INTRODUCTION
SUMMARY
Purpura, the clinical manifestation of blood extravasation into mucosa or skin, results from various conditions, including rheumatologic, infectious, dermatologic, traumatic, and hematologic disorders. This chapter does not detail purpura resulting from quantitative or functional defects in hemostasis and coagulation, such as deficiencies of platelets or coagulation factors; these causes are discussed in other chapters (e.g., thrombocytopenia in Chap. 117; coagulation factor deficiencies in Chaps. 123 and 124).
The differential diagnosis of the disparate causes of noncoagulopathic purpura is best approached by stratifying purpura into three types of lesions: (1) palpable or retiform and noninflammatory, such as hyperglobulinemic purpura of Waldenström; (2) palpable or nonpalpable but inflammatory, such as Henoch-Schönlein purpura; and (3) nonpalpable and noninflammatory, such as senile purpura. By accounting for palpability, presence of inflammation, size, and shape, the differential diagnosis of a particular lesion can be significantly reduced.
Acronyms and Abbreviations
ANCA, antineutrophil cytoplasmic antibody; APS, antiphospholipid syndrome; CSS, Churg-Strauss syndrome; DIC, disseminated intravascular coagulation; HCV, hepatitis C virus; HHT, hereditary hemorrhagic telangiectasia; HP, hypergammaglobulinemic purpura; HSP, Henoch-Schönlein purpura; MELAS, mitochondrial encephalopathy, lactic acidosis, stroke-like; SLE, systemic lupus erythematosus; WG, Wegener granulomatosis.
DEFINITION AND DIAGNOSTIC APPROACH
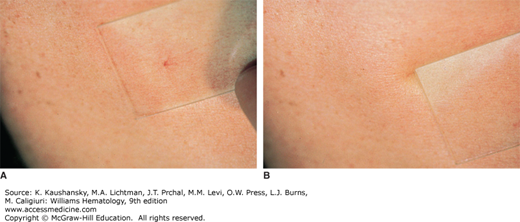
Purpura refers to visible hemorrhage into mucous membranes or skin, which corresponds to extravasation of red blood cells around small dermal vessels and chronic hemosiderin deposition.1 Purpuric lesions, by definition, do not blanch completely upon compression, as opposed to erythema. Blanching is commonly tested by compression of skin lesions with a glass slide, referred to as diascopy (Fig. 122–1). Certain conditions give rise to lesions that mimic purpura with incomplete blanching upon diascopy, but are not purpura because no hemorrhage has occurred. Examples include disorders that impede on the red cell flow, such as tortuous veins.1
Assessing lesion palpability is the first step in evaluating purpuric lesions (Fig. 122–2). The causes for palpability are varied and include fibrin deposition, localized edema, significant cellular infiltration, and subcutaneous extravasation of red blood cells.
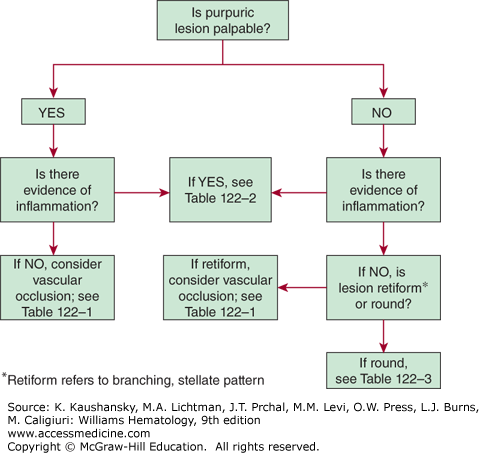
Inspecting the lesion for inflammatory changes is the next step in evaluating purpuric lesions. The presence of pain, erythema, and palpation for warmth and localized swelling are signs of inflammation and suggest a vasculitis or immune complex disorder.
