations of the MYH9 gene have been reported: Sebastian syndrome, Fechtner syndrome, and Epstein syndrome.2 Sebastian syndrome is inherited as an autosomal dominant disorder characterized by large platelets, thrombocytopenia, and granulocytic inclusions. Similar abnormalities are observed in Fechtner syndrome and are accompanied by deafness, cataracts, and nephritis. In Epstein syndrome, large platelets are associated with deafness, ocular problems, and glomerular nephritis.3 TAR syndrome is a rare autosomal recessive disorder characterized by severe neonatal thrombocytopenia and congenital absence or extreme hypoplasia of the radial bones of the forearms with absent, short, or malformed ulnae and other orthopedic abnormalities. The defects are presumed to occur as a result of a specific type of fetal injury at about 8 weeks of gestation. TAR syndrome is one of the inherited impaired deoxyribonucleic acid (DNA) repair disorders. Because exposure to radiation causes cellular DNA damage, it is also considered to be a radiation sensitivity syndrome.4 In addition, patients tend to have cardiac lesions and a high incidence of transient leukemoid reactions with elevated white blood cell (WBC) counts (sometimes with counts above 100,000/mcL) in 90% of patients.5 Platelet counts are usually 10,000 to 30,000/mcL in infancy. Interestingly, platelet counts usually increase over time, with normal levels often achieved within 1 year of birth. Signaling through the thrombopoietin (TPO) receptor appears to be abnormal, although the deficit has not been clearly identified. Fanconi anemia is also associated with thrombocytopenia, although other abnormalities are extensive, including bony abnormalities, abnormalities of visceral organs, and pancytopenia. Chapter 21 contains a more detailed description. Congenital amegakaryocytic thrombocytopenia is an autosomal recessive disorder reflecting bone marrow failure.6 Affected infants usually have platelet counts of fewer than 20,000/mcL at birth, have petechiae and evidence of bleeding, and frequently have physical anomalies. About half of the infants develop aplastic anemia in the first year of life, and there are reports of myelodysplasia and leukemia later in childhood. Allogeneic stem cell transplantation is considered curative for infants with clinically severe disease or aplasia.7 This disorder is caused by mutations in the c-mlp gene resulting in complete loss of thrombopoietin receptor function. This loss of function results in reduced megakaryocyte progenitors and high thrombopoietin levels.8 Autosomal dominant and X-linked thrombocytopenia are well documented. In autosomal dominant thrombocytopenia, bleeding is usually mild, platelet function is normal, and the megakaryocytes seem to be normal in number and morphology.9,10 X-linked thrombocytopenia can result from mutations in WASP (Wiskott-Aldrich syndrome protein) or mutations in the GATA1 gene.11–13 X-linked thrombocytopenias range from mild thrombocytopenia and small platelets to macrothrombocytopenia with severe bleeding. Causes of neonatal megakaryocytic hypoplasia include infection with Toxoplasma, rubella, cytomegalovirus (CMV), and herpes (TORCH), human immunodeficiency virus (HIV) infection, and in utero exposure to certain drugs, particularly chlorothiazide diuretics and the oral hypoglycemic tolbutamide and other agents. TORCH infections cause thrombocytopenia with characteristically small platelets. CMV is the most common infectious agent causing congenital thrombocytopenia, with an overall incidence of 0.5% to 1% of all births,14 but only 10% to 15% of infected infants have symptomatic disease,15 which suggests that the incidence of significant neonatal thrombocytopenia caused by CMV is about 1 in 1000 infants. Although the mechanism of thrombocytopenia is not well understood, reports suggest that CMV inhibits megakaryocytes and their precursors, which results in impaired platelet production.16 About 1 in 1000 to 1 in 3000 infants are affected by congenital toxoplasmosis. About 40% of such infants develop thrombocytopenia.17 Congenital rubella is now rare in countries with organized immunization programs18,19; persistent thrombocytopenia is a prominent feature in infants with congenital rubella syndrome. Thrombocytopenia also is a feature of maternal