INTRODUCTION
SUMMARY
Thrombocytopenia is one of the most frequent causes for hematologic consultation in the practice of medicine, and may be life threatening. Although the normal platelet count in humans (150 to 400 × 109/L) far exceeds the minimal level required to avoid pathologic hemorrhage (<50 × 109/L), a number of medical conditions either increasing the destruction of platelets or reducing their production, enhance the risk of bleeding. This chapter discusses an approach to the diagnosis of thrombocytopenia, grouping various causes by mechanism of action, and describing our current understanding of the pathogenesis, treatment and prognosis. In the vast majority of patients, a cause for thrombocytopenia can be identified, and effective therapy instituted.
Acronyms and Abbreviations
ACOG, American College of Obstetricians and Gynecologists; ADP, adenosine diphosphate; AFLP, acute fatty liver of pregnancy; AML, acute myelogenous leukemia; APLA, antiphospholipid antibody; APS, antiphospholipid syndrome; ARC, arthrogryposis–renal dysfunction–cholestasis; ASH, American Society of Hematology; ATG, antithymocyte globulin; ATRUS, amegakaryocytic thrombocytopenia with radioulnar synostosis; CAMT, congenital amegakaryocytic thrombocytopenia; CAPTURE, c7E3 Fab Antiplatelet Therapy in Unstable Refractory Angina; CTP, cyclic thrombocytopenia; CVID, common variable immunodeficiency; DIC, disseminated intravascular coagulation; EDTA, ethylenediaminetetraacetic acid; EPIC, Evaluation of 7E3 for the Prevention of Ischemic Complications; EPILOG, Evaluation of Percutaneous Transluminal Coronary Angioplasty to Improve Long-term Outcome of c7E3 GPIIb-IIIa Receptor Blockade; EPISTENT, Evaluation of Platelet IIb/IIIa Inhibitor for Stenting; Flt1, fms-like tyrosine kinase-1; FPD/AML, familial platelet disorder with propensity to acute myeloid malignancy; GP, glycoprotein; HCV, hepatitis C virus; HELLP, hemolysis, elevated liver enzymes, low platelets; HIT, heparin-induced thrombocytopenia; HUS, hemolytic uremic syndrome; HPA, human platelet alloantigen; ICSH, International Council for Standardization in Hematology; IDA, iron-deficiency anemia; IPD, inherited platelet disorder; ITP, immune thrombocytopenia; IVIG, intravenous immunoglobulin; IWG, International Working Group; LTA, light transmission aggregometry; MACE, modified antigen capture enzyme-linked ımmunosorbent assay; MAIPA, monoclonal antibody-specific immobilization of platelet antigens; MDS, myelodysplastic syndrome; MHC, major histocompatibility complex; NAIT, neonatal alloimmune thrombocytopenia; PAIgG, platelet-associated immunoglobulin G; sFlt1, soluble Flt1; SLE, systemic lupus erythematosus; TAR, thrombocytopenia with absent radii; TPO, thrombopoietin; Treg, T-regulatory; TTP, thrombotic thrombocytopenic purpura; VEGF, vascular endothelial growth factor; VWD, von Willebrand disease; VWF, von Willebrand factor.
DEFINITION AND HISTORY
Platelets are anucleate blood cells produced in the marrow by polyploid cells termed megakaryocytes and were described in the 19th century after the application of the improved compound microscope allowed these very small cellules, approximately 2 μM in diameter, to be identified. Many early investigators are associated with the discovery of blood platelets, including Donné, Hayem, Bizzozero, and Osler, but it was James Homer Wright who, in 1906, using his special stain (later called Wright stain), described the morphology of platelets with their central granular area and marginal hyaline zone and established that they were the product of the fragmentation of marrow megakaryocytes. Clot retraction was discovered long before platelets, but Hayem, through a series of studies, showed retraction to be dependent on platelets. During the mid-20th century the aggregation of platelets, their adherence to collagen of damaged tissues, their acceleration of blood coagulation, and their relationship to the bleeding time and the biochemistry underlying several of these processes were described by scientists, among whom were Paul Owren, Kenneth Brinkhaus, Edwin Chargaff, Ernst Lüsher, Marjorie Zucker, and William Duke.
Platelets circulate in close contact with the endothelium, continually monitoring its integrity. When the vessel wall is damaged, platelets bind to subendothelial proteins, initiating the process of primary hemostasis. At sites of blood loss, the platelets aggregate to form a vessel sealing plug to halt bleeding. Activated platelets at sites of injury also provide a surface for assembly of coagulation reactions, resulting in the production of fibrin and consolidation of the thrombus. Both qualitative and quantitative deficiencies of the platelets cause bleeding. Platelets also have important functions in inflammation, tissue remodeling and wound healing.1
Approximately 1 × 1011 platelets are produced per day by an adult human, a number that can be increased 20-fold or more, if necessary.2 One-third of the platelets are stored in the spleen, the remaining two-thirds circulate in blood vessels.3 Disorders that increase splenic volume cause more platelets to be trapped in the spleen, lowering the concentration of circulating platelets, although alone, this redistribution rarely causes a significant bleeding diathesis.
Under normal conditions, human platelets have a mean life span in the circulation of between 7 and 10 days.4,5 Patients with thrombocytopenia secondary to platelet destruction have a markedly decreased platelet survival.6,7 Patients with thrombocytopenia from marrow failure have mildly decreased platelet survival, mostly because the body’s fixed daily consumption of platelets accounts for a progressively larger fraction of the reduced total daily production as the platelet count drops.8 Platelet turnover is a measure of the net effect of platelet production and platelet destruction under steady-state conditions.7 Several studies using 111In oxine–labeled platelets have established that, under normal conditions, platelet turnover in humans ranges from 40 to 50 × 109/L per day.7 Although a high platelet turnover is expected in patients with immune thrombocytopenia (ITP), platelet production is not always increased in this disorder.7 Low platelet production may result from binding of the antiplatelet antibodies to megakaryocytes, inhibiting their maturation or leading to their destruction, causing an inappropriately muted marrow response to the degree of thrombocytopenia.9
Every day approximately 10 to 12 percent of circulating platelets are removed by the mononuclear phagocyte system, primarily by macrophages in the spleen and liver. Although the precise mechanisms of platelet clearance are not completely understood, changes that occur as the platelets circulate are thought to lead them to be recognized by macrophages. One of these changes is the progressive loss of sialic acid from platelet surface proteins. Studies in animals and humans with anticancer drugs that inhibit apoptotic pathways have also identified a role for apoptotic proteins in platelet survival and clearance. According to these studies, a classical intrinsic apoptosis pathway regulates the life span of circulating platelets, and antiapoptotic proteins, especially Bcl-XL, maintain platelet viability by restraining apoptosis.10
The normal platelet count (defined as the values between percentiles 2.5 to 97.5 in normal individuals) is given as 150 to 400 × 109/L; classically, thrombocytopenia is defined as a platelet count of less than 150 × 109/L. However, a sustained lower platelet count (100 to 150 × 109/L) can be seen in otherwise healthy individuals.11,12 Long-term observation of individuals with platelet counts between 100 and 150 × 109/L showed that 88 percent of these individuals had subsequently reached normal platelet counts or remained stable. In those individuals, the probability of developing ITP was 6.9 percent, an autoimmune disease other than ITP 12 percent, and myelodysplastic syndrome (MDS) 2 percent, after 64 months of followup. All patients with MDS in this cohort were found to be older than age 65 years.13
Thrombocytopenia can be classified as severe (platelet count less than 20 × 109/L), moderate (platelet count 20 to 70 × 109/L), or mild (above 70 × 109/L).14 Although easy bruising occurs in patients with platelet counts less than 50 × 109/L and spontaneous life-threatening bleeding can be expected in patients with platelet counts less than 15 × 109/L, bleeding symptomatology is largely determined by comorbid conditions affecting platelets or the coagulation system, including liver cirrhosis, uremia, disseminated intravascular coagulation (DIC), or antiplatelet drug usage.