The shape of a purpuric lesion, either round or retiform (branching), is important in assessing the lesion. In the absence of accompanying inflammation, retiform purpuric lesions suggest small vessel occlusion. A retiform, inflammatory purpuric lesion supports the diagnosis of vasculitis as a result of immunoglobulin (Ig) complex formation.2 Small, focal areas of hemorrhage are referred to as petechiae (≤4 mm). Larger lesions are referred to as intermediate or midsize purpura (>4 mm, <1 cm) or ecchymosis (≥1 cm).3
Purpuric lesions frequently appear purple; however, they can take on a variety of colors, according to the age of the lesion and the oxygen saturation of the hemoglobin in the extravasated blood. Ecchymosis usually starts as blue or purple, evolves to a greenish brown (a mixture of blue and yellow), and ultimately changes with variable speed to yellow as hemoglobin degrades to bilirubin.4 These examples of hemorrhage into the dermis must be distinguished from telangiectasia, which are vascular anomalies that blanch with pressure (see Fig. 122–1). Tables 122–1,122–2,122–3 classify the etiologies for purpura discussed in this chapter.
|
|
|
PALPABLE NONINFLAMMATORY PURPURIC LESIONS
See Table 122–1.
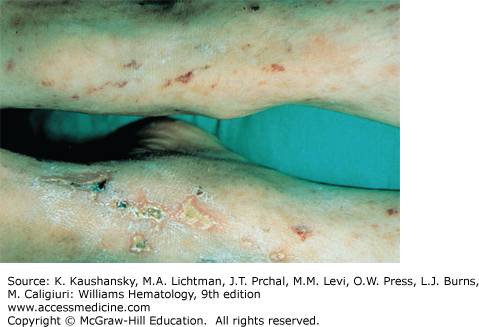
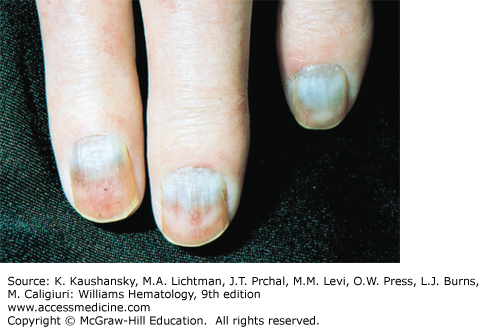
Cryoglobulinemia refers to the presence in plasma of cold-insoluble immunoglobulins,5 and is a secondary finding associated with several disease states. Cryoglobulins are commonly present in low concentrations, therefore approximately 90 percent of patients are asymptomatic or have minimal symptoms.6 Symptoms occur when the abnormal protein precipitates at the temperatures present in superficial venules in the skin and acral parts of the body. Cryoglobulinemia syndromes are divided into three main types based on the immunoglobulin composition of the precipitate. Type I cryoglobulinemia results from the accumulation of monoclonal IgG, IgM, or IgA. It is most commonly seen in association with lymphoproliferative disorders, such as myeloma, Waldenström macroglobulinemia, or lymphoma. Type II, or mixed cryoglobulinemia involves formation of complexes composed of polyclonal IgG with monoclonal immunoglobulins, typically IgM with anti-IgG specificity. Exposure to various exogenous antigens appears to cause polyclonal immunoglobulin production, with activity against bacteria, viruses, and fungi. Mixed cryoglobulinemia is commonly seen secondary to hepatitis C virus (HCV) infection,7 HIV, collagen vascular disorders, and hematologic neoplasias.8,9 In mixed cryoglobulinemia secondary to HCV infection, the presence of active cutaneous vasculitis correlates with increased levels of the B-cell–attracting chemokine 1 (CXCL13).10 This process manifests with petechiae of the legs, palpable purpura, and necrotic skin ulcerations. First-line treatment includes use of interferon-α or other antiviral agents, often with adjunct glucocorticoids or plasmapheresis.11 Direct treatment of the HCV infection with ribavirin, interferon, or other antiviral therapy, such as the protease inhibitors, ameliorates this associated lymphoproliferative disorder.12 Deposition of immune complexes on vessel walls leads to tissue damage in the vasculature, nerves, joints, and skin leading to the hallmark findings of mixed cryoglobulinemia: weakness, arthralgia, and purpura. This purpura often is palpable and is accompanied by areas of hemorrhagic necrosis (Fig. 122–3) and occasionally follicular pustular purpura. Other cutaneous manifestations include lower-extremity ulcerations, urticaria, Raynaud phenomena, and subungual purpura (Fig. 122–4). Type III cryoglobulinemia associates polyclonal IgG and IgM complexes, also resulting in symptoms of mixed cryoglobulinemia.6 It is associated with a variety of infections, systemic lupus erythematous (SLE), and poststreptococcal glomerulonephritis.