transmission of HIV to the neonate and is a sign of intermediate to severe disease.20 Maternal ingestion of chlorothiazide diuretics or tolbutamide can have a direct cytotoxic effect on the fetal marrow megakaryocytes. Thrombocytopenia may be severe, with platelet counts of 70,000/mcL and sometimes lower. Bone marrow examination reveals a marked decrease or absence of megakaryocytes. The thrombocytopenia develops gradually and is slow to regress when the drug is stopped. Recovery usually occurs within a few weeks after birth.10,21,22 A wide array of chemotherapeutic agents used for the treatment of hematologic and nonhematologic malignancies suppress bone marrow megakaryocyte production and the production of other hematopoietic cells. Examples include the commonly used agents methotrexate, busulfan, cytosine arabinoside, cyclophosphamide, and cisplatin. The resulting thrombocytopenia may lead to hemorrhage, and the platelet count should be monitored closely. Drug-induced thrombocytopenia is often the dose-limiting factor for many chemotherapeutic agents. Recombinant interleukin-11 has been approved for treatment of chemotherapy-induced thrombocytopenia, and thrombopoietin may prove to be useful for this purpose.23–25 Zidovudine (used for the treatment of HIV infection) is also known to cause myelotoxicity and severe thrombocytopenia.26 Several drugs specifically affect megakaryocytopoiesis without significantly affecting other marrow elements. Anagrelide is one such agent, although its mechanism of action is unknown. This characteristic has made anagrelide useful for treating the thrombocytosis of patients with essential thrombocythemia and other myeloproliferative disorders.27 Ingestion of ethanol for long periods (months to years) may result in persistent severe thrombocytopenia. Although the mechanism is unknown, studies indicate that alcohol can inhibit megakaryocytopoiesis in some individuals. Mild thrombocytopenia is a common finding in alcoholic patients, but other causes unrelated to ethanol use, such as portal hypertension, splenomegaly, and folic acid deficiency, should be excluded. The platelet count usually returns to normal within a few weeks of alcohol withdrawal, but thrombocytopenia may persist for longer periods. A transient rebound thrombocytosis may develop when alcohol ingestion is stopped.10 Interferon therapy commonly causes mild to moderate thrombocytopenia, although under certain circumstances, the thrombocytopenia can be severe and life-threatening. Interferon-α and interferon-γ inhibit stem cell differentiation and proliferation in the bone marrow, but the mechanism of action is unclear.28 Thrombocytopenia presumably caused by megakaryocyte suppression also has been reported to follow the administration of large doses of estrogen or estrogenic drugs such as diethylstilbestrol. Other drugs, such as certain antibacterial agents (e.g., chloramphenicol), tranquilizers, and anticonvulsants, also have been associated with thrombocytopenia caused by bone marrow suppression.29–31 Thrombocytopenia is a usual feature of the megaloblastic anemias (pernicious anemia, folic acid deficiency, and vitamin B12 deficiency). Quantitative studies indicate that, as with erythrocyte production in these disorders, platelet production is ineffective. Although the bone marrow generally contains an increase in the number of megakaryocytes, the total number of platelets released into the circulation is decreased. Thrombocytopenia is caused by impaired DNA synthesis, and the bone marrow may contain grossly abnormal megakaryocytes with deformed, dumbbell-shaped nuclei, sometimes in large numbers. Stained peripheral blood films reveal large platelets that may have a decreased survival time and may have abnormal function. Thrombocytopenia is usually mild, and there is evidence of increased platelet destruction. Patients typically respond within 1 to 2 weeks to vitamin replacement.22,32–34 Viruses are known to cause thrombocytopenia by acting on megakaryocytes or circulating platelets, either directly or in the form of viral antigen-antibody complexes. Live measles vaccine can cause degenerative vacuolization of megakaryocytes 6 to 8 days after vaccination. Some viruses interact readily with platelets by means of specific platelet receptors. Other