In clinical practice, platelet counting is automated, and includes several different technologies: impedance, optical, two-dimensional laser, and optical-fluorescence methods. Although automated cell counter technology has progressed considerably during recent decades, the analytic performances of these machines for platelet counts and platelet indices is still not perfect, especially in patients with severe thrombocytopenia and macrothrombocytopenia.15,16,17 Each step between the sampling of blood and its analysis is important: the blood sample should be obtained by a clean venipuncture without dilution with other IV solutions or drugs. Blood/anticoagulant ratio should be as recommended. The International Council for Standardization in Hematology (ICSH) recommends use of ethylenediaminetetraacetic acid (EDTA) as the anticoagulant. Adequate mixing of the blood sample with EDTA (the final EDTA concentration should be 1.5 to 2.2 mg/mL) is crucial to prevent clumping of the platelets. Blood samples should be kept at room temperature and analyzed within 6 hours of phlebotomy. If a sample is to be analyzed more than 6 hours after it is drawn, it can be kept at 4°C for 24 hours. The blood count analyzer should be cleaned according to laboratory standards.17
Although thrombocytopenia is variably attributed to single factors such as decreased platelet production, increased platelet destruction, or abnormal splenic pooling, combinations of factors are often involved in clinical settings. For instance, the thrombocytopenia seen in patients with viral infection can result from many factors, including platelet destruction (e.g., through an autoimmune mechanism or drug toxicity) or decreased platelet production because of direct megakaryocyte infection by the virus. Table 117–1 lists the multiple causes of thrombocytopenia and classifies them by pathogenesis.
|
PSEUDO (SPURIOUS) THROMBOCYTOPENIA
Pseudothrombocytopenia (or spurious thrombocytopenia) is a relatively uncommon phenomenon with multiple causes, including ex vivo agglutination of platelets, the presence of abnormally large platelets (improper counting), or improper preparation of blood samples.
The incidence of pseudothrombocytopenia reported in different studies ranges from 0.09 to 0.21 percent, which accounts for 15 to 30 percent of all cases of isolated thrombocytopenia.18,19,20,21,22,23,24,25 Pseudothrombocytopenia has been reported in association with the use of EDTA as an anticoagulant, with platelet cold agglutinins,26 and with myeloma.27 A very interesting report demonstrates pseudothrombocytopenia caused by platelet phagocytosis ex vivo in the presence of EDTA anticoagulant.28 An example of exvivo platelet clumping is shown in Chap. 1, Fig. 1–6H, accompanied by platelet–neutrophil satellitism (see “Platelet Satellitism” below).
Platelet agglutination ex vivo can be induced by antiplatelet antibodies or by activation of the platelets during collection. The responsible antibodies do not appear to be associated with a pathologic process, as they are found in normal individuals. One hypothesis put forth to explain their presence is that the antibodies are responsible for clearing aged and damaged platelets. Most antibodies implicated in pseudothrombocytopenia recognize platelet membrane glycoproteins that are modified to expose new epitopes when calcium is chelated. Typically, the artifact is most prominent in the presence of EDTA, but other anticoagulants can also cause platelet clumping, including sodium citrate, sodium oxalate, acid citrate dextrose, and heparin. The antibodies usually are of the immunoglobulin (Ig) G type; IgM and IgA antibodies also have been described.29,30,31 Most antibodies react at room temperature; thus, the reaction can be prevented by keeping the blood sample at 37°C. In 20 percent of cases, however, the antibodies, usually of the IgM type, are reactive at both 22°C and 37°C.30 Clumping usually is evident within 60 minutes after the blood is drawn, but may require incubations of 2 to 3 hours. Agglutination can be reproduced by incubating plasma from patients with pseudothrombocytopenia with blood from normal individuals in the presence of EDTA.
In most cases, the antibodies are directed against the integrin αIIbβ3 (also termed glycoprotein [GP] IIbIIIa), a conclusion supported by the observation that platelets from patients with Glanzmann thrombasthenia, who lack the integrin αIIbβ3 complex, fail to agglutinate in the presence of patient sera.32,33,34,35 Moreover, pretreatment of fresh blood with anti–integrin αIIbβ3 dramatically reduces EDTA-induced platelet agglutination.36 The responsible epitope normally is cryptic and located in the integrin αIIb subunit. Low temperature and calcium chelation combine to change the conformation of integrin αIIbβ3 and expose the epitope.33
Antibodies directed against integrin αIIbβ3 may react simultaneously with the leukocyte Fcγ receptor III (FcγRIII) and attach the platelets to neutrophils and monocytes, inducing a phenomenon known as platelet-leukocyte satellitism,32 another form of pseudothrombocytopenia (Fig. 117–1). These antibodies fail to produce satellitism in the presence of platelets from patients with type I Glanzmann thrombasthenia or in the presence of neutrophils from patients with congenital absence of FcγRIII.32 Typically, the platelets form a rosette around the periphery of leukocytes. Neutrophils are most frequently involved, but the phenomenon also is occasionally observed with monocytes.37,38 These antibodies also are naturally occurring, and their presence does not clearly correlate with any specific clinical situation, disease, or drug. As with the antibodies that induce only platelet clumping, exposure of a cryptic antigen on EDTA-treated platelets and leukocytes may trigger this phenomenon.
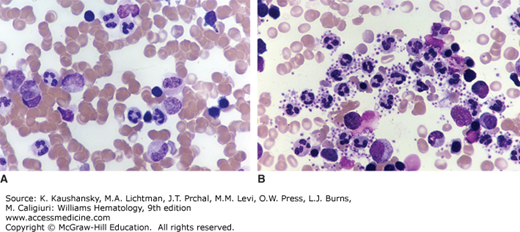
Figure 117–1.
Platelet satellitism. A. Direct (non–anticoagulated) marrow film. No platelet satellitism. B. A concentrated marrow film anticoagulated with disodium ethylenediaminetetraacetic acid (Na2EDTA) from same specimen as in (A). Note platelets are adherent to the mature neutrophil surface (satellitism) in the presence of Na2EDTA. The neutrophil precursors do not have surface features that interact with platelets, apparently a feature only present after the final steps in maturation. (Reproduced with permission from Lichtman’s Atlas of Hematology, www.accessmedicine.com.)
Some antiplatelet antibodies from patients with pseudothrombocytopenia crossreact with negatively charged phospholipids and may exhibit anticardiolipin activity.30 The sera of these patients lose their ability to clump platelets when adsorbed onto either cardiolipin or activated normal platelets, supporting the hypothesis that antibody subpopulations directed against negatively charged phospholipids can bind to antigens modified by EDTA on the platelet membrane. Another possibility is that the antigens in this case are negatively charged phospholipids on the surface of platelets.
Thrombocytopenia has been described in patients suffering from acute coronary syndromes treated with the abciximab and other integrin αIIbβ3 antagonists.39,40,41 Abciximab is associated with both pseudothrombocytopenia and true thrombocytopenia. The mechanism for platelet clumping with abciximab is unknown; the drug itself likely is not crosslinking the platelets because it is monovalent. More likely, other agglutinins bind integrin αIIbβ3 at new epitopes induced by the combination of abciximab binding and calcium chelation. True abciximab-induced thrombocytopenia occurs in approximately 0.3 to 1 percent of patients treated with the drug.42 The mechanism is incompletely understood, but likely includes reaction of preformed antibodies with a neoepitope expressed after binding of abciximab to integrin αIIbβ3 (ligand-induced binding sites) or abciximab-induced platelet activation with subsequent platelet sequestration from the circulation. In some abciximab-treated patients, high antibody titers are detected in the plasma.
The incidence of pseudothrombocytopenia and thrombocytopenia related to abciximab was determined in four large placebo-controlled trials:40 c7E3 Fab Antiplatelet Therapy in Unstable Refractory Angina (CAPTURE), Evaluation of 7E3 for the Prevention of Ischemic Complications (EPIC), Evaluation of Percutaneous Transluminal Coronary Angioplasty to Improve Long-term Outcome of c7E3 GPIIb-IIIa Receptor Blockade (EPILOG), and Evaluation of Platelet IIb/IIIa Inhibitor for Stenting (EPISTENT). In these studies, pseudothrombocytopenia accounted for more than one-third of low platelet counts in patients undergoing coronary interventions and treated with abciximab. These studies demonstrated that pseudothrombocytopenia is a benign laboratory condition not associated with increased bleeding, stroke, transfusion requirements, or the need for repeat revascularization.
Some studies suggest that platelet agglutinins occur more frequently in hospitalized patients and in association with medical conditions such as autoimmune diseases, malignancy, liver disease, and sepsis.25,43,44,45,46 However, others found no association with any particular pathology or with use of specific drugs.30
One study showed that antibodies from patients with pseudothrombocytopenia can induce agglutination of donor platelets in the presence of EDTA. This agglutination was prevented by warming the donor platelets to 37°C or by pretreating the platelets with aspirin, prostaglandin E1, apyrase, and monoclonal antibodies against integrin αIIbβ3 that block the binding site for fibrinogen and von Willebrand factor (VWF), or arg-gly-asp (RGD) peptide, which binds the site on integrin αIIbβ3 that recognizes cytoadhesive proteins.33 Whether the same reaction occurs in vivo is not known, but in that case the antibodies should have a slow reactivity, or else bleeding would sometimes occur.
