The Taxanes, Vinca Alkaloids, and Their Derivatives
INTRODUCTION
In the past decade, the taxanes have emerged as one of the most powerful classes of anticancer drugs (1). Two unmodified taxanes, paclitaxel and docetaxel, are approved for clinical use in multiple tumors. An albumin-stabilized paclitaxel (abraxane) is also available for treatment of breast cancer (2), and a new analogue, cabazitaxel, is approved for hormone refractory prostate cancer. Despite their similar structures and a common mechanism of action (disruption of microtubule function), the taxanes differ in their pharmacological profiles, toxicity, and their patterns of clinical activity. Taxanes are predominantly employed in solid tumor chemotherapy in combination with platinum derivatives, with other cytotoxics, or with monoclonal antibodies such as Herceptin (trastuzumab). Both unmodified taxanes act synergistically with trastuzumab against HER2/neu overexpressed breast cancer cells in vitro and in vivo, and the combination of taxane and trastuzumab improves survival against HER2/neu amplified breast cancer in the adjuvant setting. The two original taxanes differ in their interaction with doxorubicin, paclitaxel potentiating the anthracycline’s cardiac toxicity, while docetaxel and doxorubicin are well tolerated and highly active in combination (3). The taxanes are also primary agents for treating other malignancies, including ovarian, lung, and bladder cancer.
A closely related antimitotic agent, ixabepilone, is approved for second-line breast cancer treatment after taxanes, and differs from taxanes in its greater neurotoxicity and its lack of cross-resistance in MDR-positive tumors.
STRUCTURE
Paclitaxel was first isolated from the bark of the Pacific yew, Taxus brevifolia. Paclitaxel and its analogue, docetaxel, are now synthesized from 10-deacetylbaccatin III, a precursor found in the leaves of the European yew, Taxus baccata (4). Both molecules are composed of a 15-member taxane ring system linked to a 4-member oxetan ring at the C-4 and C-5 positions of the molecule. The structures of paclitaxel and docetaxel differ in substitutions at the C-10 ring position and in the configuration of an ester side chain attached at C-13. Docetaxel is slightly more water soluble than paclitaxel and a more potent inhibitor of tubulin in cell-free systems. The side chain substitutions at C-13 position are essential for antimicrotubule activity. The chemical structures of paclitaxel and docetaxel are shown in Figure 3–1. Abraxane is identical to paclitaxel, but is formulated within a microalbumin particle that eliminates the hypersensitivity caused for the lipid excipient used to deliver paclitaxel. Cabazitaxel retains the taxane nuclear ring system but has multiple side chain modifications to increase its solubility and decrease susceptibility to multidrug resistance.
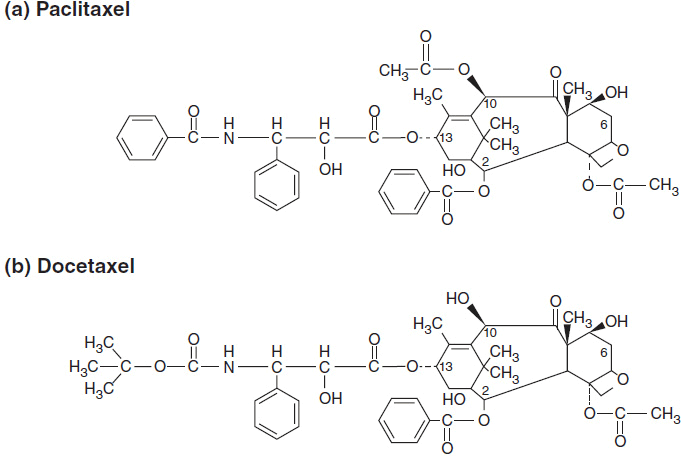
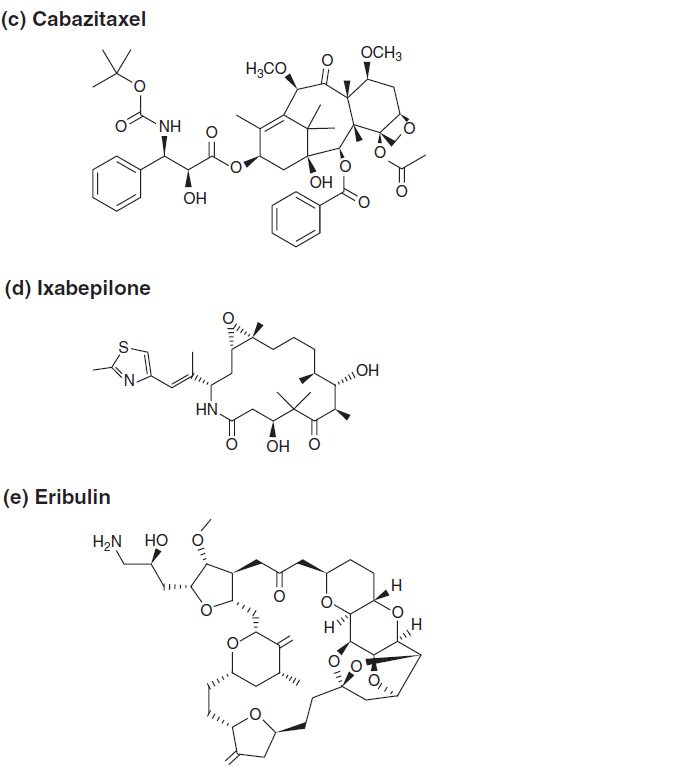
FIGURE 3-1 The chemical structure of antimitotics: (A–C) taxanes, (D) ixabepilone, and (E) eribulin.