A polyclonal increase of immunoglobulins, most commonly IgG1, appears to be responsible for the varied cutaneous findings seen in this hypergammaglobulinemic purpura (HP). Waldenström first described a hyperproteinemic syndrome characterized by hypergammaglobulinemia, recurrent purpura, elevated erythrocyte sedimentation rate, and anemia.13 Most commonly seen in young women, this syndrome is associated with a large number of autoimmune disorders, including rheumatoid arthritis, Sjögren syndrome, SLE, hepatitis C, polymyositis, and sarcoidosis. Discrete to confluent collections of lower limb petechiae are its most common skin findings (Fig. 122–5), but lesions can occur in various body locations.14 Although lesions are usually self-limited and resolve in 7 to 10 days, recurrence of purpura is common and is associated with exposure to cold temperatures or increases in hydrostatic pressure, such as with the use of tight stockings or prolonged standing.15 Clinical manifestations consist of palpable purpura or diminutive macular erythematous lesions occurring on the lower legs. A reticulate pattern of purpura has been described.16 Development of edema and arthralgia has also been described.17
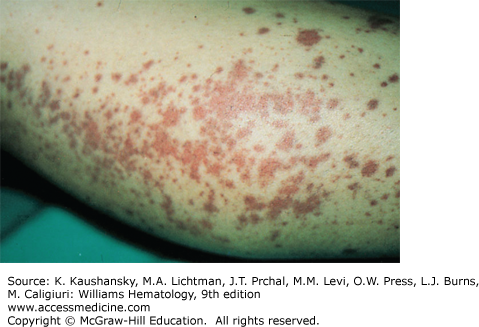
Common histologic findings include perivascular infiltrates, hemorrhage, and vascular necrosis. In addition to a polyclonal increase in either IgA, IgM, or IgG, serology may reveal cryoglobulinemia, rheumatoid factor, or antinuclear antibodies.18 Imbalances in IgG subclass expression, usually because of a decrease in IgG2, appear to be associated with recurrent infections.17 Development of antilymphocyte antibodies results in lymphopenia. Anti-Ro/SSA antibodies occur in up to 78 percent of HP patients, suggesting that screening for anti-Ro/SSA should be considered in cases suspicious for Waldenström.19
Precipitates of immunoglobulin light chains that form crystalline deposits in the skin cause hemorrhagic palpable purpura. A nonamyloid monoclonal light chain of predominant κ type is involved in two-thirds of the cases.20,21 Crystalline deposits are present in the skin and other tissues. Although the clinical presentation may mimic a systemic vasculitis, no histologic signs of inflammation are seen. Light-chain vasculopathy with cutaneous findings has also been described in association with multiple myeloma. Intravascular deposition of crystals containing IgG and λ light chains were found on immunohistochemical analysis and manifested with gangrene of the feet and intestinal perforation.22
First described by Korst and Kratochvil in 1955, cryofibrinogenemia is a form of serum dysproteinemia characterized by formation of an abnormal cold-precipitable fibrinogen. Cutaneous manifestations include cyanosis, erythema, Raynaud phenomenon, and palpable purpura of the nose, ears, and distal extremities.23 Tissue ischemia and gangrene may result. Pathogenesis of cryofibrinogenemia may involve an inhibition of normal fibrinolysis produced by a high plasma level of α1-antitripsin and α2-macroglobulin proteases.24 Cryofibrinogenemia is commonly secondary to thromboembolic disorders, metastatic malignancies, infections, and collagen vascular disease.25 Treatment modalities include avoidance of cold, plasmapheresis, and danazol, an anabolic glucocorticoid, or immunosuppression with glucocorticoids or cytotoxic agents.