An (unexpected) low platelet count reported by automated cell counters should be confirmed by microscopic examination of the blood film. Automated cell counters identify platelets merely based on their small volumes in comparison to those of other blood cells, generally defined as volumes between 2 and 20 fL. Because platelet clumps tend to exceed 20 fL, the clumps may be counted as leukocytes,18 and even if counted as platelets, several platelets are counted as one. Thus, pseudothrombocytopenia may be accompanied by pseudoleukocytosis.5,21,24,47 The greater the delay in processing of anticoagulated blood, the greater is the degree of platelet clumping and the greater the potential for artifact.21 Platelet clumping can be prevented by collecting the sample in EDTA and maintaining its temperature at 37°C. Even with these measures, however, clumping will still occur in approximately 20 percent of cases.30
Another alternative is use of sodium citrate, which chelates calcium more weakly than does EDTA but still causes platelet clumping in approximately 10 to 20 percent of cases with EDTA-induced clumping. In some patients, an accurate platelet count can be obtained only by sampling blood directly into ammonium oxalate and manually counting the platelets using a Bruker chamber.30 Flow cytometry may help for determining exact platelet number by immunostaining of the platelets.
Platelet agglutinins are not associated with bleeding or thrombosis, so they appear to have no clinical implications, except that they may lead to unnecessary therapy because of misdiagnosis. Transplacental transmission of agglutinins has been documented, but the pseudothrombocytopenia induced by these antibodies in the neonate resolves spontaneously.48,49 No complications have been reported when platelet agglutinins are discovered during pregnancy.48,50 Transfusion of blood products from patients with pseudothrombocytopenia produces an acceptable corrected count increment in the recipient, again supporting its benign nature.23 Thus, the clinical importance of pseudothrombocytopenia concerns conditions with which it is confused rather than any pathology associated with the condition. It is important that this syndrome be recognized promptly to avoid unnecessary diagnostic tests and treatment.
INHERITED PLATELET DISORDERS
Megakaryopoiesis and thrombopoiesis are regulated by a number of hematopoietic growth factors and transcription factors (Chap. 113). Any genetic defect affecting platelet production, function or morphology may cause inherited platelet disorders (IPDs; Chap. 121). In recent decades, knowledge of normal megakaryocyte and platelet physiology has grown enormously,51 aided in part by the study of IPDs.52,53
IPDs are a very heterogeneous group of disorders. Some disorders, such as Bernard-Soulier syndrome, appear to be restricted to platelets,54 whereas others appear as a part of a complex pathology, as seen in thrombocytopenia with absent radii (TAR) syndrome (Fig. 117–2). In some IPDs, the platelet count may be normal despite severely impaired platelet function, such as in Glanzmann thrombasthenia. Other disorders are accompanied by abnormal platelet numbers, usually thrombocytopenia. Table 117–2 summarizes the inherited thrombocytopenias.
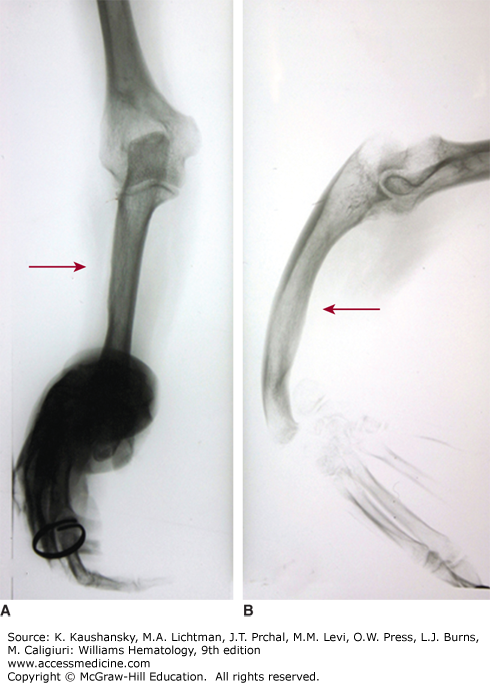
Figure 117–2.
Thrombocytopenia with absent radii (TAR) syndrome. Radiograph of right forearm. A 48-year-old woman with repeated platelet counts in the range of 85 to 100 × 109/L. Bleeding time was 11 minutes. No laboratory evidence of von Willebrand disease. Marrow examination was normal. Both forearms were short and bowed with angulated wrists and normal hands. No family history of forearm deformity. A. Anterior-posterior film of right arm. B. Lateral film of right arm. Absence of radius and bowed, hypertrophied ulna (arrows). Angulation deformity at wrist. (Reproduced with permission from Lichtman’s Atlas of Hematology, www.accessmedicine.com. Kindly provided for the Atlas by Timothy J. Woodlock, Unity Health Systems, Rochester, NY.)
|
Severe forms of IPDs that present as a bleeding tendency early in childhood are rare. IPD patients usually present with mucocutaneous bleeding, such as with purpura, epistaxis, and/or gingival bleeding. Menorrhagia and bleeding during pregnancy and labor are common problems in female patients. Spontaneous life-threatening bleeding is rare, including intracranial hemorrhage, massive gastrointestinal or genitourinary bleeding. Recent molecular investigations of IPD patients and their families with bleeding diathesis demonstrated that most IPDs cause mild bleeding tendencies, and IPDs may be more prevalent than previously thought.55 In these milder cases, a bleeding diathesis may only be diagnosed after an episode of excessive bleeding, such as during surgery or following trauma.
Diagnosis of IPD presents a significant challenge because of the heterogeneity of clinical and laboratory findings of the patients with the same disorder, even in the same family. IPD patients with isolated macrothrombocytopenia share common clinical and basic laboratory features with certain acquired platelet disorders and are sometimes misdiagnosed. It is very important to distinguish IPD patients from those with acquired platelet disorders, such as ITP, to avoid unnecessary or potentially harmful treatments. Helpful in this regard is information obtained during the history, including a family history of bleeding and consanguinity in the family, because the majority of IPDs are inherited as autosomal recessive traits. Because some IPDs are associated with increased risk of developing myeloid malignancies, the patient and family should be asked about a family history of myeloid malignancies. The presence of skeletal, facial, ocular, audiologic, neurologic, renal, cardiac, and immune problems associated with platelet disorders may also suggest IPD.51,56
Laboratory evaluation of a potential IPD should start with a careful blood film investigation, which could be helpful for patients with MYH9-related diseases (giant platelets and Döhle-like inclusion bodies within leukocytes; Fig. 117–3), Bernard-Soulier syndrome (macrothrombocytopenia), Gray platelet syndrome (pale platelets), and sitosterolemia (giant platelets surrounded by a circle of vacuoles, stomatocytosis). Platelet function analyzer (PFA-100) occlusion times are usually found to be prolonged. The skin bleeding time is not recommended for screening, because it is invasive and poorly reproducible. Although the PFA-100 test is very sensitive in detecting Bernard-Soulier syndrome and platelet-type von Willebrand disease (VWD), it may be normal in patients with variant forms of these disorders, or patients with storage pool deficiencies.57 Light transmission aggregometry (LTA) using different concentrations of adenosine diphosphate (ADP), collagen, ristocetin, epinephrine, and arachidonic acid is accepted as a gold standard in diagnosing IPDs, but, again, may be normal in variant forms of IPDs and in some patients with storage pool diseases. Measurement of platelet nucleotide content and release is recommended in patients with platelet granule deficiencies. Flow cytometric analysis is very informative in patients with platelet surface GP deficiencies such as Bernard-Soulier syndrome. Marrow biopsy is needed in patients who have pancytopenia or severe thrombocytopenia, as in Fanconi anemia and congenital amegakaryocytic thrombocytopenia (CAMT), respectively. Unfortunately, these tests may help diagnose only a small portion of the IPD patients. Further tests are only available in specialized centers, and include electron microscopy, Western blotting, and others. Electron microscopy is able to define characteristic ultrastructural abnormalities; Western blotting, enzyme-linked immunosorbent assay (ELISA), or radioimmunoassay can be used for qualitative and quantitative analysis of specific platelet proteins.51,56 Even with these expensive, complicated, and time-consuming tests, the results are inconclusive in nearly half of patients being evaluated for IPD.56 Genetic analysis is often able to determine the underlying molecular pathology, but the very large number of candidate genes limits the traditional target gene approach. Within the past decade, next-generation sequencing techniques have not only improved the speed and cost of genetic investigations, but have also begun to generate very interesting data about the genetic causes of IPD.52,53
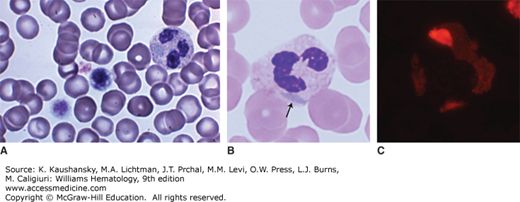
Figure 117–3.