viruses associated with thrombocytopenia include CMV, varicella-zoster virus, rubella virus, Epstein-Barr virus (which causes infectious mononucleosis), and the virus that causes Thai hemorrhagic fever.10 Certain bacterial infections commonly are associated with the development of thrombocytopenia. This may be the result of toxins of bacterial origin, direct interactions between bacteria and platelets in the circulation, or extensive damage to the endothelium, as in meningococcemia. Many cases of thrombocytopenia in childhood result from infection. Purpura may occur in many infectious diseases in the absence of thrombocytopenia, presumably because of vascular damage (see Chapter 44).10,35 A common cause of unexplained thrombocytopenia is infiltration of the bone marrow by malignant cells with a progressive decrease in marrow megakaryocytes as the abnormal cells replace normal marrow elements. Inhibitors of thrombopoiesis may be produced by these abnormal cells and may help to account for the thrombocytopenia associated with conditions such as myeloma, lymphoma, metastatic cancer, and myelofibrosis.22,32,36 The infection is most often a nonspecific upper respiratory tract or gastrointestinal tract viral infection, but acute ITP also may occur after rubella, rubeola, chickenpox, or other viral illnesses and may follow live virus vaccination.37 The incidence of acute ITP is estimated to be 4 in 100,000 children, with a peak frequency in children between 2 and 5 years of age. There is no sex predilection. In about 10% to 15% of the children initially thought to have acute ITP, the thrombocytopenia persists for 6 months or longer, and these children are reclassified as having chronic ITP.38 The observation that acute ITP often follows a viral illness suggests that some children produce antibodies and immune complexes against viral antigens and that platelet destruction may result from the binding of these antibodies or immune complexes to the platelet surface. In mild cases of acute ITP, patients may have only scattered petechiae. In most cases of acute ITP, however, patients develop fairly extensive petechiae and some ecchymoses and may have hematuria or epistaxis or both. About 3% to 4% of acute ITP cases are considered severe, and typically generalized purpura is present, often accompanied by gastrointestinal bleeding, hematuria, mucous membrane bleeding, and retinal hemorrhage. Of patients with severe disease, 25% to 50% are considered to be at risk for intracranial hemorrhage, which is the primary complication that contributes to the overall 1% to 2% mortality rate for patients with acute ITP.38 Most patients with life-threatening hemorrhage have a platelet count of fewer than 4000/mcL.39 Hemorrhage is rarely experienced by patients whose platelet count exceeds 10,000/mcL. Most patients with acute ITP recover with or without treatment in about 3 weeks, although for some, recovery may take 6 months. In a few children, recurrent episodes of acute ITP are occasionally seen after complete recovery from the first episode.40 Most patients with acute ITP have relatively mild symptoms, and no treatment is needed. The most severe cases may need to be treated, however, and intravenous immunoglobulin (IVIG), platelet transfusions, and splenectomy (or some combination of these) seem to offer the most immediate benefit.37,38 Chronic ITP can be found in patients of any age, although most cases occur in patients between the ages of 20 and 50 years. Females with this disorder outnumber males 2 : 1 to 3 : 1, with the highest incidence in women between 20 and 40 years of age. The incidence of chronic ITP ranges from 3.2 to 6.6 cases per 100,000 per year.41 Chronic ITP usually begins insidiously, with platelet counts that are variably decreased and sometimes normal for periods of time. Presenting symptoms are those of mucocutaneous bleeding, with menorrhagia, recurrent epistaxis, and easy bruising (ecchymoses) being most common. Platelet destruction in chronic ITP is the result of an immunologic process. The offending antibodies attach to platelets, and as a result, the antibody-labeled platelets are removed from the circulation by reticuloendothelial cells, primarily in the spleen. Autoantibodies that recognize platelet surface glycoproteins such as glycoprotein IIb (GP