Cutaneous reactions to heparin administration vary greatly from a type I urticarial rash to purpuric plaques with cutaneous ulceration or necrosis.26 The syndrome occurs after both subcutaneous and intravenous administration of unfractionated heparin, but it has also been rarely described after low-molecular-weight heparin.27 A delayed-type hypersensitivity reaction to the medication is involved. Skin lesions appear within 1 to 2 weeks after treatment initiation and include necrotic purpuric lesions.28 Development of cutaneous lesions is closely related to heparin-induced thrombocytopenia (Chap. 118), which involves anti–platelet factor 4 antibody–mediated platelet aggregation with development of thrombosis and microvascular occlusion.27
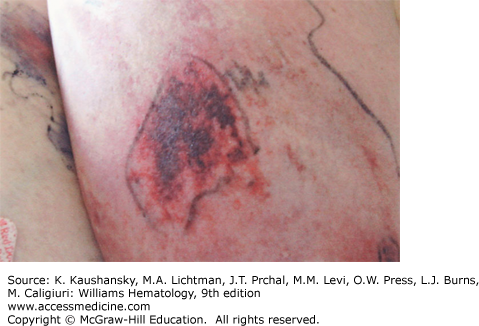
The development of painful erythematous plaques and nodules is a potential complication of warfarin therapy (Fig. 122–6). These lesions can rapidly become hemorrhagic and necrotic, leading to large areas of infarct with black eschar formation and subsequent skin sloughing. Purpura, vesicular, maculopapular, or urticarial eruptions can be encountered.1 Warfarin-induced necrosis has a prevalence between 0.01 and 0.1 percent and presents typically 3 to 10 days after initiation of anticoagulant treatment.29,30 However, an atypical presentation can occur much later, for example, in a patient with protein S deficiency.31,32 Although warfarin necrosis tends to develop in areas of greatest fat deposition, such as breasts, thighs, and buttocks, acral areas, including penis, fingers, and toes, can also be involved.33 Warfarin necrosis results from the rapid decrease of vitamin K–dependent coagulation factors of relatively short half-life, such as proteins C and S, while longer-lasting coagulation factors, such as factor II and factor X, are not yet decreased, resulting in a net procoagulant state. Microvascular occlusion of small dermal and subcutaneous vessels by fibrin deposits is seen on histologic analysis, but true vasculitis is infrequent.29 Treatment involves prompt cessation of the vitamin K antagonist, along with administration of heparin and vitamin K, and occasionally surgical debridement. Because patients with protein C or S deficiency are at increased susceptibility to warfarin necrosis, heparin should always be administered in these patients prior to initiation of Coumadin.34
Figure 122–6.
Coumadin necrosis. Develops in acral areas and areas of fat deposition such as buttocks or breast. Typically, lesions develop 3 to 10 days after initiation of anticoagulant treatment and are caused by rapid clearing of protein C. The lesions are characterized microscopically by small-vessel thrombosis.
Clinical manifestations of proteins C and S deficiencies include venous thromboembolism, warfarin-induced skin necrosis, and neonatal purpura fulminans (Chap. 129). Congenital and acquired deficiencies in these proteins can lead to palpable necrotic purpura and ecchymosis.35,36 Erythematous purpuric lesions associated with homozygous protein C deficiency can develop within hours of birth and can rapidly progress to hemorrhagic necrosis.37 Acquired deficiencies of protein C are associated with autoantibodies to protein C, antibiotics administration, septic shock, HIV, and liver disease (Chap. 127).38 Acquired protein S deficiency may occur after varicella infection, when it is associated with the generation of antiprotein S immunoglobulins.39 Protein repletion with fresh-frozen plasma or protein C concentrate is effective as initial treatment for protein C deficiency to help clear both cutaneous lesions and venous occlusion, while lifelong anticoagulant treatment is used to prevent recurrence.34,40
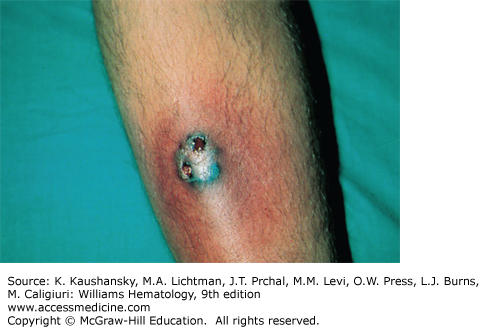
Paroxysmal nocturnal hemoglobinuria (Chap. 40) is a hematopoietic clonal disorder resulting in defective production of cell surface-binding proteins.41 Cutaneous manifestations are secondary to a hypercoagulable state and include palpable purpura, petechiae, ecchymosis, leg ulcers, plaques, necrosis, and hemorrhagic bullae.42 Parvovirus B19 may play an etiologic role in the development of cutaneous necrosis.43 An association with pyoderma gangrenosum (Fig. 122–7)44 and occurrence of purpura fulminans45 have been described. Histology reveals formation of microvascular fibrin thrombi.42
Figure 122–7.