MYH9 abnormality. A. Blood film. May-Hegglin anomaly. Macrothrombocytes, thrombocytopenia, and light-blue cytoplasmic inclusions in neutrophils. Note two giant platelets approximately the diameter of red cells. The neutrophil has a large gray-blue inclusion in the cytoplasm at the 9 o’clock position. B. Blood film. Neutrophil of an individual with a mutation (E1841K) in exon 38 of the MYH9 gene. This mutation results in macrothrombocytopenia and Döhle-body–like inclusions in neutrophils (arrow). C. Blood film. Immunofluorescent analysis with antibodies to the A heavy chain of nonmuscle myosin in the neutrophils of the same patient as in (B). The fluorescent body in the neutrophil indicates that the inclusion contains precipitated nonmuscle myosin heavy chains, characteristic of this family of disorders. (Reproduced with permission from Lichtman’s Atlas of Hematology, www.accessmedicine.com. Images B and C kindly were provided for the Atlas by Dr. Shinji Kunishima, the Japanese Red Cross Aichi Blood Center, Nagoya, Japan.)
NUTRITIONAL DEFICIENCIES AND ALCOHOL-INDUCED THROMBOCYTOPENIA
Iron, vitamin B12, and folic acid deficiencies are the nutrient deficiencies most widely recognized to impair blood cell production. Severe nutritional deficiencies primarily cause anemia, rarely causing bicytopenia or pancytopenia. Isolated thrombocytopenia is rare in patients with nutritional deficiencies.
Iron is present in all human cells, and mediates electron transfer reactions. Iron is a key component of hemoglobin, and iron deficiency causes a hypochromic and microcytic anemia (Chap. 42). Iron-deficiency anemia (IDA) generally develops after acute or chronic bleeding, and is usually accompanied by thrombocytosis rather than thrombocytopenia. Thrombocytopenia associated with IDA is relatively rare, reported in only 2.3 percent and 2.4 percent of pediatric and adult IDA patients, respectively.58,59
Cobalamin (vitamin B12) and folate are both required for DNA synthesis and repair, but humans can synthesize neither vitamin. Dietary deficiencies, impaired absorption, or inhibition with drugs (as seen in methotrexate therapy) of these vitamins can cause megaloblastic anemia (Chap. 41). Mild thrombocytopenia occurs in approximately 20 percent of patients with megaloblastic anemia resulting from vitamin B12 deficiency in the United States.60 The frequency may be higher in patients with folic acid deficiency because of the high frequency of concomitant alcohol abuse (Chap. 41). One large study of 139 patients examined the rates of cytopenias associated with megaloblastic anemia in India.61 In this study, 76 percent had isolated vitamin B12 deficiency, 7 percent had isolated folate deficiency, 9 percent had a combined deficiency, and 8 percent had normal vitamin levels. All were anemic by definition, and 80 percent had thrombocytopenia with mild to moderate depression of the platelet count. More than half of those with thrombocytopenia were also neutropenic. The authors of this study suggested that the cytopenias tended to progress from isolated anemia, to anemia plus thrombocytopenia, to pancytopenia, with the degree of cytopenia related to the severity of vitamin deficiency. Occasionally, thrombocytopenia is severe in patients with megaloblastic anemia and, when accompanied by fever, hepatomegaly, and splenomegaly, and may suggest a diagnosis of acute leukemia. In these syndromes, the primary mechanism of thrombocytopenia is ineffective platelet production62; marrow megakaryocyte number usually is normal or increased. Abnormalities of megakaryocyte morphology are much less distinctive than the characteristic erythroid and myeloid defects, but often nuclear abnormalities are seen, with nuclei of larger size and dispersed nuclear segments, rather than single polyploid nuclei.63 Thrombocytopenia may be seen in association with vitamin B12 deficiency when the latter results from autoantibodies against parietal cells or intrinsic factor and is associated with ITP.64,65 Various other autoimmune disorders can coexist with pernicious anemia, including autoimmune vitiligo and autoimmune thyroiditis.66 Abnormalities of platelet function are sometimes seen associated with vitamin B12 deficiency.67,68 Diminished platelet aggregation and reduced release of ADP and ATP from granule stores in response to different agonists have been reported, and vitamin deficiency has been suggested to induce an acquired storage pool disease (Chap. 41).68
Copper deficiency is usually seen in patients who have undergone gastric bypass surgery, and may cause anemia, leukopenia, and thrombocytopenia associated with neurologic deficits resembling vitamin B12 deficiency. Patients with copper deficiency also may be misdiagnosed as having MDS, because increased ring sideroblasts and dysplastic precursor cells can be seen on marrow smears.69,70
Acute and chronic alcohol (ethanol) consumption affects hematopoiesis and blood cell survival both directly and indirectly. Alcohol is one of the leading causes of thrombocytopenia in Western countries. Acute ethanol intoxication in healthy volunteers induces thrombocytopenia.71 Platelet counts in these cases are usually mildly decreased (generally more than 100 × 109/L); severe thrombocytopenia is quite rare. Acute ethanol-induced thrombocytopenia usually resolves within 5 to 21 days with cessation of ethanol ingestion, sometimes with a transient rebound thrombocytosis that may reach up to 1,000,000 × 109/L.72 Although the mechanism of acute alcohol-related thrombocytopenia is not clear, it has been suggested that metabolites of ethanol, especially acetaldehyde, impair the late stages of platelet production and increase platelet destruction.73 Thus, thrombocytopenia associated with acute alcohol ingestion would be expected to be more frequent in those with poor nutrition (delayed oxidation of acetaldehyde) and those with partial acetaldehyde dehydrogenase defiance. Thrombocytopenia induced by alcohol ingestion is accompanied by a decreased number of marrow megakaryocytes. Vacuolated proerythroblasts and granulocyte precursors are sometimes seen, as are multinuclear erythroblasts and megaloblasts.74 Vacuolization of the periphery of mature megakaryocytes has been reported.75 Alcoholism (chronic ethanol consumption, which is defined as consumption of more than 80 g of ethanol per day), on the other hand, may cause thrombocytopenia by other mechanisms, such as alcoholic liver cirrhosis (both splenomegaly and thrombopoietin deficiency), folic acid deficiency, and alcohol-induced marrow suppression.74,75,76,77,78
ACQUIRED PURE AMEGAKARYOCYTIC THROMBOCYTOPENIA
Thrombocytopenia attributable to pure aplasia or hypoplasia of megakaryocytes is rare.79 More common are instances in which amegakaryocytic thrombocytopenia anticipates the development of full-blown MDS or aplastic anemia and is associated with subtle abnormalities of other lineages, such as macrocytosis and dyserythropoiesis.80,81,82,83,84 Most commonly the disorder is caused by autoimmune suppression of megakaryocyte development, either idiopathic,85 associated with autoimmune disorders such as systemic lupus erythematosus (SLE)86 and eosinophilic fasciitis, or associated with infections such as hepatitis C.87 Antibodies against thrombopoietin (TPO)88 have been described to cause the disorder, as have antibodies against the TPO receptor.89 Patients may achieve durable remission with therapies designed to blunt the autoimmune response, such as cyclosporine or antithymocyte globulin (ATG).90
Table 117–3 summarizes the various types of ITP.
|
ITP, formerly known as autoimmune thrombocytopenic purpura, is the most common cause of isolated thrombocytopenia in clinical practice. ITP is characterized by immune-mediated platelet destruction and impaired platelet production. ITP occurs in every age group. Childhood ITP typically is acute in onset, often developing after a viral infection or vaccination. Although thrombocytopenia may be severe, it usually resolves spontaneously, within a few weeks up to 6 months.91 In contrast to childhood ITP, adult ITP generally is a chronic disease of insidious onset and rarely resolves spontaneously.
“Purpura” was recognized by Hippocrates (c. 460 to c. 370 BC) and Galen (AD 129 to c. 200/c. 216) as a sign associated with fever. Chronic purpura was first described in details by Ibn-i Sina (Avicenna, c. 980 to c. 1037) in his famous book “The Canon of Medicine.” In 1705, Werlof suggested that purpura was related to infections and described it as “morbus maculosus haemorrhagicus.” Patients with purpura were diagnosed as having “Werlof disease” for centuries. After the discovery of platelets and their role in hemostasis, the relationship between purpura and low-platelet count was understood.92
In 1915, Erich Frank renamed this disorder as “essential thrombocytopenia,” and suggested that platelet production from megakaryocytes was impaired because of a toxic substance produced by the spleen.93 Kaznelson, inspired by Frank’s theory, proposed splenectomy for a patient with chronic thrombocytopenic purpura. The treatment was successful, and splenectomy was first-line therapy for ITP until the introduction of glucocorticoids in 1950s.