MECHANISM OF ACTION
The taxanes stabilize microtubules. They bind to the interior surface of the β-microtubule chain and enhance microtubule assembly by promoting the nucleation and elongation phases of tubulin polymerization. In solution they reduce the critical tubulin subunit concentration required for microtubule assembly. Unlike the vinca alkaloids, which prevent microtubule assembly, the taxanes decrease the lag time to assembly and dramatically shift the dynamic equilibrium from tubulin dimers to microtubule polymers (5).
The taxane binding site on β-tubulin is distinct from those of vinca alkaloids, podophyllotoxin, colchicines, maytansine, and the maytansine-like antimitotic attached to anti CD30 antibody (brevituximab vendotin). Paclitaxel and docetaxel bind reversibly to both the N-terminal residues and the internal amino acid residues at 217–233 positions. This binding increases the rate of tubulin polymerization, disrupts the orderly formation of mitotic spindles and segregation of chromosomes, halts progression through mitosis, and promotes apoptosis. Taxanes block the anti-apoptotic effects of the BCL-2 gene family, and induce p53 gene activation with consequent mitotic arrest, formation of multinucleated cells, and cell death.
In addition to their direct cytotoxic effects, taxanes potently inhibit vascular endothelial cell proliferation, and enhance the cytotoxic effects of radiation at clinically achievable concentrations.
DRUG RESISTANCE
Two major mechanisms of taxane resistance have been characterized in cells selected in vitro (6). Taxanes are one of many natural product drugs affected by multidrug resistance (MDR) as mediated through increased expression of the 170-kD p-glycoprotein, an efflux pump encoded by the MDR-1 gene. The p-glycoprotein promotes rapid efflux of taxanes, anthracyclines, and vinca alkaloids, as well as other natural products. MDR resistance can be reversed in vitro and in animal test systems by calcium channel blockers, tamoxifen, hormones, cyclosporine A, and even cremaphor, the principal lipid used to formulate paclitaxel. The precise role of MDR-1 in conferring resistance to the taxanes in the clinical setting is not firmly established. For example, clinical observations to date suggest that in breast cancer, there is incomplete cross-resistance between taxanes and anthracyclines, implying that MDR-1 expression is not responsible for drug resistance in all cases. A second form of resistance to taxanes is seen in cells that express an altered β-tubulin phenotype, either through mutations or due to minor polymorphisms that modify taxane binding. Paclitaxel-resistant, β-tubulin mutant cells have an impaired ability to polymerize tubulin dimers into microtubules. Amplification of β-tubulin encoding genes, mutation of the β-tubulin binding sites, and isotype switching of β-tubulin all have been reported in taxane-resistant cell lines.
An additional mechanism responsible for taxane resistance has been attributed to increased expression of MCL-1 and BCL-2, both of which inhibit apoptosis. Ixabepilone and cabazitaxel are less susceptible to MDR-mediated resistance, as compared to the original taxanes. β-Tubulin mutations have been linked to ixabepilone resistance in preclinical experiments.
CLINICAL PHARMACOLOGY AND METABOLISM
The taxanes are active only in their parent form, and all are administered intravenously. Oral bioavailability of either paclitaxel or docetaxel is poor due to high-level expression of p-glycoprotein and other ATP-binding cassette (ABC) transporters in intestinal epithelium, and first-pass drug metabolism in the liver.
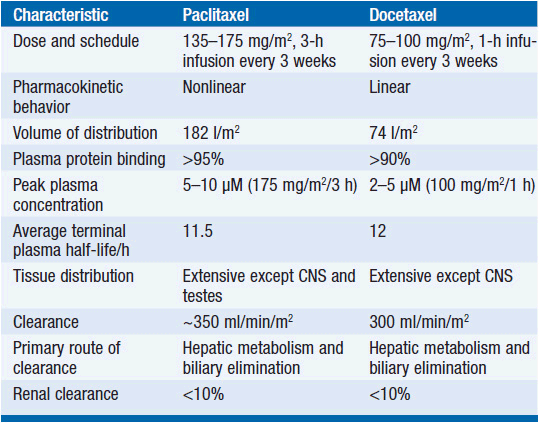
The metabolism of all clinically approved taxanes is mediated through hepatic cytochrome p450 mixed-function oxidases. Paclitaxel is inactivated to hydroxylated metabolites through stepwise catalysis by cytochrome 2C8, producing 6α-OH, and CYP3A4, producing 6α-OH-3′OH, and finally to the dihydroxyl product (7). Docetaxel is oxidized at C13 by CYP3A4. The involvement of cytochrome enzymes in taxane biotransformation has two important implications: first, co-medications capable of inducing or inhibiting cytochromes influence the rate of inactivation and the metabolic fate of taxanes (8). Second, polymorphisms affecting enzymatic function have been described for both CYP2C8 and CYP3A4, thereby leading to interpatient variability of pharmacokinetics. Pharmacokinetic data for paclitaxel and docetaxel are shown in Table 3-1.
 PACLITAXEL
PACLITAXEL
Pharmacokinetic studies have disclosed substantial interpatient variability and nonlinearity of the relationship between paclitaxel dose and drug concentration in plasma (Table 3-2). Nonlinearity is particularly prominent with shorter (1- to 3-h) drug infusion schedules, and may indicate variability of tissue binding and clearance mechanisms.
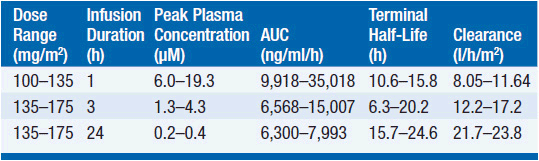
The pharmacokinetics of this drug have been evaluated in doses ranging from 100 to 300 mg/m2 infused in time periods of 1, 3, and 24 h. Following intravenous administration, the drug exhibits a biphasic decline in plasma concentration, reaching peak concentrations between 5 and 10 μM for 1-3 h infusions, and remaining in the inhibitory range for myelopoiesis (above 50 nM) for 12–24 h. Both the terminal half-life of 10-24 h and the mean clearance of paclitaxel appear to either remain unchanged or slightly increase as the infusion time is increased. Approximately 80% of paclitaxel is excreted in feces in the form of CYP2B8 and 3A4 metabolites, the 6α-hydroxy-paclitaxel, the C3′-hydroxy paclitaxel, and the dihydroxy products accounting for the bulk of the dose. Renal clearance of paclitaxel and its metabolites is minor, accounting for about 15% of administered dose and only 5% is excreted unchanged. The dose should be reduced by 50% in patients with a bilirubin greater than 1.5 mg/dl, and the drug should be withheld in patients with severe hepatic dysfunction.
 DOCETAXEL
DOCETAXEL
The pharmacokinetic behavior of docetaxel on a 1-h schedule at doses of 75–115 mg/m2 or less displays a linear relationship between dose and drug concentrations in plasma. The terminal half-life is about 17 h. As with paclitaxel, docetaxel is cleared by CYP-mediated metabolism and is widely distributed among tissues except for central nervous system.
DRUG INTERACTIONS
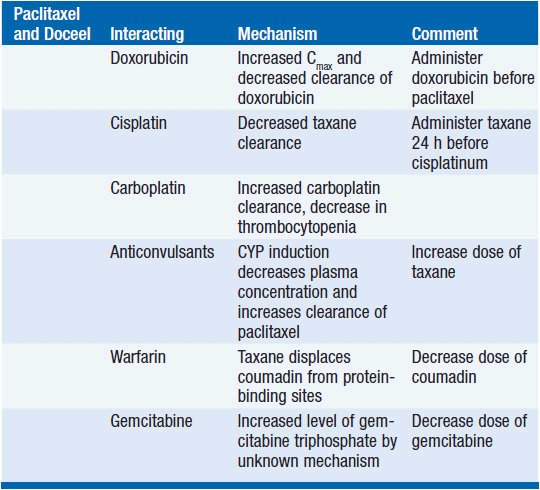
Because of its reliance for clearance upon the cytochrome system, taxanes have pharmacokinetic interactions with other cancer drugs (Table 3-3).
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