Pyoderma gangrenosum. A large number of systemic diseases are associated with pyoderma gangrenosum, including inflammatory bowel diseases, hematologic and solid malignancies, and rheumatologic disorders. Microscopically, the lesions are characterized by central necrotizing, neutrophilic infiltration, and a surrounding perivascular and intramural lymphocytic infiltration.
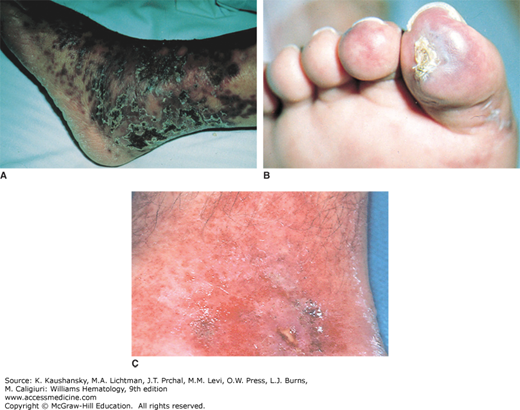
Antiphospholipid syndrome (APS) is a disease characterized by hypercoagulability associated with the presence of antibodies against phospholipids, such as anticardiolipin and lupus anticoagulant (Chap. 131).46 Approximately 40 percent of patients with APS present with cutaneous lesions secondary to both large-vessel and microvascular thrombosis.47 Skin manifestations include ecchymosis, livedo reticularis and racemosa, leg ulcerations, bullae, splinter hemorrhages, livedoid vasculopathy, superficial venous thrombosis, atrophie blanche, and extensive necrosis (Fig. 122–8).47,48 Presence of livedo reticularis is frequently the presenting symptom of APS, most commonly when the syndrome is secondary to SLE, and its presence commonly precedes vascular events.49 Development of acute bullous purpura has been described.50 Treatment includes anticoagulant agents with immunosuppressant administration for associated thrombocytopenia. Prevention of thromboembolic events with aspirin is of uncertain value.51
Figure 122–8.
A. Antiphospholipid antibody syndrome. A number of skin lesions can be seen, including ecchymosis, livedo reticularis and racemosa, leg ulcerations, bullae, splinter hemorrhages, superficial venous thrombosis, atrophie blanche, and, as shown here, extensive necrosis. B. Anticardiolipin antibody. C. Lupus anticoagulant.
Livedoid vasculitis (segmental hyalinizing vasculitis) is a chronic recurrent thrombo-occlusive disorder characterized by the initial development of erythematous purpuric lesions with telangiectasis and peripheral petechiae, and lower-extremity ulcerations. Subsequent healing leads to atrophie blanche, a term that refers to the appearance of ivory-white stellate scars commonly surrounded by hyperpigmented areas and telangiectasia. These lesions appear to be caused by small-vessel fibrin thrombi in the middle and lower dermis as a result of a procoagulant tendency.52 Although most commonly arising without associated cause, livedoid vasculitis is associated with polyarteritis nodosa, APS, and SLE.53,54 Although not consistently beneficial, common therapies include discontinuation of oral contraceptives, anticoagulation and antiplatelet medications, glucocorticoids, and dapsone. Ketanserin, an S2 serotoninergic receptor blocker, psoralen plus ultraviolet A therapy, and intravenous immunoglobulins also have been used successfully.55
Also known as atheroemboli, cholesterol crystal emboli are responsible for a syndrome characterized by lower extremity pain and livedo reticularis with preservation of peripheral pulses. Other common cutaneous findings include gangrene, purpura, ulcerations, cyanosis, and nodules (Fig. 122–9).56 Clinical symptoms include fever, myalgia, and altered mental status. Laboratory features include an elevated erythrocyte sedimentation rate, eosinophilia, and acute renal failure. Onset of symptoms varies from immediate after physical dislodgement of plaque, up to months later when caused by anticoagulant therapy.1 A blue toe syndrome is, in fact, rare, and most atheroemboli are clinically silent.57 Atherosclerotic lesions in the descending aorta are the most common source of cholesterol emboli. This explains the propensity for lower-extremity findings during intravascular procedures or initiation of thrombolytic or anticoagulant therapy.56 Histologic evaluation can offer a definitive diagnosis with findings of intraluminal birefringent cholesterol crystals within blood vessel lumen, in the absence of vasculitis.58 No effective treatment is available. Nevertheless, supportive care with proper hydration and dialysis may lessen the potential for end-organ damage.