IIb) and GP IIIa (αIIb/β3), GP Ia/IIa, and others can be demonstrated in 50% to 60% of ITP patients.42,43 Because megakaryocytes also express GP IIb/IIIa and GP Ib/IX on their membranes, these cells are obvious targets of the antibodies. Platelet turnover studies have shown impaired platelet production in ITP. Overall, the life span of the platelet is shortened from the normal 7 to 10 days to a few hours, and the rapidity with which platelets are removed from the circulation correlates with the degree of thrombocytopenia. If plasma from a patient with ITP is infused into the circulation of a normal recipient, the recipient develops thrombocytopenia. The thrombocytopenia-producing factor in the plasma of the ITP patient is an immunoglobulin G (IgG) antibody that can be removed from serum by adsorption with normal human platelets. In addition, cytotoxic T cell–mediated lysis of platelets has been shown in vitro using CD3+CD8+ lymphocytes from patients with active chronic ITP, although the in vivo significance of this mechanism is not known.44 The only abnormalities in the peripheral blood of patients with ITP are related to platelets. In most cases, platelets number between 30,000/mcL and 80,000/mcL. Patients with ITP undergo periods of remission and exacerbation, however, and their platelet counts may range from near-normal to fewer than 20,000/mcL during these periods (Figure 43-3). Morphologically, platelets appear normal, although larger in diameter than usual. This is reflected in an increased mean platelet volume as measured by electronic cell counters. The marrow typically is characterized by megakaryocytic hyperplasia. Megakaryocytes are increased in size, and young forms with a single nucleus, smooth contour, and diminished cytoplasm are commonly seen. In the absence of bleeding, infection, or other underlying disorder, erythrocyte and leukocyte precursors are normal in number and morphology. Coagulation tests showing abnormal results include tests dependent on normal platelet function, such as bleeding time and clot retraction time. Although platelet-associated IgG levels are increased in most patients,10,21,45 it has not been shown conclusively that any method of testing for platelet antibodies is sensitive or specific for ITP. The initial treatment of chronic ITP depends on the urgency for increasing the platelet count. In ITP patients with platelet counts greater than 30,000/mcL who receive no treatment, the expected mortality rate is equal to that of the general population. Unless there are additional risk factors, ITP patients with platelet counts greater than 30,000/mcL should not be treated. If additional risk factors are present, such as old age, coagulation defects, recent surgery, trauma, or uncontrolled hypertension, the platelet count should be kept at 50,000/mcL or higher depending on the clinical situation. In patients in whom the need is considered urgent, IVIG remains the treatment of choice. For most patients, however, the initial treatment of chronic ITP consists principally of prednisone. About 70% to 90% of patients respond to this therapy with an increase in platelet count and a decrease in hemorrhagic episodes. Although reported response rates vary widely, about 50% of patients have a long-term beneficial effect from corticosteroid treatment.46 If the response to corticosteroids is inadequate or no response is seen, steroid therapy can be supplemented with IVIG or, in some cases, anti-D immunoglobulin.47,48 For patients in whom prednisone is ineffective, intravenous rituximab should be tried. Responses to rituximab are usually seen within 3 to 4 weeks. In some patients splenectomy may become necessary. Splenectomy eliminates the primary site of platelet removal and destruction, but it also removes an organ containing autoantibody-producing lymphocytes. Splenectomy is an effective treatment for adult chronic ITP, with 88% of patients showing improvement and 66% having a complete and lasting response.49 Vaccination with pneumococcal, meningococcal, and H influenza vaccines should be performed 2 weeks prior to surgery. In the most severe refractory cases, immunosuppressive (chemotherapeutic) agents such as azathioprine given alone or with steroids may be necessary. In such patients, platelet transfusions may be of transient