In the first issue of the journal Blood (in 1946) Damashek and Miller reviewed the megakaryocyte count and marrow morphology of patients with “idiopathic thrombocytopenic purpura.”94 They showed that most ITP patients had an increased number of megakaryocytes, but very few of them were producing platelets, so “actual platelet-producing tissue” might be decreased.94
Although Marino first showed that antiplatelet antibodies might cause thrombocytopenia in animal studies in 1905, the Harrington-Hollingsworth experiment (1951) was an important milestone in the understanding of autoantibody-directed platelet destruction in the pathophysiology of ITP. In this pioneering work, normal volunteers (including Harrington himself, who received the highest dose) were infused with the plasma from patients with ITP, resulting in severe thrombocytopenia in the recipients, and they postulated that ITP could be caused by antiplatelet antibodies.95,96 Subsequently, Shulman and coworkers97 showed that the thrombocytopenic effect of ITP plasma was dose-dependent and associated with the globulin fraction. In the 1950s, glucocorticoids began to be used to treat ITP, and they became first-line therapy for adults. Shortly thereafter other immunosuppressive agents were introduced for the treatment of chronic ITP.92
In the early 1970s, two groups showed that platelets from chronic ITP patients had elevated levels of platelet-associated immunoglobulin G (PAIgG).98,99 In 1982, the first platelet target was identified: autoantibodies from patients with ITP failed to bind platelets deficient in the integrin αIIbβ3 complex (i.e., from patients with Glanzmann thrombasthenia).100 In the late 1980s, two specific assays for the target antigens were described: the immunobead assay101 and the monoclonal antibody-specific immobilization of platelet antigens (MAIPA) assay.102 These assays showed that the majority of antiplatelet antibodies in patients with ITP are directed against integrin αIIββ3(GPIIβ-IIIα)(approximately 80 percent), and the remainder are against the GPIb-IX-V complex and other platelet GPs such as GPIV and integrin α2β1 (GPIa-IIa).103,104 Some sera contain antibodies that recognize several antigens. Most antiplatelet autoantibodies are IgG; the remainder are IgM and IgA. Unfortunately, elevated levels of PAIgG later were found in patients with non-ITP. Therefore, PAIgG could not be used as a specific laboratory test for ITP in the same way that the direct antiglobulin test is used for the diagnosis of autoimmune hemolytic anemia.105,106 To date there is still no specific laboratory test for ITP, the diagnosis of ITP being based on exclusion of other causes.
Antibody-coated platelets bind tissue macrophages through Fcγ receptors, leading to their destruction primarily in the spleen and, to a lesser extent, in the liver and marrow.97,107,108 In 1981, Imbach reported successful treatment of pediatric ITP with intravenous immunoglobulin (IVIG) and suggested that the mechanism could involve blockade of macrophage Fc receptors. IVIG became first-line therapy in children, and now is also used in adults when a prompt increase is the platelet count is desired.109
Early studies of PAIgG reported that the antibodies in ITP were polyclonal.110 However, later studies showed that at least some ITP patients had clonal B-cell proliferation, as determined by DNA analysis for immunoglobulin heavy- and light-chain rearrangements and by flow cytometry of B cells from blood and spleen for surface Ig light chains.111,112 This led to the use in ITP of the chimeric anti-CD20 monoclonal antibody, rituximab, which was designed for the treatment of CD20-positive B-cell lymphomas. The rapid elimination of B cells with rituximab encouraged the use of this agent in the treatment of ITP.
Numerous abnormalities in cell-mediated immunity have been described in patients with ITP, including abnormalities in antigen-presenting cells, T lymphocytes, and cytokine release. Under normal conditions, antigen-presenting cells recognize and process foreign antigens and express the antigens on their surface in association with major histocompatibility complex (MHC) molecules. MHC–antigen complexes activate resting (naïve) CD4+ T cells to differentiate into a variety of phenotypes such as T-helper 1 (Th1) and T-helper 2 (Th2), Th17, and T-regulatory (Treg) cells. Th1 cells are involved in cell-mediated immunity and host defense against intracellular bacteria and protozoa. Th2 cells are involved in humoral immunity and host defense against extracellular parasites. Th17 cells are involved in host defense against extracellular bacteria and fungi. Treg cells (formerly known as suppressor T cells) play an important role in self-tolerance by inhibiting autoimmune responses. Abnormal T-cell responses drive the differentiation of autoreactive B-cell clones and autoantibody secretion. In patients with ITP, both Th1 and Th17 cells have been found to be upregulated, whereas the number and the suppressor functions of the Treg cells were found to be decreased.113,114,115 This imbalance is believed to induce an autoimmune responses against the platelets. It is unclear whether these abnormalities are causative or represent an epiphenomenon.114,115 In addition, CD8+ cytotoxic T cells might be involved in the pathogenesis of ITP through cell-mediated destruction of platelets and megakaryocytes and through suppression of megakaryocytes, impairing platelet production.115,116,117
Antiplatelet autoantibodies may also activate platelet destruction by activating complement through the classical complement pathway. Increased platelet-associated C3, C4, and C9 have been demonstrated on the platelets from patients with ITP.118,119 In vitro studies show that, in the presence of antiplatelet antibodies, C3 and C4 can bind platelets, increase the phagocytosis of the platelets by macrophages, and can cause their lysis by stimulating assembly of the membrane attack complex.120,121
Early studies demonstrated that platelet survival is shortened in ITP patients and returns to normal after splenectomy-induced remission.122 Platelet transfusion only transiently increases a patient’s platelet count, and the transfused platelets also have a shortened survival, reflecting the fact that the major problem in ITP is platelet destruction. However, later studies showed that platelet life span was not short enough to account for the observed thrombocytopenia on the basis of destruction alone, again suggesting a concomitant defect in platelet production.123 Potential mechanisms for this observation were provided by later studies that autoantibodies against platelet surface GPs might interfere with the maturation of megakaryocytes, resulting in reduced platelet production, contributing to the severity of thrombocytopenia in some ITP patients.124 Antibodies that target the GPIb–IX–V complex may induce thrombocytopenia by decreasing platelet production, as GPIb autoantibodies inhibit megakaryopoiesis in vitro,124 and GPIb monoclonal antibodies inhibit proplatelet formation in vitro.125
In 1958, a hematopoietic growth factor regulating platelet production was proposed and named TPO by Kelemen.126 Although interleukin (IL)-3, IL-6, IL-11, granulocyte-macrophage colony-stimulating factor, and c-KIT ligand increase megakaryocyte or platelet counts in vivo and in vitro, animal studies of these factors proved that they are not the main regulator of megakaryopoiesis.127 In 1994, TPO was first characterized by five independent groups. TPO binds to its receptor MPL (formerly known as c-MPL), enhances megakaryocyte colony formation, and increases the size, number, and ploidy of megakaryocytes, and platelet production (Chap. 113).128,129,130 TPO is synthesized in greatest quantity in the liver but is found in other organs (kidney, muscle, and marrow stromal cells).128 TPO is also required to maintain the viability of hematopoietic stem cells.131 The regulation of TPO production is complex. Hepatic production of TPO is both constitutive (in the steady state) and inducible (by inflammation), and the concentration of TPO to which megakaryocytes are exposed is also determined by the platelet concentration. Platelets, bearing TPO receptors, remove the hormone from the circulation, at least partially accounting for the inverse relationship between TPO and platelet levels. TPO levels are markedly elevated in patients with thrombocytopenia associated with megakaryocytic hypoplasia, including disorders such as aplastic anemia or acute leukemia. In most reports, ITP patients have normal or slightly elevated TPO levels whether measured in plasma or serum, but the levels are always lower than the concentrations found in thrombocytopenias resulting from megakaryocytic hypoplasia.128,129,130,132,133 Initial studies with recombinant and pegylated TPO molecules showed successful responses in patients with thrombocytopenia, but development of autoantibodies against these molecules restricted their use in clinical settings. Based on the success of creating erythropoietin receptor agonist peptides, a number of screening efforts were undertaken to design small peptides or organic molecules that might bind to the TPO receptor and stimulate thrombopoiesis. One such molecule contains four copies of a 14-amino-acid peptide grafted onto an Ig Fc domain, forming a “peptibody” termed romiplostim. This agent, which binds to a region of the TPO receptor that overlaps that bound by authentic TPO, was shown to increase platelet counts in patients with ITP who had failed other modalities,134 and was approved by the FDA for this indication in 2008. Another small organic thrombopoietic molecule, eltrombopag, was developed almost simultaneously135 and approved in 2008 by FDA for the same indications.127 This agent activates TPO receptor signaling by binding to the transmembrane domain of the receptor, a site quite distinct from the binding site for TPO and romiplostim. Both TPO-receptor agonists are currently being evaluated for additional clinical indications in clinical trials.136
Some patients with ITP appear to display a genetic predisposition. ITP has been documented in monozygotic twins137 and shown to be highly prevalent in some families.138 In addition to contributing to the development of ITP, like in other autoimmune disorders heredity may also affect the response to ITP therapy. Human leukocyte antigen (HLA) class I and class II allele frequencies in patients with ITP have been studied by several investigators, with inconsistent results. Some investigators reported an increased frequency of HLA-Aw32, -DRw2, and -DRB1*0410.108,139,140,141 Investigation has focused on genetic differences associated with dysregulation of immune tolerance and humoral immunity, but results have been inconclusive. For example, genetic polymorphisms of cytotoxic T-lymphocyte antigen (CTLA)-4, tumor necrosis factor, and Fcγ receptors IIA and IIIA have been suggested to influence the development of ITP and the response to therapy,141,142,143 but as yet no strong association has been found.