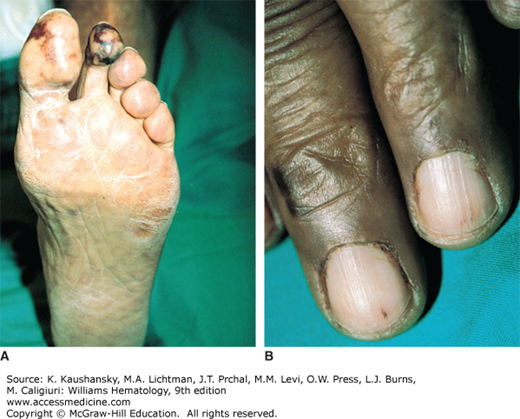
Calciphylaxis (calcific uremic arteriolopathy)59 is a thrombo-occlusive disorder involving formation of cutaneous, subcutaneous, and vascular calcifications. It is most commonly seen in patients with end-stage renal disease, classically caused by the development of secondary hyperparathyroidism.60 Approximately 4 percent of hemodialysis-dependent patients suffer from calciphylaxis. Survival is less than 50 percent at 5 years after diagnosis.61 Other etiologies include primary hyperparathyroidism, malignancy, alcoholic liver disease, and collagen tissue disorders.62 Cutaneous lesions present initially as reddish-purple plaques, evolving to tender, gangrenous ulcers or reticular hemorrhagic necrosis. Treatment involves a combination of medical and surgical interventions, such as parathyroidectomy, renal transplantation, wound debridement, and amputation.61
Acral purpuric lesions secondary to emboli arise from left atrial myxomas or right atrial clots through paradoxical embolization.63 These purpuric lesions include palpable purpura, livedo reticularis, erythematous macules and papules, cyanosis, petechiae, splinter hemorrhages, ulcerations, and cutaneous necrosis. Cyanosis, livedo reticularis, and lower-extremity ulcerations can also be seen.64
Purpuric lesions are not uncommon after arthropod bites. Bites from bed bugs, Cimex lectularius, can give rise to localized purpuric macules or papules, while bites from kissing bugs, Reduviidae, often manifest as urticaria with hemorrhagic bulla.65 Cutaneous findings after envenomation from a brown recluse spider, Loxosceles reclusa, include purpuric necrosis with surrounding erythema evolving to ulcer formation.
PALPABLE AND NONPALPABLE INFLAMMATORY PURPURIC LESIONS
See Table 122–2.
Pyoderma gangrenosum is an idiopathic inflammatory skin condition characterized by early follicular erythematous papules and pustules or tender, fluctuant nodules with surrounding erythema that spread peripherally and ulcerate, surrounded by a violaceous rim (see Fig. 122–7).66 In 50 percent of cases of pyoderma gangrenosum, there is an associated disorder, such as inflammatory bowel disorders (classically ulcerative colitis), arthritis, hematologic disorders, and solid tumors.67
Related posts:
 Hematopoietic Stem Cells, Progenitors, and Cytokines
Examination of the Marrow
Therapeutic Apheresis: Indications, Efficacy, and Complications
Hemolytic Anemia Resulting from Infections with Microorganisms
Structure and Composition of the Erythrocyte
Disorders of Hemoglobin Structure: Sickle Cell Anemia and Related Abnormalities
Hematopoietic Stem Cells, Progenitors, and Cytokines
Examination of the Marrow
Therapeutic Apheresis: Indications, Efficacy, and Complications
Hemolytic Anemia Resulting from Infections with Microorganisms
Structure and Composition of the Erythrocyte
Disorders of Hemoglobin Structure: Sickle Cell Anemia and Related Abnormalities
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