benefit in treating severe hemorrhagic episodes but should not be given routinely.45 IVIG given alone or just before platelet transfusion also may be beneficial.37,45 Chronic ITP occurring in association with HIV infection, with hemophilia, or with pregnancy presents special problems in diagnosis and therapy. Unexplained thrombocytopenia in otherwise healthy members of high-risk populations may be an early manifestation of acquired immune deficiency syndrome (AIDS).36,45 The differences between acute and chronic ITP are summarized in Table 43-1. Acute ITP occurs most frequently in children 2 to 9 years of age and in young adults, whereas chronic ITP occurs in patients of all ages, although most frequently in adults aged 20 to 50 years and more commonly in women. Of patients with acute ITP, 60% to 80% have a history of infection, usually viral (rubella, rubeola, chickenpox, and nonspecific respiratory tract infection), occurring 2 to 21 days before ITP onset. Acute ITP also may occur after immunization with live vaccine for measles, chickenpox, mumps, and smallpox. TABLE 43-1 Clinical Picture of Acute and Chronic Immune Thrombocytopenic Purpura Data from Triplett DA, editor: Platelet function: laboratory evaluation and clinical application, Chicago, 1978, American Society of Clinical Pathologists; Quick AJ: Hemorrhagic diseases and thrombosis, ed 2, Philadelphia, 1966, Lea & Febiger; and Bussel J, Cines D: Immune thrombocytopenia, neonatal alloimmune thrombocytopenia, and post-transfusion purpura. In Hoffman R, Benz EJ Jr, Shattil SJ, et al, editors: Hematology: basic principles and practice, ed 3, New York, 2000, Churchill Livingstone, pp 2096-2114. Acute ITP usually is self-limited, and spontaneous remissions occur in 80% to 90% of patients, although the duration of the illness may range from days to months. In chronic ITP, there is typically a fluctuating clinical course, with episodes of bleeding that last a few days or weeks, but spontaneous remissions are uncommon and usually incomplete.45 Symptoms of acute ITP vary, but petechial hemorrhages, purpura, and often bleeding from the gums and gastrointestinal or urinary tract typically begin suddenly, sometimes over a few hours. Hemorrhagic bullae in the oral mucosa are often prominent in patients with severe thrombocytopenia of acute onset. Usually the severity of bleeding is correlated with the degree of thrombocytopenia.45 In contrast, presenting symptoms of chronic ITP begin with a few scattered petechiae or other minor bleeding manifestations. Occasionally, a bruising tendency, menorrhagia, or recurrent epistaxis is present for months or years before diagnosis. Platelet counts range from 5000/mcL to 75,000/mcL and are generally higher than those in acute ITP. Giant platelets are commonly seen. Platelet-associated immunoglobulin levels are elevated in most patients, but the test lacks sensitivity and specificity.45 Treatment also varies for acute and chronic ITP. In chronic ITP, initial therapy often consists of glucocorticoids (corticosteroids), which interfere with splenic and hepatic macrophages to increase platelet survival time. If the ITP does not respond to corticosteroids or the patient cannot tolerate them because of the resultant immunosuppression and toxicity, splenectomy may be necessary. In acute ITP, treatment for all but the most severely thrombocytopenic and hemorrhagic patients is contraindicated. When treatment is necessary, a good response to IVIG or corticosteroids or both usually can be obtained, and splenectomy is rarely required.37,38 As can be seen from Box 43-2, many drugs can induce acute thrombocytopenia. Drug-induced immune-mediated thrombocytopenia can be divided into several types based on the interaction of the antibody with the drug and platelets. Mechanisms of drug-antibody binding are shown in Figure 43-4. The initial studies of quinidine-induced thrombocytopenia suggested that the drug first combines with the antibody and that the antigen-antibody (immune) complex then attaches to the platelet in an essentially nonspecific manner (the “innocent bystander” hypothesis). It now seems clear, however, that the antibodies responsible for drug-induced thrombocytopenia bind to the platelets by their Fab regions, rather than by attaching nonspecifically as immune