Accumulating data indicate that the pathophysiology of ITP is more complex than previously thought, with ITP comprising a heterogenous group of disorders with different etiologies and responding to different treatment modalities. The identification of the different subsets of ITP patients will help to better define treatment options.
Although ITP has been recognized for centuries, there is as yet no consensus on either the definition or management of the disease. In 1996, the American Society of Hematology (ASH)144 published practice guidelines for the diagnosis and management of ITP. In 2003, the British Committee for Standards in Haematology published its own guidelines.145 In spite of these guidelines, the heterogeneity of the definitions and clinical criteria used in different studies has made it difficult to interpret the data regarding the incidence, pathogenesis, and treatment of ITP. In 2008, the International Working Group (IWG) proposed a standardization of terminology, definitions, and outcome criteria for ITP patients.146 In 2010, an international consensus report on the investigation and management of ITP was published.147 Shortly thereafter, in 2011, ASH updated its 1996 ITP guidelines.148
The IWG definition proposed use of the term “immune thrombocytopenia” instead of “idiopathic thrombocytopenic purpura” as the basis for the ITP acronym, because the immune nature of ITP is clear but most ITP patients do not have purpura. A platelet count of 100 × 109/L was proposed as the threshold level to entertain the diagnosis of ITP, because a sustained lower platelet count (100 to 150 × 109/L) can be seen in otherwise healthy individuals,11,12 and long-term observation of these indicate that 88 percent reach normal platelet counts or remain stable.13 ITP is classified based on the absence or presence of other diseases as “primary” or ‘”secondary.” “Primary ITP’” denotes the absence of any other identified pathology. All other autoimmune thrombocytopenias are classified as “secondary ITP” (see Table 117–3), and the associated primary disorder is indicated in parenthesis, for example “secondary ITP (SLE-associated)” or “secondary ITP (drug-induced).” Heparin-induced thrombocytopenia (HIT) or alloimmune thrombocytopenias are not classified as ITP, and maintain their standard classifications.146
The IWG described three phases of ITP: (1) newly diagnosed ITP (within 3 months of diagnosis); (2) persistent ITP (patients who do not achieve a stable remission between 3 and 12 months after diagnosis); and (3) chronic ITP (continuing for more than 12 months). ITP was formerly classified as mild, moderate, and severe depending on the platelet counts. However, the degree of thrombocytopenia does not always correlate with bleeding. The IWG proposed that the term “severe ITP” only be used for patients with clinically significant bleeding requiring additional therapy regardless of platelet count.146
One of the major problems with comparing ITP studies had been the definition of response to therapy. The IWG proposed the following terms and criteria for response to ITP treatment: “complete response, CR” (platelet count exceeding 100 × 109/L and no bleeding symptoms), “response, R” (platelet count higher than 30 × 109/L or at least a twofold increase from the baseline count and no bleeding symptoms), “no response, NR” (platelet count below 30 × 109/L or less than a twofold increase from the baseline count, or presence of bleeding symptoms). “Duration of response” is measured from the time between first measured CR or R to relapse. “Corticosteroid dependence” is defined as the need for ongoing or repeated glucocorticoid use for at least 2 months to maintain CR or R. Patients who relapsed after splenectomy (failure to maintain CR or R) and required therapy are classified as “refractory ITP.” “On-demand therapy” is a term used for therapies employed to temporarily increase the platelet count in special situations such as trauma or surgery. “Adjunctive therapies” are treatments that are not designed to increase platelet counts, but that may decrease bleeding symptoms by other means, for example, treatment with oral contraceptives or antifibrinolytic drugs.146
ITP is relatively common, but demographic studies have yielded a wide range of incidence rates largely because of differences in the age and gender distribution of the populations studied and differences in cutoff platelet counts used to define the disease. ITP can affect males and females of any age. In one detailed study, the reported incidence of ITP was 3.9 per 100,000 per year. Although the overall incidence was higher in women than in men, a male predominance was seen in patients younger than 18 years of age and older than 65 years of age.149
ITP is of acute onset in children, often developing after vaccination or after a viral illness, and resolves spontaneously in 90 percent of cases. In adults, however, ITP usually is a chronic disease. Table 117–4 highlights the differences in ITP in children and adults. Approximately 25 percent of adult ITP patients are diagnosed incidentally on routine complete blood counts. Symptoms and signs of ITP depend not only on the platelet count, but also on the nature of coexisting conditions that can increase the tendency to bleed, such as uremia, trauma, and ingestion of drugs that affect platelet function (Table 117–5). Approximately one-third of patients have platelet counts greater than 30 × 109/L at diagnosis and no significant bleeding.150 Common bleeding signs include purpura (ecchymoses and petechiae), epistaxis, menorrhagia, and gingival bleeding. Hematuria, hemoptysis, and gastrointestinal bleeding are less common. Intracerebral hemorrhage is rare and generally occurs in patients with platelet counts less than 10 × 109/L and usually is associated with trauma or vascular lesions. The incidence of life-threatening complications is highest in patients older than age 60 years.150,151,152,153,154 The majority of ITP patients have a good prognosis, the mortality rate being only slightly higher than that of the general population. However, ITP patients who present with severe thrombocytopenia (<30 × 109/L) and do not respond to any therapy within 2 years, have a fourfold increased risk of death compared to the general population.155
| Children | Adults | |
|---|---|---|
| Occurrence | ||
| Peak age (years) | 2–4 | 15–40 |
| Sex (Female-to-Male) | Equal | 1.2–1.7 |
| Presentation | ||
| Onset | Acute (most with symptoms lasting <1 week) | Insidious (most with symptoms lasting >2 months) |
| Symptoms | Purpura (<10% with severe bleeding) | Purpura (typically bleeding not severe) |
| Platelet count | Most cases <20,000/μL | Most cases <20,000/μL |
| Course | ||
| Spontaneous remission | 83% | 2% |
| Chronic disease | 24% | 43% |
| Response to splenectomy | 71% | 66% |
| Eventual complete recovery | 89% | 64% |
| Morbidity and mortality | ||
| Cerebral hemorrhage | <1% | 3% |
| Hemorrhagic death | <1% | 4% |
| Mortality of chronic refractory disease | 2% | 5% |
| Drugs: Anticoagulants, antiplatelet drugs, nonsteroidal antiinflammatory drugs, chemotherapy |
| Gastrointestinal pathologies that may cause bleeding (e.g., active peptic ulcer, inflammatory bowel disease) |
| Miscellaneous disorders that disturb hemostasis (e.g., congenital bleeding disorders, hepatic cirrhosis, uremia) |
| Older age (>60 years) |
| Nutritional factors such as herbal teas, kinin, and tonic water |
| Previous history of bleeding |
| Sport and occupational activities that increase bleeding risk |
| Trauma, surgery, and childbirth |
| Uncontrolled hypertension |
The purpuric lesions seen in ITP are not palpable, do not blanch with pressure, and often develop on distal regions of the extremities and on skin areas exposed to pressure (e.g., around tight belts and stockings and at tourniquet sites). Hemorrhagic bullae, which may develop in the buccal mucosa, generally reflect acute, severe thrombocytopenia. Bleeding after surgery, trauma, or tooth extraction is common.
Besides the physical findings associated with platelet-type bleeding, the history and physical examination are usually unremarkable, except for the possibility of similar symptoms in other family members. Family history is especially important to discriminate familial thrombocytopenic syndromes from ITP. The spleen usually is not enlarged but may be palpable in some patients, a finding considered to occur with the same incidence as in normal adults.156 Constitutional symptoms, such as fever, significant weight loss, marked splenomegaly, hepatomegaly, and lymphadenopathy provide evidence that the thrombocytopenia has another cause. The presence of skeletal, cardiac, renal abnormalities, hearing loss, albinism. or immune deficiencies in patients with thrombocytopenia should trigger suspicion of IPDs.
Fatigue is one of the common, but often neglected, complaints of patients with primary ITP. In a survey including UK and U.S. ITP cohorts, the prevalence of fatigue was found to be significantly higher in adult primary ITP patients (39 percent and 22 percent for the UK and U.S. cohorts, respectively) compared with healthy controls.157 Fatigue has also been described in 20 percent of pediatric patients with ITP; fatigue resolved with the elevation of platelet counts.158 Although glucocorticoids and immunosuppressive agents may induce fatigue, fatigue can occur in untreated ITP patients. The mechanism of fatigue in patients with ITP is unknown.
Patients with ITP are at slightly increased risk of venous and arterial thrombosis.159 A recent retrospective study evaluating 986 patients with ITP showed the cumulative incidences of venous and arterial thrombosis to be 1.4 percent and 3.2 percent, respectively. This study found that increased thrombotic risk was associated with splenectomy, older age (>60 years), with the presence of more than two thrombotic risk factors at the time of diagnosis, and with glucocorticoid therapy.160
In ITP patients the blood film usually demonstrates isolated thrombocytopenia without erythrocyte or leukocyte abnormalities. Platelet anisocytosis is a common finding. Mean platelet volume and platelet distribution width are increased. Platelets may be abnormally large or abnormally small. The former reflect accelerated platelet production,161 and the latter represent platelet fragments associated with platelet destruction.162 The observation of giant platelets should trigger consideration of IPDs, which often are misdiagnosed as ITP.163 The bleeding time correlates inversely with platelet count if the count is less than 50×109/L, but may be normal in patients with mild or moderate thrombocytopenia,164 making it an unreliable test for use in such patients. The ultrastructure of ITP platelets viewed by electron microscopy is similar to that of normal platelets.165
Hemoglobin concentration and hematocrit are generally normal in patients with ITP. Anemia that is not easily explained (e.g., resulting from iron deficiency in bleeding patients or associated with thalassemia minor in endemic areas) must be investigated further. Autoimmune hemolytic anemia with a positive direct antiglobulin (Coombs) test and reticulocytosis may accompany ITP; this association is termed Evans syndrome.166 Neither erythrocyte poikilocytosis nor schistocytes should be present. Total leukocyte counts and differential are generally normal. Although atypical lymphocytes and eosinophilia may occur in children with ITP, leukocytosis and leukopenia with immature cells are not consistent with the diagnosis.
Marrow examination, which is not always required to make a diagnosis of ITP in adults, generally reveals a normal or increased number of megakaryocytes of normal morphology, although a decreased number of megakaryocytes does not rule out ITP. Erythropoiesis and myelopoiesis are normal. The international consensus report states that a marrow examination should usually be reserved for patients older than age 60 years, for those with systemic symptoms or other signs, and for those for whom splenectomy is contemplated. Biopsy for morphologic examination should be carried out, along with aspirate for flow cytometric and cytogenetic analysis.147 The ASH 2011 guidelines, however, conclude that a marrow examination is unnecessary when the presentation is typical, even if the patients are older or being considered for splenectomy.148
In ITP patients, initial workup should be targeted to exclude secondary causes of thrombocytopenia (see Table 117–3). Testing for viral etiology (hepatitis C virus [HCV], HIV, and in endemic areas hepatitis B virus [HBV]) and Helicobacter pylori is also recommended.147,148 Quantitative immunoglobulin assessment should be considered for pediatric cases to rule out common variable immunodeficiency (CVID).147 Mild thrombocytopenia has been reported in patients with hypo- or hyperthyroidism, which returns to normal after appropriate therapy. Thyroid-stimulating hormone (TSH) and antithyroid antibodies may help to evaluate thyroid status in those patients.147 Other tests to consider include blood group analysis and a pregnancy test for female patients of childbearing age, antiphospholipid antibodies, antinuclear antibody (ANA), viral polymerase chain reaction (PCR) for parvovirus, and cytomegalovirus (CMV). The results of these tests can change the treatment strategy.147 On the other hand, the ASH 2011 guidelines do not recommend routine testing for antiphospholipid antibodies and ANAs in the initial workup of ITP,148 unless signs or symptoms of an autoimmune disorder are present in the patient. Other tests, such as TPO levels, reticulated platelets, PAIgG, platelet survival studies, bleeding time, and serum complement levels are not recommended for the diagnosis and management of ITP patients in either of these guidelines.147,148
What little is known of the natural course of moderate or severe ITP derives from before the glucocorticoid era, and suggests that left untreated, ITP in adults typically is a chronic disease, in contrast to ITP in children. In adults, the rate of spontaneous remission is reported as 9 percent,167 and can occur even after 3 years in patients who present with severe thrombocytopenia.168 Although ITP is a benign disease, side effects of the therapies can cause serious morbidity and even mortality. Treatment for patients with ITP should be based on bleeding signs and symptoms and on the presence of factors that increase the bleeding risk (see Table 117–5). Possible side effects of the drugs and other treatments used in ITP should always be considered.
Observation Because a significant portion of ITP patients are diagnosed incidentally in routine evaluation, signs and symptoms of bleeding are important in determining whether any treatment is required. The primary therapeutic goal is not simply to increase the platelet count, but to reach a safe platelet count where the risk of bleeding is minimal. Patients with no bleeding and consistent platelet counts in excess of 30×109/L do not require treatment and can be observed periodically. These patients are at low risk for clinically important bleeding. Simple observation is not recommended for patients with platelet counts lower than 10 × 109/L, in those with platelet counts between 10 and 30 × 109/L and significant mucosal bleeding, or in those with risk factors for bleeding (see Table 117–5).169 The presence of extensive purpura or hemorrhagic bullae in mucosal tissues (wet purpura) should be regarded as a harbinger of life-threatening bleeding and treated as such. Because ITP patients often have large platelets that may not be recognized by automated cell counters, a blood film should be evaluated before starting therapy in ITP patients with very-low platelet counts who are not bleeding. Identification of secondary ITP cases is very important, and management of these patients should include treatment of the underlying pathology, if possible.
Emergency Treatment of Acute Bleeding Resulting from Severe Thrombocytopenia Bleeding symptoms generally are not severe in adult patients with ITP, even with very-low platelet counts. However, life-threatening bleeding can occur, especially after trauma. Emergency treatment should be instituted in patients with intracranial or gastrointestinal bleeding, massive hematuria or internal hematoma, or those in need of emergency surgical intervention or about to go into labor. Patients who experience significant bleeding should be hospitalized and monitored closely. Recommended treatment includes IVIG and parenteral glucocorticoids in combination. IVIG is given as 1 g/kg per day for 2 days, and high-dose parenteral glucocorticoid therapy includes high-dose prednisone or methylprednisolone (1 g/day for 1 to 3 days). In most patients, IVIG increases the platelet count within 2 to 3 days.147,148 Although platelet transfusions may not increase the platelet counts because the transfused platelets are destroyed rapidly, they nevertheless may contribute to the formation of platelet plugs at sites of bleeding and improve hemostasis. Platelet transfusion following IVIG infusion may increase the platelet count because IVIG may improve platelet survival.147,148,170 Aminocaproic acid, which inhibits fibrinolysis, can be used to reduce bleeding170 and is safe except in the presence of hematuria, in which case it can cause thrombi of the glomeruli, renal pelves, and ureters. This agent does not affect platelet count or function. Aminocaproic acid is usually administered intravenously (initial dose 0.1 g/kg over 30 minutes, then given either by continuous infusion at 0.5 to 1.0 g/h or as an equivalent intermittent dose every 2 to 4 hours). Aminocaproic acid also can be administered orally in a similar dose in emergency situations because it is absorbed very rapidly from the gastrointestinal tract.147,148 Vincristine can be used in combination with glucocorticoids and IVIG in older patients.108 Other hemostatic therapies, such as recombinant factor VIIa and fibrinogen infusions, have been reported to be effective in some ITP patients with life-threatening bleeding, but the risk-to-benefit ratio needs to be evaluated in controlled studies.171,172 Emergency splenectomy has been reported to be successful in refractory ITP with bleeding, but reports of its use in this situation are rare.173 Because of this, this therapy should only be considered in the most dire circumstances. Although there are some case reports describing successful results with plasmapheresis, this treatment is not recommended in current ITP guidelines.147,148
Glucocorticoid Therapy Glucocorticoids are accepted as the standard therapy for initial treatment in adult patients with ITP.147,148 Glucocorticoids increase the platelet count in several ways, including by inhibiting phagocytosis of antibody-coated platelets by macrophages, decreasing autoantibody production, and improving marrow platelet production.174,175 These agents also appear to reduce capillary leakage, thereby decreasing blood loss.176 The major drawback of glucocorticoid therapy is that often the adverse effects of the treatment are worse than the disease itself. Important side effects, which can be severe, include facial swelling (chipmunk or moon facies), weight gain, folliculitis, hyperglycemia, hypertension, cataracts, osteoporosis, aseptic bone necrosis, opportunistic infections, and behavioral disturbances.177,178
Still under investigation is which glucocorticoid and dosing regimen is best for raising the platelet count. Prednisolone, dexamethasone and methylprednisolone are all used. Generally, oral prednisone 1 to 2 mg/kg per day (or methylprednisolone at equivalent doses) is preferred as first-line therapy.147,148 Patients usually respond to prednisone therapy within 3 weeks. In approximately two-thirds of patients, platelet counts increase to greater than 50 × 109/L within 1 week, but decrease again when the prednisone dose is decreased.152,177 Although no consensus exists regarding the duration of initial therapy, treatment should continue until platelet counts reach a safe range. In patients who respond, the recommendation is to continue glucocorticoid therapy 1 mg/kg per day for a total of 3 weeks before initiating a slow tapering of doses.148 Sustained remission rates with glucocorticoid therapy are variable, reported rates ranging from 5 to 50 percent.108,155,177 If the patient does not respond to 3 weeks of prednisone therapy, other therapeutic options should be considered.
In addition to the standard 1 to 2 mg/kg per day dose of prednisone, lower179,180 and higher doses181,182,183,184 of prednisone, dexamethasone, and methylprednisolone have been investigated, with good results. The major aim of the high-dose glucocorticoid regimes is to reduce duration of therapy, and therefore reduce the side effects of the glucocorticoids. Studies with dexamethasone 40 mg/day for 4 consecutive days for one course, or with the same dose for four courses given every 2 weeks have been reported to produce responses in 50 percent and 89.2 percent of newly diagnosed ITP patients, respectively.185,186 High-dose methylprednisolone therapy has also been shown to be effective, with an 80 percent response rate.187 Despite the favorable results of these studies, high-dose glucocorticoid regimens as first-line therapy still have not been validated with randomized controlled trials. ASH 2011 guidelines recommend longer courses of standard doses of glucocorticoids (prednisone 1 to 2 mg/kg per day) as a first-line treatment of ITP.148
Splenectomy Splenectomy was demonstrated to be an effective treatment for patients with ITP a century ago188 and after the glucocorticoid era, it has been used for decades as a standard second-line therapy. The spleen is the major site both for synthesis of antiplatelet antibodies and for destruction of antibody-coated platelets. Splenectomy will decrease antibody production and platelet destruction, and will be effective in patients in whom antibody-mediated platelet destruction rather than platelet production is the major cause of thrombocytopenia. Although splenectomy has been reported to be less preferred in recent ITP cohorts because of the emergence of new therapies such as TPO receptor agonists and rituximab,189 splenectomy still produces the highest cure rates for ITP patients compared to all other therapies. Approximately 85 percent of patients with persistent or chronic ITP respond well to splenectomy, and 60 to 66 percent of the patients remain in remission after 5 years.189,190,191 These high cure rates makes splenectomy an important therapeutic option in the treatment of chronic ITP. The duration of the disease prior to splenectomy does not affect the outcome of the procedure, as it can be effective even years after ITP is diagnosed.192,193 Splenectomy can be performed during pregnancy (preferably during the second trimester), and does not affect the response rates to other treatments except anti-D therapy in chronic ITP patients. Also, the cost of splenectomy is lower than that of newer treatments such as rituximab and TPO-receptor agonists.191
On the other hand, splenectomy is an invasive procedure, causes the permanent loss of an organ, and increases the risk of serious bacterial infection, bleeding and thrombosis. Because ITP can remit spontaneously, splenectomy should be postponed at least 6 to 12 months after diagnosis if possible.147,148 Splenectomy is not recommended in patients with CVID, with chronic infections such as chronic hepatitis and HIV, or with known thrombophilia.
No validated clinical or laboratory tests exist that can predict whether splenectomy will be effective in elevating platelet counts in ITP patients. Although it has been suggested that ITP patients with predominant splenic sequestration (as determined by radioisotope techniques) have better response rates than patients with predominantly nonsplenic sequestration, these data have not been validated in other studies189 and the required radioisotope techniques are not widely available.
Over the past decade minimally invasive laparoscopic splenectomy has gained preference over open splenectomy. Modern laparoscopic approaches reduce mortality rates (<1 percent), even in patients with severe thrombocytopenia.194 The mortality rate increases in older patients, in patients with severe thrombocytopenia, and in the presence of coexisting illnesses.177,195 Postsplenectomy sepsis is a major cause of morbidity and mortality in ITP. Extended steroid or other immunosuppressive therapy preceding splenectomy may increase the risk of perioperative infection. To minimize the risk of sepsis, patients should be immunized at least 2 weeks before splenectomy with polyvalent pneumococcal vaccine, Haemophilus influenzae type B vaccine, and quadrivalent meningococcal polysaccharide vaccine.196 Interestingly, newer studies of ITP patients undergoing splenectomy show enteric organisms to be responsible for most of the cases of postsplenectomy sepsis, probably because of the widespread vaccination of ITP patients.191 Splenectomized patients should be informed to be alert for the symptoms and signs of infection and be prepared for an emergency situation. Any fever should be carefully evaluated, and the patient treated with broad-spectrum antibiotics.
Splenectomy also increases the risk of thrombosis in ITP patients. In a large cohort of 9976 ITP patients, in whom 1762 underwent splenectomy; the cumulative incidences of abdominal venous thromboembolism and deep vein thrombosis/pulmonary embolism were increased in splenectomized patients compared to nonsplenectomized patients (1.6 percent vs. 1 percent for abdominal venous thrombosis, 4.3 percent vs. 1.7 percent for deep vein thrombosis–pulmonary embolism, respectively).197 Several mechanisms may contribute to this enhanced risk for thrombosis, including postsplenectomy thrombocytosis and a failure to clear platelets, other cells and microparticles that express the procoagulant lipid phosphatidylserine. Perioperative measures such as antiembolic stockings and anticoagulant prophylaxis should be considered in those cases.
Both the time required to reach a normal platelet count and the magnitude of platelet recovery are accepted as useful predictors of the long-term efficacy of splenectomy. In most cases, platelet counts recover within 10 days. Patients who attain a normal platelet count within 3 days of splenectomy generally have a good long-term response.198 In patients refractory to splenectomy, the presence of accessory splenic tissue should be suspected, particularly if the blood film shows no evidence of splenectomy (i.e., pitting and Howell-Jolly bodies are absent in the erythrocytes; Chap. 55). Such patients should be screened with sensitive radionuclide or magnetic resonance scans to identify residual or accessory splenic tissue.
Intravenous Immunoglobulin IVIG was first shown to be effective in childhood ITP in 1981,109 then later in adult patients.199 IVIG rapidly increases the platelet count in more than 75 percent of patients with chronic ITP and normalizes the platelet count in approximately 50 percent of the patients.177,178 The effect of IVIG is similar whether or not the patient has undergone splenectomy and is transient, generally lasting only 3 to 4 weeks. Postulated mechanisms for the action of IVIG include blockade of macrophage Fc receptors, which slows clearance of antibody-coated platelets, antiidiotype neutralization of antiplatelet autoantibodies, cytokine modulation, immunomodulation (increased suppressor T-cell function and decreased autoantibody production), complement neutralization, and dendritic cell priming.178,200,201 The recommended total dose of IVIG is 2 g/kg administered either as 0.4g/kg per day on 5 consecutive days or as 1 g/kg per day on 2 consecutive days. If the need to increase the platelet count is urgent, the preferred dosing is 1 g/kg per day for 2 days combined with glucocorticoids.148 For maintenance therapy, 0.5 to 1.0 g/kg as a single dose may be used, administered every 3 to 4 weeks, or as needed. Although the annual total world consumption of IVIG exceeds 100 tons, the cost of IVIG is still high, and this also limits the use of IVIG in adults.202 Adverse effects of IVIG therapy include headache, backache, nausea, fever, aseptic meningitis, alloimmune hemolysis, hepatitis, renal failure, pulmonary insufficiency, and thrombosis. Anaphylactic reactions may occur in patients with congenital IgA deficiency.177 The patient may become refractory to the effect with repeated infusions of IVIG.203 IVIG is used as a first-line therapy in childhood ITP, because the thrombocytopenia is usually transient. In adult ITP, however, IVIG is usually reserved for patients with life-threatening bleeding, when a prompt increase in platelet count is needed,147,148 or as first-line therapy when glucocorticoids are contraindicated.148
Anti-(Rh)D Anti-(Rh)D is a polyclonal γ-globulin containing high titers of antibodies against the Rho(D) antigen of erythrocytes. It is administered intravenously for treatment of ITP. Anti-(Rh)D binds Rh-positive erythrocytes and leads to their destruction in the spleen. Because splenic Fc receptors are blocked, more antibody-coated platelets survive in the circulation.204,205 Anti-(Rh)D also can also modulate Fcγ receptor expression and regulate the production of various cytokines, including IL-6, IL-10, and tumor necrosis factor-α.206 A positive direct antiglobulin test, a decrease in serum haptoglobin levels, and mild and transient hemolysis occur in all Rh-positive patients after anti-(Rh)D infusion, generally without requiring a blood transfusion.205 The rate of serious hemolytic reactions has been estimated as one in 1115 patients; any reaction occurs within 4 hours of administration in almost all cases.207 Anti-(Rh)D therapy is not effective in patients who have undergone splenectomy or in Rh-negative patients, and is not recommended in patients with a positive direct antiglobulin test.148
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