complexes. The innocent bystander/immune complex explanation for this type of drug-induced thrombocytopenia should be abandoned. The Fab portion of the antibody binds to a platelet membrane constituent, usually the GP Ib/IX complex or the GP IIb/IIIa complex, only in the presence of drug.50,51 The mechanism by which the drug promotes binding of a drug-dependent antibody to a specific target on the platelet membrane without covalently linking to the target or the antibody remains to be determined, however. Because the Fc portion of the immunoglobulin is not involved in binding to platelets, it is still available to the Fc receptors on phagocytic cells. This situation may contribute to the rapid onset and relatively severe nature of the thrombocytopenia. Most drug-induced platelet antibodies are of the IgG class, but in rare instances, IgM antibodies are involved.45 A second mechanism of drug-induced thrombocytopenia is induction of hapten-dependent antibodies. Some drug molecules are too small by themselves to trigger an immune response, but they may act as a hapten and combine with a larger carrier molecule (usually a plasma protein or protein constituent of the platelet membrane) to form a complex that can act as a complete antigen.45 Penicillin and penicillin derivatives are the primary offending agents causing drug-induced thrombocytopenia by this mechanism. Drug-induced thrombocytopenia of this type is often severe. The initial platelet count may be fewer than 10,000/mcL and sometimes fewer than 1000/mcL. The number of bone marrow megakaryocytes is usually normal to elevated.45 Bleeding is often severe and rapid in onset, and hemorrhagic bullae in the mouth may be prominent. Heparin-induced thrombocytopenia (HIT) is a good example of another type of drug-induced thrombocytopenia. Heparin binds to platelet factor 4 (PF4), a heparin-neutralizing protein made and released by platelets (Figure 43-5). Binding of heparin by plasma PF4 or platelet membrane–expressed PF4 causes a conformational change in PF4, resulting in the exposure of neoepitopes. Exposure of these neoepitopes (“new antigens”) stimulates the immune system of some individuals, which leads to the production of an antibody to one of the neoepitopes. In HIT, heparin and PF4 form a complex on the platelet surface or circulating free complexes to which the antibody binds. The Fab portion of the immunoglobulin molecule binds to an exposed neoepitope in the PF4 molecule; this leaves the Fc portion of the IgG free to bind with the platelet FcγR receptor, which causes platelet activation.52,53 Because the Fc portions of the IgG molecules bind to platelet FcγR receptors, they are not available to the Fc receptors of the cells of the reticuloendothelial system. This may explain the less severe decline in platelet count in this thrombocytopenia. That does not mean, however, that the consequences are less serious. The opposite may be true. Because platelets are activated by occupancy of their FcγR receptors, in vivo platelet aggregation with thrombosis is possible. HIT sometimes is referred to as heparin-induced thrombocytopenia and thrombosis.
Thrombocytopenia and Thrombocytosis
Thrombocytopenia: Decrease in Circulating Platelets
Impaired or Decreased Platelet Production
Congenital Hypoplasia
Congenital Amegakaryocytic Thrombocytopenia
Autosomal Dominant and X-Linked Thrombocytopenia
Neonatal Hypoplasia
Acquired (Drug-Induced) Hypoplasia
Drug-Induced Hypoplasia
Ineffective Thrombopoiesis
Miscellaneous Conditions
Increased Platelet Destruction
Immune Mechanisms of Platelet Destruction
Immune (Idiopathic) Thrombocytopenic Purpura: Acute and Chronic
Differentiation of acute versus chronic idiopathic thrombocytopenic purpura
Characteristic
Acute
Chronic
Age at onset
2-6 yr
20-50 yr
Sex predilection
None
Female over male, 3 : 1
Prior infection
Common
Unusual
Onset of bleeding
Sudden
Gradual
Platelet count
<20,000/mcL
30,000-80,000/mcL
Duration
2-6 wk
Months to years
Spontaneous remission
90% of patients
Uncommon
Seasonal pattern
Higher incidence in winter and spring
None
Therapy
Steroids
70% response rate
30% response rate
Splenectomy
Rarely required
<45 yr, 90% response rate; >45 yr, 40% response rate
Immunologic Drug-Induced Thrombocytopenia
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree