4.1
Introduction
The lineage relationships and kinetics of the cellular components of bone are the focus of this chapter. Tissues in mammals are generally maintained by one of two schemas, mature cell replacement by mature cell division or mature cell replenishment from a more immature, stem or progenitor population. It is the latter model that applies to bone, and we will review here the definition of the skeletal stem cell (SSC), its relationship to descendent semicommitted osteoprogenitors and the terminally differentiated osteogenic cells that make up the mature bone. In addition, we will discuss the molecular and functional heterogeneity of SSCs in adult bones, their migration and kinetics in regulation of lifelong bone production, and the interaction of bone cells with hematopoietic cells in the bone marrow (BM).
4.2
Defining skeletal stem cells
The human skeleton is formed during embryogenesis and is estimated to undergo complete turnover four to five times in an average human lifetime . Lifelong maintenance of human skeletal tissues requires a continuous supply of bone-forming cells, which is thought to be accomplished by a pool of SSCs. SSCs in adult bones are known to be a slow-cycling heterogeneous population that resides in multiple locations of bone and BM. Upon stimulation, they orchestrate and supply various cell types in skeletal tissues that include cartilage chondrocytes, bone-forming osteoblasts, marrow adipocytes, and marrow stromal cells. Although in vivo identity and distinctive properties of SSCs in different tissue locations are not well understood, the primary function of SSCs in postnatal bones is a reservoir of bone-forming cells for bone growth, bone remodeling (lifelong bone turnover), and bone repair. Therefore SSCs like other tissue stem cells are defined by two cardinal features in vivo: their ability to self-renew, thereby providing durable replenishment of themselves and their descendants, and their ability to differentiate to achieve specific mature cell fates.
The most rigorous experimental definition of SSCs requires that they achieve these two cardinal functions in vivo and on a clonal (single-cell) level. This has been achieved with relatively few stem cell types to date and is best exemplified by the hematopoietic system. In that context, single cells have been shown on transplantation to repopulate all blood lineages in a recipient, including the production of new stem cells that can repopulate a second recipient. It is serial transplantation that is used as an indicator of in vivo self-renewal. The accomplishment of the experimental definition of a stem cell is not possible in humans, and therefore much of what we understand about stem cells in general and SSCs are particularly derived from animal models. The mouse has been the most common animal model used either by examining mouse cell functions in vivo or studying human cells transplanted into immunodeficient mice. The information discussed in this chapter should therefore be viewed with these caveats in mind and recognition that not all conclusions that will be presented from experimental work can be rigidly assumed to hold true in the human.
It is important to also realize that the term “stem cells” is often applied to cells that have not met the rigorous definitions indicated above. This is particularly problematic with bone- or tissue-derived mesenchymal cells (MCs) where the term “mesenchymal stem cells” (MSC) has been applied to a wide range of human and mouse cells. Many cells called MSCs have been so labeled because of their functions in vitro, not in vivo. Such assessment may or may not reflect the cell’s function in the body as MCs are notoriously plastic when cultured. A note of caution to the reader then on equating an MSC with true, functionally defined SSCs: these cells cannot and should not be assumed to be the same. MCs that are grown ex vivo have extensive medical testing and interest but lack rigorous confirmation that they reflect an in vivo stem cell population, they will be termed here as MCs.
When considering SSCs, it should also be recognized that there may be different types or subsets of SSCs. Multiple cell types comprise the bone, and they may be derived from shared or distinct stem cell types. For example, stem cells may produce all the mesenchymal lineages of the bone, as is proposed for putative “MC” or they may produce only a single-cell type, such as osteolineage cells. In this chapter, we will define an “SSC” as a cell that is harvested from bone and meets the experimental criteria for a stem cell in vivo. Where rigorous experimental definition is lacking, we will point out the limitations. The cells will often also be further delineated as “multipotent SSCs,” those capable of giving rise to two or more skeletal tissues, including osteogenic, adipogenic, chondrogenic, and BM stromal cells, or “osteogenic-SSCs,” those restricted to osteogenic differentiation, but maintaining stem cell features.
The distinction between stem cells and progenitor cells also merits comment. Progenitor cells are often highly proliferative components of a tissue that are intermediate in their differentiation state. They are destined to differentiate and do not generally have self-renewal capability. Progenitor cells are therefore distinct from stem cells in their self-renewing ability. Because they are intermediate in differentiation state and are often capable of rapid proliferation, progenitors are referred to as a transit, amplifying population. Clear boundaries between the stem and progenitor pools of cells exist conceptually but are often difficult to define experimentally or by cell markers; therefore it is common for primitive cells to be clumped together in one heterogeneous description as stem/progenitor cells.
4.2.1
Developmental origin of bone and skeletal stem cells
The skeletal system derives from the mesodermal layer of the embryonic disc. Intraembryonic bone is formed either by direct ossification of embryonic connective tissue (intramembranous ossification) or by replacement of hyaline cartilage (endochondral ossification) . Intramembranous ossification forms the skeletal infrastructure of the head (roof of the skull and most bones of the face) and begins in the primordial mesenchyme. As the primordial mesenchyme becomes increasingly vascularized, proliferating MCs give rise to osteoprogenitors, which subsequently develop into mature osteoblasts. As osteoblasts aggregate, they begin to lay down osteoid, an organic unmineralized portion of bone matrix. The osteoid is then mineralized with hydroxylapatite crystals brought by the blood vessels in a centrifugal manner beginning at the center of ossification. Concurrently, the spicules of spongy bone develop and group together to form the trabeculae. As the bone compacts, osteoblasts become entrapped in the newly formed bone and are then referred to as osteocytes. Simultaneously, osteoprogenitor cells at the leading edges of separate membranous bones start to proliferate and develop the growing osteogenic fronts. When the osteogenic fronts of two bones are adjacent to each other, they develop fibrous cellular joints called sutures where undifferentiated MCs and SSCs reside in the center of suture and contribute to postnatal bone formation in the expanding skull.
Endochondral ossification takes place in most mammalian bones, including the base of the skull, vertebrae, hips, and limbs through the replacement of a cartilaginous rudiment with bone. In mice the process begins with condensation of lateral plate mesodermal cells expressing Prx1, which initiates limb bud formation at embryonic day 9 (E9) . On E10 a subset of limb bud MCs begins to express Sox9 and undergoes chondrogenic differentiation to form the cartilage primordia and perichondrium . These Prx1 -Cre and Sox9 -Cre marked cells become all limb MCs, including chondrocytes, perichondrial cells, osteoblasts, and stromal cells at later stage. On E12.5–13.5, chondrocytes in the center of the cartilage primordia stop proliferation and become hypertrophic chondrocytes and simultaneously the surrounding chondrocytes also develop a hierarchical columnar structure originating at the primary growth plate. At this time a subset of perichondrial progenitor cells adjacent to hypertrophic chondrocytes begins osteogenic differentiation and produces bone matrix making the bone collar as a support structure for the elongation of the cortical bone. Simultaneously, perichondrial cells in the outer layer of the bone collar develop a primitive periosteum. Subsequently, vessel endothelial cells and a subset of periosteal progenitor cells that can be labeled by osterix ( Osx -CreER) respond to vascular endothelial growth factor (VEGF) signaling from hypertrophic chondrocytes, migrate into the central areas of hypertrophic chondrocytes, and form a primary ossification center. Certain areas of the calcified matrix disintegrate and open cavities where hematopoietic cells and stromal cells begin to infiltrate through blood vessels and develop BM structure (E15.5–16.5). These Osx + progenitors in the BM are transiently present during embryogenesis (as early as E13.5), contributing to nascent bone tissue and stromal cells that are replaced in the adult marrow (~13 weeks of age) . When Osx + progenitor cells are pulse labeled on postnatal day 5, these cells initially contribute a significant portion of osteogenic cells, and slowly transition to stromal cells . Lepr + SSCs, on the other hand, appear first at E17.5 and are localized to the outer bone surface and trabecular bone regions. Unlike Osx + progenitor cells, Lepr + SSCs are present but do not make a significant contribution to osteogenic differentiation until the end of skeletal growth (2 months, 10% preosteoblasts and mature osteoblasts) . Thus during embryonic skeletal development and early postnatal bone growth, there is a transition between developmental SSCs and postnatal SSCs, and several postnatal SSC populations remain and perform specialized functions for skeletal development and maintenance. Once bone growth is completed, multiple postnatal SSC subsets reside not only in the BM but also in the periosteum and sutures and continuously contribute to remodeling of the skeleton throughout life . We will mention the periosteum and adult stem cell heterogeneity in later sections.
4.2.2
Isolation and identification of skeletal stem cells
The first experiments demonstrating that postnatal SSCs might exist were performed in the late sixties, showing that transplantation of BM-derived cells in heterotopic sites was capable of generating a bone template that was then repopulated by marrow cells, thus recapitulating embryonic development . In 1970 Friedenstein et al. identified that osteogenic potential was specifically attributable to a small subset of BM cells that were adherent, spindle shaped, and capable of forming fibroblast colonies (colony-forming unit–fibroblast, CFU-F) . Much like studies exploring hematopoietic stem and progenitor cells (HSPCs) at the time, the identification of a CFU-F allowed for the establishment of clonogenicity, demonstrating that resultant experimental effects were due to a single cell. These cells, after in vitro culture forming CFU-F, were implanted subcutaneously in mice and demonstrated the ability to produce bone (ossicles) . These cells were also capable of serial transplantation into mice with the formation of new ossicles, presumably demonstrating their self-renewal ability . More recently, it has been demonstrated that the cell type in the human that forms colonies, can be transplanted and generates bone and BM and is capable of secondary colony formation, is marked by the expression of the antigen, CD146 . These cells were capable of serial transplantation and ossicle formation at heterotopic sites at a clonal level in immunodeficient mice . This is the most compelling evidence for an SSC in humans. The only caveat in interpreting these studies is that the cells were assessed after culture rather than as freshly isolated cells. It is therefore possible that characteristics of the cells changed during the time of in vitro culture. Also of note, the cells that were generated both in vivo and in vitro were of the osteolineage. Whether the cells also made other MC types in vivo was not assessed, and therefore it would be proper to consider the CD146 + cell as an osteogenic-SSC.
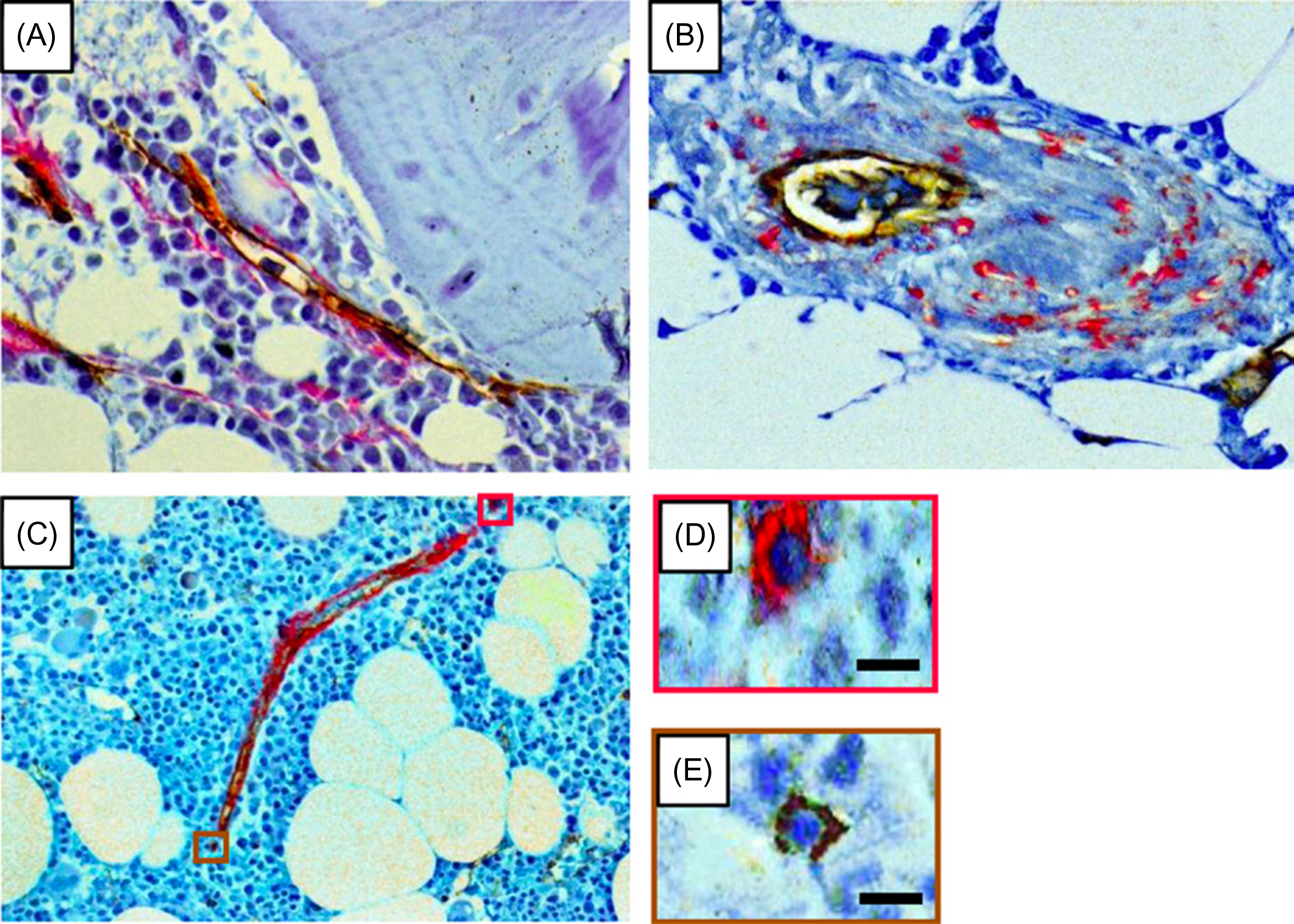
The use of multiple antigenic markers to identify cells selectively is extremely important for studies of stem cell function and number. The study of CD146 + cells therefore represents an important advance in that it validated the functions of the antigen-bearing cells. Other markers have also been studied, although with perhaps less rigor and are summarized in Table 4.1 . The reader is reminded that the antigens studied have enabled fractionation of BM cells to enrich for cells with SSC features by flow cytometric cell sorting. However, there is not a clear method by which to purify SSC to unity (each cell having SSC features). Some of these markers have also been used to evaluate the location of SSCs in bone by immunohistochemistry. For example, human cells expressing STRO-1 , CD105 , CD146, and CD271 surround blood vessels in vivo ( Fig. 4.1 ). A perivascular location for a subset of SSCs in BM is now an accepted concept in the field. It has raised the issue of whether SSCs are equivalent to, or are a subset of, so-called pericytes. It has also raised the issue of whether the cells are capable of trafficking into and out of the vasculature.
| Markers | Molecules | References (+) or (−) |
|---|---|---|
| CD4 | T4 | (−) |
| CD5 | LEU1 | (−) |
| CD9 | Nondescript tetraspanin | (+) |
| CD10 | Membrane metalloendopeptidase (neprilysin) | (+) |
| CD11a | Integrin alpha L chain | (−) |
| CD11b a | Integrin alpha M chain | (−) |
| CD13 | Aminopeptidase N | (+) |
| CD14 a | Monocyte differentiation antigen | (−) |
| CD15 | Fucosyltransferase 4 | (−) |
| CD18 | Integrin beta 2 chain | (−) |
| CD25 | Interleukin 2 receptor alpha | (−) |
| CD29 | Integrin beta 1 chain | (+) |
| CD31 | PECAM | (−) |
| CD34 a | Nondescript sialomucin | (−) (−/+) |
| CD38 | Cyclic ADP ribose hydrolase | (−) |
| CD44 | Hyaluronic acid receptor | (+) (−) b |
| CD45 | PTPRC | (−) (dim) |
| CD49a | Integrin alpha 1 chain | (+) |
| CD49b | Integrin alpha 2 chain | (+) (low) |
| CD49c | Integrin alpha 3 chain | (+) |
| CD49d | Integrin alpha 4 chain | (−) |
| CD49e | Integrin alpha 5 chain | (+) (low) |
| CD50 | ICAM3 | (−) |
| CD51 | Integrin alpha V chain | (+) |
| CD54 | ICAM1 | (+) (−) |
| CD56 | NCAM1 | (+) (−) |
| CD58 | LFA-3 | (+) |
| CD59 | MIRL; protectin | (+) |
| CD61 | Integrin beta 3 chain | (+) (low) |
| CD62E | E-selectin | (−) |
| CD62L | L-selectin | (+) |
| CD62P | P-selectin | (−) |
| CD63 | TSPAN30 | (+) |
| CD71 | Transferrin receptor | (+) |
| CD73 a | 5′-Nucleotidase, ecto | (+) |
| CD90 a | Thy-1 cell surface antigen | (+) (low) |
| CD102 | ICAM2 | (+) (−) |
| CD104 | Integrin beta 4 chain | (+) (low) |
| CD105 a | Endoglin | (+) (low) |
| CD106 | VCAM1 | (+) |
| CD109 | Nondescript GPI-linked glycoprotein | (+) |
| CD117 | C-kit; stem cell factor receptor | (−) (−/+) |
| CD119 | IFNGR1 | (+) |
| CD120a | Tumor necrosis factor receptor superfamily, member 1A | (+) |
| CD120b | Tumor necrosis factor receptor superfamily, member 1B | (+) |
| CD121a | IL-1R | (+) |
| CD123 | IL-3Rα | (+) |
| CD124 | IL-4R | (+) |
| CD126 | IL-6R | (+) |
| CD127 | IL-7R | (+) |
| CD130 | Interleukin 6 signal transducer (gp130); oncostatin M receptor | (+) |
| CD133 | Prominin 1 | (−) (+) c |
| CD140a | PDGFRα polypeptide | (+) |
| CD140b | PDGFRβ polypeptide | (+) |
| CD144 | Cadherin 5, type 2 (VE-cadherin) | (−) |
| CD146 | MCAM | (+) (−) |
| CD164 | Endolyn | (+) |
| CD166 | ALCAM | (+) |
| CD172a | SIRPA | (+) |
| CD178 | Fas ligand | (−) |
| CD200 | Orexin (OX-2) | (+) |
| CD235a | Glycophorin A | (−) |
| CD271 | NGFR | (+) |
| CD309 | VEGF-R2 | (+) |
| CD340 | Erythroblastic leukemia viral oncogene homolog 2 (ERBB2) | (+) |
| α-SMA | α-Smooth muscle actin | (+) |
| ALDH | Aldehyde dehydrogenase | (bright) d |
| CNN1 | Calponin | (+) |
| CDCP1 | CUB domain-containing protein 1 | (+) |
| D7-FIB | Orphan antigen (fibroblast) | (+) |
| DES | Desmin | (+) |
| EGFR-3 | Epidermal growth factor receptor 3 | (−) |
| FGFR | Fibroblast growth factor receptor | (+) |
| GD2 | Ganglioside | (+) |
| HSP90β | Heat-shock protein 90 beta | (+) |
| NES | Nestin | (+) |
| SSEA4 | Stage-specific embryonic antigen | (+) |
| Stro-1 | Orphan antigen | (+) |
| TGF-βIR | Transforming growth factor receptor beta I receptor | (+) |
| TGF-βIIR | Transforming growth factor receptor beta II receptor | (+) |
| TNAP | Tissue nonspecific alkaline phosphatase | (+) (−) e |
| VIM | Vimentin | (+) |
| vWF | Von Willebrand factor | (−) |
| W5C5 | Orphan antigen | (+) |
a Markers as part of the International Society for Cellular Therapy recommendations .
b CD44—Qian et al. argue that primary SSCs do not express CD44 and that the CD44 negative fraction contains the clonogenic activity and that in vitro culture results in expression of CD44.
c CD133—Gindraux et al. suggest that native SSCs express CD133 while cultured SSCs lose expression.
d ALDH—Assessed using Aldefluor staining for enzyme activity.
e TNAP—Kim et al. suggest that true multipotent SSCs are TNAP negative, and as SSCs progress down osteogenesis they express TNAP.
More recently, another subset of adult human SSCs (hSSCs) has been identified and is uniquely different than the previously reported populations. These nonhematopoietic (CD45 − CD235a − ), nonendothelial (Tie2 − CD31 − ) CD146 − hSSCs (PDPN + CD146 − CD73 + CD164 + ) cells isolated from the femoral head or BM aspirates are not only self-renewable but also generate the aforementioned CD146 + hSSC population in vitro and in vivo. Furthermore, the CD146 + hSSC population is self-renewable but is unable to generate the CD146 − hSSC population in vivo. Nevertheless, upon transplantation CD146 − and CD146 + hSSCs differentiate into chondrocytes, osteoblasts, and stromal cells making this population an osteochondrogenic-SSC . Of note, there is probably an additional multipotent SSC population since these SSCs were lineage restricted to chondrocytes, osteoblast, and stromal cells but lacked the ability to form adipocytes.
As might be expected from the interest in SSC and the relative ease of using the model, much work has been done on the identification of SSCs in the mouse. In one such example, green fluorescent protein (GFP) was driven by the type I collagen promoter in a genetically engineered mouse. Whole BM taken from the animal was transplanted into lethally irradiated recipient mice to test whether transplanted SSC could engraft in a manner similar to blood stem cells . GFP positive cells were tracked within the bone of the transplanted animals and could be seen lining endosteal and trabecular bone, suggesting that SSC transfer had occurred. There was no evidence for the cells progressing to osteocytes and when the cells were cultured, they did not form mineralized bone nodules, but these studies were indicative of a transplantable osteolineage cell . Others have used flow cytometry and subsequent ectopic transplantation (e.g., kidney capsule) to define subpopulations of BM cells and to evaluate their ability to form osteoblasts and adipocytes as evidence for SSC. Using the markers, PDGFRα + , Sca-1 + , CD45 − , and TER119 − , it was possible to demonstrate that the Fluorescence-activated cell sorting (FACS)-isolated population could engraft and form osteogenic and adipogenic tissue, again supporting the notion of a transplantable SSC . Subsequent studies have identified that long-term repopulating SSCs were a subset of BM perivascular cells and can be prospectively isolated by their expression of CD140a (PDGFRα), V-cam1, and CD200 in mice . Other independent studies revealed that the Nestin -GFP + ( Nestin + ) stromal cells in mouse BM (Nestin + CD31 − CD45 − ) contain clonal sphere-forming ability in nonadherent culture conditions and long-term repopulation and bone ossicle forming ability after serial transplantation . More recently, additional studies have identified multipotent SSCs and hierarchical populations using the markers, CD45 − Ter119 − Tie2 − AlphaV + Thy − 6c3 − CD105 − CD200 + (CD200 + ). This CD200 + population is capable of self-renewal as well as generating two additional multipotent skeletal stem/progenitor cell (SSPC) populations, a pre–bone, cartilage, and stromal progenitor (pre-BCSP; CD45 − Ter119 − Tie2 − AlphaV + Thy − 6c3 − CD105 − CD200 − ) and the bone, cartilage, and stromal progenitor (BCSP; CD45 − Ter119 − Tie2 − AlphaV + Thy − 6c3 − CD105 + ) cell populations capable of self-renewal. Transplantation of the CD200 + , pre-BCSP, or BCSP cell populations generated bone, cartilage, stromal progenitors and BM stromal capable of supporting hematopoiesis . Although these studies clearly defined the presence and markers of multipotent SSCs during embryonic bone development and early postnatal stage, they raise an issue of whether such SSCs are prolonged throughout life or become more lineage-restricted SSCs in adult bones. Further, it should be noted that many of these experimental conditions require ectopic transplantation of sorted cells under kidney capsule or mouse fat pad, and therefore it is not clear whether endogenous SSCs have such multipotential function during bone regeneration and repair.
4.2.3
Heterogeneity of skeletal stem cells in adult bones
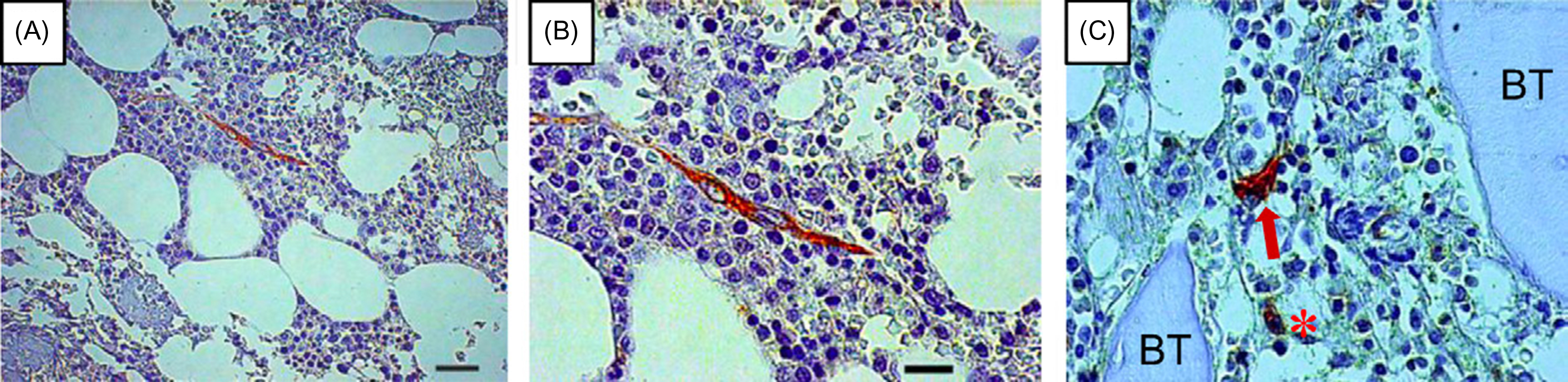
The existence of diverse stem cell pools characterized by distinct molecular markers and context-dependent functions has been reported in multiple tissues . Tissue-resident SSCs are also known to be a heterogeneous population with different locations . In mouse BM, attempts have been made to use in vivo labeling of defined subsets of MCs to track whether they are capable of functioning as a distinct SSC. These so-called lineage tracing studies have indicated that BM perivascular cells with characteristics of SSCs can be genetically marked by Cxcl12 -DTR-GFP , Nestin -GFP , Prx1 -Cre , Lepr- Cre , or Mx1 -Cre . Several of these perivascular SSPCs can be further identified by their association with arteriolar ( Nestin -GFP high ) or sinusoidal ( Lepr + , Nestin -GFP low ) vessels. These BM perivascular cells exhibit all SSC characteristics and express high levels of hematopoietic stem cell (HSC) niche factors, and their ablation impairs the osteogenic and adipogenic differentiation potentials of BM cells . Interestingly, when the progeny of adult Nestin-expressing BM cells were tracked by using a genetically engineered mouse, the cells were found to contribute to skeletal remodeling through differentiation into cartilage- and bone-forming cells . However, their contribution to osteoblasts and chondrocytes required 8 months to be visualized, suggesting that these cells may be quiescent and that there may be distinct stem/progenitors that more readily and rapidly contribute to osteogenesis. Perivascular Nestin + cells have also been described in histological sections of human BM biopsies ( Fig. 4.2 ). Like Nestin + cells, Lepr -labeled cells are present perinatally but do not have a significant contribution toward osteogenic differentiation until skeletal growth (~8 weeks of age). Unlike Nestin + cells, Lepr + cells do not contribute to cartilage-forming cells, but rather most postnatal osteoblasts, BM stromal cells, and adipocytes . Mx1 is an interferon-inducible promoter that has been used to label cells that contribute to durable osteogenesis over time and to be serially transplantable. This may, therefore, also be a marker for endogenous SSCs . A difference between these two models is that, unlike the delayed appearance of osteoblastic progeny from Nestin – or Lepr -labeled BM cells, Mx1 -labeled cells can rapidly replenish the vast majority of osteogenic cells and can do so durably over time . Therefore Mx1 -labeled cells may be more rapidly dividing cells. However, Mx1 -labeled cells do not appear to contribute to chondrogenesis, suggesting that Mx1 can label active osteogenic-SSCs that supply the majority of new osteoblasts over time, whereas Nestin may mark more quiescent, multipotent SSCs. Interestingly, a nonperivascular SSC population with in vivo osteogenic and chondrogenic potential that is distinctly different from perivascular SSCs is labeled by Gremlin 1 (Grem1) . Another population of cells, including Gli1 -Cre , Sox9 -CreER , Acan -CreER , and Col 2-CreER , is present in the metaphyseal growth plate.
Similar to BM, osteo/chondrogenic progenitors have also been found in the periosteum . These periosteal progenitors can be labeled with early mesenchymal lineage markers such as Prx1 -Cre, Axin2 -CreER, and Ctsk -Cre during fetal bone development and contribute to outer bone shaping, cortical thickness, and fracture repair . In addition, these periosteal cells are particularly important in the lifelong regeneration of bones with limited or no BM, suggesting that SSC subsets reside in the adult periosteum and that heterogeneous populations of SSCs are regionally distributed throughout the adult skeleton to cooperatively support skeletal maintenance. Heterogeneity among stem cells within other tissues has also been recently defined and may reflect a general principle for how the stem cell compartment has distinctive subsets of cells performing related, but different functions.
4.3
Migration of skeletal stem cells
If SSCs are capable of migration, they may participate in bone repair at a distance, opening the possibility of a systemically administered SSC therapy. As early as the beginning of the 20th century, the presence of circulating mesenchymal progenitors was suggested by the presence of CFU-F in the peripheral blood (as reviewed in Ref. ). Numerous reports confirmed the presence of circulating fibroblastic progenitors in mouse , adult humans , and human umbilical cord blood . One study reported that some of these circulating cells express osteocalcin and form bone nodules in vitro, although their functional contribution to bone remodeling in vivo is not clear . Understating the molecular signals that direct circulating mesenchymal progenitors to target tissues is of great interest. Several factors that induce SSPC migration have been identified from the resulting bone tissue response to fracture. During the normal progression of bone healing, several inflammatory factors [interleukin (IL)-1β, IL-6, TNF-α, receptor activator of nuclear factor-κB ligand (RANKL), stromal-derived factor (SDF)-1, and transforming growth factor beta (TGF-β) superfamily bone morphogenetic proteins (BMP-2, BMP-4, BMP-6)] are released during the inflammatory phase (as reviewed in Ref. ). Several studies have evaluated MSC circulatory and homing capabilities. Specifically, in a study where skin flaps between animals are joined, so-called parabiotic animals, it is possible to test if cells from one animal can traverse the circulatory system and populate in the parabiotic partner. This experimental system was used to test whether osteogenic stem/progenitors can traffic from one animal to a BMP-2 treated collagen scaffold implanted in the partner . Another known BM MSC chemoattractant is SDF-1, which is necessary for homing of BM stem cells to injuries and endochondral bone repair . When femoral critical size defects were implanted with tissue grafts expressing SDF-1 and BMP-2 there was a significant greater homing ability of intravenously injected MSCs to injury sights than SDF-1 or BMP-2 alone . However, these studies used the transplanted SSCs and their contribution to the formation of ectopic bone induced by implantation of a BMP-2-containing collagen pellet or injury. In addition, a recent study reported that this systemic recruitment of endogenous SSCs does not occur during bone fracture healing and their recruitment is mainly mediated by local translocation. Therefore whether endogenous osteogenic stem/progenitor cells move via the circulation and respond to bone defects remains a question.
In addition to intravascular migration of osteogenic-SSPCs, there is supporting evidence for local migration of SSPCs. In human, many cell migration experiments are conducted in vitro, making extrapolation to an in vivo system challenging. To date the majority of in vivo based assessments of SSPC migration has been conducted in mice. In one such study the release of TGF-β during bone resorption has been shown to induce the migration of SSPCs to bone resorptive sites , suggesting the breakdown of the bone matrix can also induce SSC migration. Due to recent advances in live animal, intravital microscopy and genetic labeling of SSCs in reporter mice, in vivo tracking of individual cells is now possible. In the adult mouse, periosteal SSCs localize to niches in the calvarial sutures. Using in vivo intravital microscopy, we have tracked the expansion of suture SSCs 24- and 48-hour post calvarial injury. We observed significant induction of migrating SSCs out of the coronal suture and toward the injury sites . Within 4–7 days, SSCs localize and repopulate the outer layer of the soft callus . Therefore tissue-resident SSCs indeed play an integral role in skeletal maintenance, and their early response to injury signals and subsequent local migration is a key mechanism for the initiation of bone injury repair.
4.4
Kinetics of skeletal cell turnover
4.4.1
Osteoblast kinetics
Early studies of osteoblastic cell kinetics were estimated by the labeling of proliferating cells, and the persistence of labeled cells on growing bone surfaces before cells became embedded in matrix. This was done by simultaneous single-dose injection of tritiated glycine (3H-glycine) and tritiated thymidine (3H-thymidine) into growing rabbits . With this method newly produced extracellular matrix proteins incorporated 3H-glycine labeling in new bone surfaces, and 3H-thymidine is incorporated into newly formed DNA during the cell replicative process. Autoradiographic analysis of femur bone sections revealed that labeled cells spend about 10 days on the bone surface before becoming embedded in the bone as osteocytes . Later, a study defining the lifetime of osteoblasts in the mouse periodontium demonstrated that periodontal proliferating progenitors go through a single S-phase and become osteoblasts that last for ~20 days . Although these studies did not define with precision the cell types involved in proliferation and had limited ability to measure the turnover rate of nonproliferating cells, they strongly suggested that a fraction of cells in growing bones have a relatively short duration on the bone surface and rapidly become bone-embedding osteocytes (10–20 days).
Human osteoblast function and average life span have been determined independently. After double tetracycline labeling, dividing the total wall width (the amount of bone made by osteoblasts) by the mineral apposition rate was used to examine the osteoblast life span in humans. These very indirect studies concluded that the average life span of normal human osteoblasts is ~150 days . The estimates of cell durability in these studies were based on a calculation derived from the measurements of new bone synthesis that is highly variable in different types of bone and different species, possibly accounting for the large differences seen in mouse and human studies. In addition, these studies did not precisely define cell types, broadly classifying all cuboidal cells localized along the bone surface as osteoblasts. However, despite the limitations of these studies and the discrepancies between species, the data indicated that osteoblasts have a limited life span and implied that they are postmitotic, therefore, replaced by unspecified immature cells .
More modern techniques have taken advantage of the ability to genetically label specific subsets of cells at a particular time and follow them by sequential in vivo imaging . This technology is obviously limited to mice at this time but indicates that mature osteoblast populations have a high rate of basal turnover with a durability of ~60 days. These studies also evaluated the cell populations that replenish the osteoblasts and will be discussed next. Overall, it appears that mature osteoblasts in adult bones turn over rapidly in homeostasis.
4.4.2
Osteoblasts are postmitotic
Osteoblasts are short-lived cells, and the basis for their replenishment is a topic of some interest in understanding how the tissue is organized as a system. Multiple studies have suggested the possibility that mature osteoblasts are capable of proliferation in bones exposed to stress conditions or under early developmental processes. For example, when mice were irradiated and proliferation of the cells along the endosteal surface assessed by 5-bromo-2-deoxyuridine (Brd-U) administration (a pyrimidine analog incorporated into newly synthesized DNA), over 10% of the collagen I–expressing cells were Brd-U positive compared with ~2% of controls, suggesting a rapid expansion of the resident cell population . Interestingly, most of the increases in collagen I– and osteocalcin-expressing cells were observed in the metaphyses of bones. Similar results have been reported in developing bones . Therefore under certain stress conditions and at particular ages, osteoblasts along endosteal surfaces appear to gain proliferative potential. However, the studies must be interpreted with caution as Brd-U labeling may occur in proliferating osteoprogenitors upstream of the mature osteoblasts, and the observed increase in the Brd-U positive collagen I–expressing osteoblasts could be the outcome of rapid differentiation from a progenitor pool, rather than from mature osteoblast proliferation. This is raised in particular because other studies using genetic marking where only mature, osteocalcin-expressing osteoblasts were labeled, no observable proliferation of mature osteoblasts was noted . This was confirmed in long-term homeostasis in adult animals and in models of injury providing fairly definitive evidence for the osteoblast as a postmitotic cell. However, it cannot be excluded that under particular kinds of stress not tested in the model or at particular ages, a subset of mature osteolineage cells can divide.
4.4.3
Osteocyte kinetics
Osteocytes comprise over 90% of bone cells and have long been considered inactive “old” osteoblasts embedded within the bone matrix that they secrete. The estimated life span of osteocytes in human bones is very variable (from 1 to 50 years) depending on the type of bone and age. This wide range is most likely because some bone envelopes are continuously remodeled (especially trabecular bones) while other envelopes such as intracortical bone are more durable. It has been suggested that only the osteocytes in intracortical bone can complete their full life span. However, it has now become clear that the transition from osteoblasts to osteocytes is an active process rather than passive process. In addition, the matrix-embedded osteocytes are capable of sensing mechanical stress, regulate mineralization, and produce multiple factors that can regulate the recruitment of osteoblasts and osteoclasts. Indeed, osteocytes are the essential cellular sources of sclerostin and RANKL for osteoclast and osteoblast regulation during bone remodeling. In the ovariectomized rodent model, estrogen loss dramatically increased osteocyte apoptosis over the 2 weeks postovariectomy corresponding with the peak osteoclastic activity . Another study revealed that conditional deletion of osteocytes in cortical bone generated a robust activation of osteoclasts, leading to severe bone loss . Both human and rodent studies have further demonstrated that osteoblast/osteocyte apoptosis can be induced by genotoxic agents, oxidative stress [reactive oxygen species (ROS)] , hypoxia , or loss of various growth factors and hormones (estrogens and androgens) , leading to bone loss phenotypes. Taken together, a new paradigm has emerged that osteocytes undergo apoptosis in response to skeletal unloading, fatigue damage, estrogen deficiency, or at a specific site where bone must be removed, apoptotic osteocytes stimulate the production of RANKL by neighboring viable osteocytes. The local increase in RANKL in turn initiates the development of osteoclasts to remove the defective bone and stimulate modeling or remodeling . Hence, the maintenance of the existing healthy osteocyte pool is a critical factor in bone integrity, and further understanding of the regulatory mechanism of osteocyte activation and turnover may help to minimize the effects of degenerative bone diseases.
4.4.4
Turnover of other osteolineage cell types
Evaluation of what cells divide to replace osteoblasts in part depends upon the ability to define osteolineage cells. In bone development, osteoblast differentiation from osteogenic progenitors can be distinguished by changes in stage-specific genes. Runx2 is one of the earliest genetic markers of osteo/chondrogenic progenitors, followed by osterix ( Osx ) in osteoprogenitors and type I collagen ( Col1 ) and osteocalcin ( Ocn ) in maturing osteoblastic cells . Unfortunately, in adult animals, these genetic markers do not correlate with reliable surface expressed phenotypic markers to clearly label SSCs and progenitors. Furthermore, while the temporal expression of these genetic markers makes it tempting to place these populations of cells in a linear relationship to each other, with Runx2 expression representing an early stem cell to Ocn expression representing a mature cell, with various restricted progenitors in between, the relationship amongst these populations may be more complex. Therefore it is difficult to measure definitively the subsets of cells involved in the replenishment of osteoblasts in adults. However, a number of studies have provided useful information.
The anatomic sites where bone-forming cells are thought to reside include the inner layer of the periosteum (inner cambial layer) , the endosteum , osteonal (harvarsian) canals, and perivascular locations within BM . Engineered mice where cells can be fluorescently tagged at a particular time and followed kinetically (so-called cellular pulse-chase experiments) have provided information as to which cells divide and replace mature cells in adult animals. In the example provided earlier, the promoter that is only activated at the level of the mature osteoblast (osteocalcin) was used to drive activation of a cell label and indicated that osteocalcin-expressing osteoblasts do not divide and do not replace themselves . Rather, they appeared to be replaced by cells expressing osterix as evident by another similarly engineered mouse using the osterix promoter to drive a fluorescent tag. The osterix-expressing cells were also minimally proliferative, however, as indicated by Ki-67 staining. Therefore a more primitive population of cells appeared to be required. These could be marked by induction of Mx1 in mice engineered such that the Mx1 promoter would activate a fluorescent tag . These Mx1 -labeled cells generated new osteoblasts under settings of both homeostasis and after microfracture. Mx1 -labeled stem/progenitors appeared at the site of fracture within 2–5 days and dramatically increased in number at 12 days. They did differentiate into fully mature osteoblasts as evident by osteocalcin expression and the ability to lay down new bone in vivo. Similar dynamics and contributions of osteogenic progenitors have been observed in injury models using a smooth muscle α-actin promoter ( αSMA ) transgenic mouse model . Lepr and other promoters have also been reported to denote SSCs. Therefore osteoblast replenishment in adults appears to be mediated by the stem/progenitor pool without evidence for mature cells replacing other mature cells.
4.5
Potential use of skeletal stem cells as therapy
If bone cells depend upon a dynamic population of SSCs that can migrate interstitially and by the circulation, it is biologically feasible to consider cell-based bone therapies. This issue has prompted much of the attention on the putative SSC that has been called MSC by in vitro features as discussed previously. While the in vitro cultured population that is termed MSC or here, MC, may not have close correspondence with bona fide endogenous SSCs, it may be valuable as a source of cells with osteolineage potential. The latter has been extensively shown through the use of the cells in vitro in bone constructs, in vivo by homotopic transplant into bone and even by systemic administration of an allogeneic BM graft in children with osteogenesis imperfecta (OI) . Several aspects of the biology of these cells are important considerations for cell therapy.
Practical clinical utility of cell therapy will likely require intravenous infusion and SSC homing to the BM microenvironment, although several animal models of osteoporosis have shown some success with intrabone injections of MC . The homing ability of primary or ex vivo expanded MC has been rigorously studied . These studies used varying approaches to isolate and expand MCs in vitro and found that the engraftment efficiency toward BM or target tissues of ex vivo expanded MC was variable and dependent on the source of cells, prior culture conditions, passage number, and route of delivery. In general, studies utilizing cultured MC showed poor engraftment in target tissues after systemic administration, confirmed by a comparative study demonstrating that primary murine SSCs have significantly higher efficiency of homing to the BM compared to cultured MC . The downregulation of cell adhesion molecules and/or chemokine receptors (CXCR4, receptor for SDF-1α) in culture-expanded MC is a possible cause of poor migration and homing. To overcome these limitations of SSCs to home and migrate appropriately in vivo, and thus improve the engraftment of SSCs in a potential clinical setting, multiple approaches have been employed to enhance the homing response of MC. Since the SDF-1α–CXCR4 axis is a key mechanism of SSC homing, retroviral overexpression of CXCR4 in MC resulted in increased MC homing to inflammatory sites or ischemic myocardium . Similarly, studies employing either an adeno-associated viral vector to transiently cause ectopic expression of the α4 integrin on murine MC or by chemical modification of the cell surface with a peptidomimetic ligand against α4 integrin on the MC cell surface coupled to bisphosphonate , leading to substantially increased homing of MC into the BM of recipient mice and a greater than 10-fold increase in new bone formation compared to control. Sackstein et al., using human MC preparations, chemically modified the cell surface of MC via an alpha-1,3-fucosyltransferase preparation to convert the native CD44 glycoform into hematopoietic cell E-selectin/L-selectin ligand . This allowed for the chemically modified MC to have potent E-selectin binding without effects on cell viability or multipotency. When transplanted into immunocompromised mice, the chemically modified MC were capable of infiltrating the marrow within hours of infusion, with foci of endosteal localized cells and human osteoid generation . These novel approaches may eventually lead to therapeutic strategies allowing for the prospective harvesting of SSCs and directed targeting of those cells to sites of injury or disease to coordinate bone formation and repair.
As continued advancements are made in directing SSCs to the BM after systemic delivery, the next phase of clinical therapy will be how best to apply SSCs to treat disease. SSCs from aged patients or donors are undoubtedly reduced in number and have a reduced capacity for osteogenic potential , and hence therapeutic efficacy, compared to younger SSCs. Therefore autologous transplantation of SSCs, without modification, may have little long-term value. Peripheral, circulating osteogenic-SSCs are in five times greater abundance in adolescents than the aged and are also present in higher numbers after injury . In a mouse study, aged mice transplanted with MC from young donors had increased bone density and a longer period of survival . Therefore allogeneic transplantation from younger, healthier donors might be preferred, but graft tolerance and immunosuppression issues create additional complications. Therefore if SSCs from patients can be modified or corrected, then autologous transplantation of modified cells, absent of graft rejection issues, may represent the best option. One example where this approach is moving toward clinical testing is in the treatment of OI.
Clinical trials using allogeneic BM transplants or SSC enriched transplants have demonstrated improvement in children with OI . Given that OI is normally associated with a mutation in a single gene, type I collagen, it is a highly attractive target for gene therapy. Using an adeno-associated viral (AAV) vector approach, Chamberlain et al. have demonstrated the ability to correct gene defects in OI patient SSCs and restore osteogenic potential in vitro . In a similar approach the laboratory-generated induced pluripotent stem cells (iPSCs) from AAV corrected SSCs and demonstrated that they were able to form bone in immunocompromised mice . The transformation to an iPSC state theoretically allows for unlimited expansion of the cells before differentiation to acquire sufficient numbers for a robust transplant, and the iPSC reprograming transgenes could be removed by activating Cre recombinase . As gene-targeting therapies evolve, and our understanding of the genetic and molecular pathways governing bone degenerative diseases increases, a combinatorial gene therapy/SSC transplant strategy may offer new opportunities for bone therapeutics.
4.6
Interactions with the hematopoietic system
4.6.1
The endosteal hematopoietic niche
The BM has long been recognized as the primary source of hematopoiesis in adult mammals. The very notion of a BM transplant relies on the fact that bone is a source of HSPCs capable of repopulating myeloablated hosts. Intriguingly, one of the very first studies describing cells that behaved like osteogenic-SSCs was actually seeking to determine why hematopoiesis was located in the bone . Tavassoli and Crosby placed BM fragments into nonbony tissues to see if the addition of nonhematopoietic bone cells could enable engraftment of hematopoietic cells in those tissues. Hematopoiesis occurred only if a bony ossicle with microcirculation formed. They observed that a “reticular” cell that could differentiate into osteoblasts was required for the bone and BM formation, thus presaging the recognition of a bone-forming SSC.
Within BM the endosteal surfaces of bone were recognized as a rich source of primitive hematopoietic cells by studies where tissue at progressive distances from the endosteum was preserved and tested . These studies suggested that endosteum was a site of enrichment for hematopoietic stem/progenitors, and therefore, the location of the HSC niche. This issue is controversial, however. A number of histologic studies have indicated that HSC is perivascular in the unmanipulated mouse BM (see next). This may be different than the BM in the setting of stem cell transplantation. Several different imaging approaches have demonstrated that transplanted HSCs localize to endosteal surfaces, while more mature hematopoietic progenitor cells (HPCs) localize further away . HSCs that were closely associated with the endosteum were also shown to have greater transplantation activity than those HSC contained in the central marrow cavity . Given that osteolineage cells line the endosteum, they were theorized as a potential supportive mediator of the HSC niche. When HSCs were acquired from aged donors, it was found that they localize to sites further away from the endosteum than HSCs from younger donors , suggesting that HSC localization within the bone may be a mediator of the difference in HSC function observed with the aging process. Studies by Taichman et al. demonstrated that osteolineage cells produce many supportive growth factors, including granulocyte colony-stimulating factor (G-CSF), granulocyte macrophage colony-stimulating factor, IL-1, IL-6, and TGF-β. Later, in vivo studies using various genetic approaches demonstrated that osteolineage cells are a significant regulatory component of hematopoiesis . In one of these studies where a constitutively active parathyroid hormone (PTH)/PTH related peptide (PTHrP) receptor was driven by an osteolineage promoter, the effect of osteolineage cells on HSC expansion was mediated, at least in part, by the Notch ligand Jagged1. Notch signaling regulates cell fate decisions, including HSC self-renewal , thus by changing self-renewal versus differentiation decisions, Notch can increase HSC number without differentiation or increase in HPC or mature cells . This regulatory relationship of Notch ligand expression by osteolineage cells may only be important with PTH/PTHrP receptor activation, however, as studies on Notch in homeostasis in adult animals do not support an important role in HSC function .
Within the endosteal niche, HSCs are thought to be retained through a variety of adhesion molecule interactions, many of which are likely redundant systems. Early studies exploring the role of osteolineage cells in maintaining HSCs suggested that N-cadherin interactions mediated the positive effects on HSCs ; however, more recent studies have contradicted these findings . Numerous other adhesion molecules have been implicated as contributing to HSC and HPC tethering, including, but not limited to, the integrins α4β1—very late antigen (VLA)-4 , α5β1—VLA-5 , α4β7—lymphocyte Peyer’s patch adhesion molecule-1 (LPAM-1) , the alpha 6 integrins (laminins) , CD44 , E-selectins , the angiopoietin receptor tyrosine kinase with immunoglobulin-like and EGF-like domains-2 (Tie-2) , endolyn (CD164) , the calcium-sensing receptor (CaR) , SDF-1α , and osteopontin (OPN) . Intriguingly, OPN is also a negative regulator of HSC pool size within the BM niche , and knockout of OPN in mice results in endogenous hematopoietic mobilization to the periphery and increases the mobilization response to G-CSF . Recently, osteolineage cells were also found to express agrin that mediates cell–cell contact with short-term HSCs and initiates proliferation, perhaps functioning as an opposing signal to OPN . These results suggest that OPN, a target for modulation of the endosteal HSC niche, and that agrin receptors, a target on HSCs, may serve as future therapeutic strategies to modify and enhance hematopoietic transplant and recovery, in line with previously described strategies targeting other adhesion interactions in the niche .
4.6.2
Hematopoietic regulation of the endosteal niche
Up to this point the focus has been on the role of osteolineage cells and their associated regulation of hematopoietic cells. However, cells of hematopoietic origin also support and regulate osteolineage cells. Cell-to-cell contact between osteolineage cells and HPCs has been shown to be important for HPC survival . Using an in vitro system, Gillette et al. demonstrated that human CD34 + cells (an HPC/HSC population of cells) cocultured with osteolineage cells made cell–cell contact and exchanged a portion of their membrane with the osteolineage cells, creating a signaling endosome . This signaling endosome caused osteolineage cells to downregulate Smad signaling and increase the production of SDF-1α, suggesting that hematopoietic cells may instruct osteolineage cells to create a more habitable environment, allowing them to directly affect their own microenvironment. These results may suggest that HSCs help to create their own niches, perhaps by instructing neighboring stromal cells to produce supportive factors.
Mature hematopoietically derived cells also have the ability to alter the endosteal osteolineage cells. One therapeutic manipulation of the hematopoietic niche involves treatment with G-CSF to “mobilize” HSCs and HPCs out of the BM and into the peripheral blood, where they can subsequently be collected by apheresis and used in hematopoietic transplantation. Treatment with G-CSF causes significant suppression of osteolineage cells , causing apoptosis and a characteristic “flattening” of osteolineage cells . Recently, Winkler et al. reported that a population of macrophage cells, characterized as F4/80 + Ly-6G + CD11b + and termed “osteomacs” , line the endosteal surfaces of bone, and that G-CSF treatment reduced the number of endosteal associated osteomacs . Presumably, this reduction in osteomacs was responsible for the attenuation of function of osteolineage cells. To confirm this hypothesis, macrophages were depleted using Mafia transgenic mice or with clodronate-loaded liposomes (Clo-lip) treatment, causing significant mobilization of HSC and HPC. A similar report from Chow et al. also demonstrated that depletion of macrophages with Clo-lip treatment results in hematopoietic mobilization , describing a Gr-1 negative F4/80 + CD115 mid CD169 + macrophage population as a regulator of osteolineage cells. Similar to the osteomac population, the authors suggest that the CD169 + macrophages express a soluble, yet to be identified, factor(s) that positively supports niche cells. In comparable studies by Christopher et al. a soluble factor produced by monocyte/macrophage populations supported osteolineage function , suggesting that manipulation of monocyte/macrophage populations or the factor(s) produced by them is a future therapeutic strategy to manipulate the endosteal niche and possibly suggest a novel strategy to affect bone cells therapeutically.
Osteoclasts are also monocyte-derived cells, which are present along endosteal surfaces of bone. Osteoblasts and osteoclasts regulate both bone formation and bone resorption, respectively, within the BM niche, and preosteoclasts cause retraction of osteoblasts , theoretically creating transient “holes” in the HSC supporting endosteal niche. Given that treatment with G-CSF causes increases in osteoclast number and activity , it was hypothesized that osteoclasts may regulate the endosteal niche and alter HSC retention and localization. Kollet et al. treated mice with RANK ligand that increased osteoclast activity and correlated with a moderate increase in HPC mobilization , consistent with a recent independent report . Correspondingly, stress models such as bleeding or lipopolysaccharide (LPS) treatment also increased the number of osteoclasts in the endosteal niche and HPC in the peripheral blood, supporting a role for osteoclasts in niche regulation. Kollet et al. suggested that osteoclast-derived proteolytic enzymes, such as cathepsin K, degraded important niche retention molecules, thereby facilitating egress of HSC and HPC . A more recent study by the same laboratory showed decreased osteoclast maturation and activity in CD45 knockout mice, with reduced mobilization in response to RANK ligand and G-CSF , further suggesting that osteoclasts are important in regulation of the endosteal niche. In contrast to these studies, Takamatsu et al. earlier reported that while G-CSF treatment increased osteoclast number and bone resorption in both BALB/c mice and humans, the increase in osteoclasts did not occur until 10–15 or 6–8 days, respectively, after treatment with G-CSF , which has also been observed by other groups using similar systems . Since niche clearance by G-CSF is typically observed 4–5 days after treatment, the importance of osteoclasts remains unclear. Similarly, treatment of mice with osteoclast inhibiting bisphosphonates does not result in an impaired mobilization response to G-CSF and in fact increases HSC egress . Intriguingly, the endosteal surface of bone, particularly underneath resorbing osteoclasts, is a significant source of soluble extracellular calcium, and previous studies by our laboratory demonstrated that HSC expression of CaRs mediates a chemoattraction to soluble Ca 2+ . When the CaR was knocked out, mice had reduced HSC content within the BM niche and increased HSC in peripheral blood. These results might suggest that the bone resorbing, and subsequent Ca 2+ releasing activity of osteoclasts, may be involved in niche retention.
Hematopoietic transplantation normally requires conditioning of the BM niche, or a clearance of resident HSC and HPC, to allow for successful engraftment. Conditioning is normally accomplished by treatment with chemotherapeutic agents and/or irradiation to remove HSC and HPC; however, these agents can also affect niche cells themselves. A recent study from Dominici et al. demonstrated that following a lethal dose of irradiation, megakaryocytes migrated toward the endosteal surfaces of bones and induced a twofold expansion in osteolineage cells, presumably increasing the available niches for subsequent HSC engraftment . These results are in close alignment with other studies in which mice deficient in GATA-1 and NF-E2 transcription factors had significantly increased megakaryocyte production, leading to a sixfold expansion of osteolineage cells , a process requiring direct cell–cell contact between megakaryocytes and osteolineage cells . Further support for blood cells affecting bone biology is the observation that excessive blood loss, causing stress on the hematopoietic system, leads to osteoporosis in mice and that splenectomy in mice results in an expansion of megakaryocytes with reduced GATA-1 expression and increases in bone formation . These intriguing findings suggest that there is much yet to be learned about the bidirectional regulatory relationships of blood and bone. Further exploration of these relationships is clearly warranted particularly since there is a substantial prevalence of bone disorders following hematopoietic transplantation and its associated conditioning regimens .
4.6.3
Perivascular skeletal stem cells—the perivascular niche
The endosteal surface of bone, as described earlier, is a dynamic tissue with a high rate of turnover of osteoblasts that are replenished by earlier osteogenic precursors and SSCs. Several reports, using in vitro coculture studies, have shown that HSCs cultured on more immature osteolineage cells have a higher activity and repopulating ability than HSCs cultured on more mature osteoblasts , suggesting that the maturation status of osteolineage cells may be a key determinate of HSC niche support. Indeed, several in vivo reports described niche cells responsible for HSC maintenance that resemble SSCs and their immediate downstream precursors , and in studies in which terminally differentiated osteoblasts were ablated, hematopoiesis was unaffected . These findings suggest that the HSC niche is composed of more immature osteolineage cell subsets. The aforementioned Nestin -GFP + multipotent SSCs express high levels of hematopoietic supportive molecules, kit ligand (also referred to as stem cell factor or SCF) and CXCL12 (also known as SDF-1), and overlap with CXCL12-abundant reticular (CAR) cells, suggesting that these cells may form a unique BM niche . Intriguingly, these Nestin -GFP + cells have a perivascular location by wrapping around arterial and sinusoidal endothelial cells within the BM, suggesting the presence of an alternate HSC niche localization in addition to the endosteum. Further studies defined that these perivascular cells can be marked by constitutive expression of Cre driven by the Lepr – , osterix- , or Prx-1 promoter , and they contribute to HSC maintenance via synthesis of CXCL12 and SCF, whereas the deletion of the same factors in committed osteoblasts (using Osteocalcin -Cre) did not reveal a significant HSC phenotype. The selective Cxcl12 deletion from arteriolar NG2 + perivascular cells caused HSC reductions and altered HSC localization in BM. By contrast, the deletion of Scf in Lepr + sinusoidal perivascular cells led to reductions in BM HSC numbers . These results suggest distinct contributions of cytokines derived from different perivascular cells in separate vascular niches to HSC maintenance.
4.6.4
The vascular niche
While it is clear that HSCs reside and thrive within the endosteal BM niche, evidence, including the localization of CAR cells and Nestin + SSCs , suggests that the endosteum is not the exclusive and may not even be the predominant niche for hematopoiesis. Stem cell localization studies utilizing in vivo imaging out of our laboratory and others found that while HSCs were near the endosteum, they were not exclusively adjacent to osteolineage cells. Studies assessing the localization of signaling lymphocyte activation molecule (SLAM; CD150 + , CD48 − and CD244 − ) cells, which are highly enriched for HSC, have shown a greater proportion of SLAM cells adjacent to BM sinusoidal blood vessels, suggesting a “vascular niche” for HSC . This is supported by the fact that endothelial cells, very similar to osteolineage cells, can support hematopoiesis both in vitro and in vivo . We have also described small endothelial microdomains that express E-selectin and CXCL12 and act as a point of entry for HSC after transplantation . Regeneration of sinusoidal endothelial cells has been reported to be essential for hematopoietic reconstitution following myelosuppression , and BM endothelial cells have been reported to support the growth and expansion of HSC through angiocrine factors . Indeed, genetic deletion of SCF in arterial endothelial cells, but not sinusoidal endothelial cells, significantly reduced functional HSCs . Interestingly, similar to the HSC cell-to-cell contact with osteolineage cells altering the endosteal niche function , Slayton et al. reported that donor hematopoietic cells migrated to sites of vascular sinusoidal injury within bones and contributed to repair following transplantation , further highlighting the complexity and interconnectedness of the BM microenvironment.
While it is not completely clear what factors influence the particular niche location of HSCs, the fact that reticular cells with high production of CXCL12 are present in both the endosteal and vascular niches may provide a common mechanism of HSC support in both niches . A recent report has demonstrated that HSC, shortly after transplantation into nonmyeloablated recipients, preferentially localize to the endosteum in metaphyseal regions of bone in close association with blood vessels, and that this requires hyaluronic acid . Given that the endosteal region is itself highly vascularized may suggest that the HSC niche actually encompasses overlapping regions within the bone.
4.6.5
The bone marrow environment is hypoxic
Several reports have demonstrated that HSCs are present in regions of hypoxia within the BM , and those HSCs contained in hypoxic areas have greater hematopoietic repopulating ability than those in more perfused areas , suggesting that perhaps the overlapping regions of the endosteal and vascular biomes contain the right mix of atmospheric (oxygen and other chemical mediators) conditions to support hematopoiesis. Cellular exposure to hypoxia results in stabilization of hypoxia inducible factor 1-α (HIF-1α), which is a transcriptional regulator of erythropoietin production , numerous cell proliferation and survival genes , the angiogenic growth factor VEGF , and other genes. In mice where the HIF-1α-responsive element is mutated on the Vegfa promoter, thereby preventing upregulated expression in hypoxic conditions, HSCs were found to have defects in their function , supporting a role of hypoxia-induced gene expression in the niche for maintenance of HSCs. HIF-1α also increases production of CXCL12 and CXCR4 receptor expression , and prevents hematopoietic cell damage caused by overproduction of ROS . Therefore HSCs residing in areas of low oxygen will have increased activity of HIF-1α, thereby maintaining their fitness and stemness , a hypothesis that is supported by the fact that in vitro hypoxic conditions expand human HSC and HPC populations . Recently, using HIF-1α-deficient mice, Takubo et al. demonstrated that HSC cell cycle quiescence was lost, and HSC numbers diminished after stressful insults . Additional studies exploring the protein stabilization of HIF-1α demonstrated that a precise regulation of HIF-1α activity was necessary for optimal HSC function. Miharada et al. also reported that HIF-1α-deficient mice have reduced expression of Cripto, also known as teratocarcinoma-derived growth factor-1, on endosteal osteolineage cells and a reduction of one of Cripto’s cognate receptors, GRP78, on HSCs . The authors demonstrate that the Cripto/GRP78 signaling axis is an important regulator of HSC quiescence downstream of HIF-1α signaling and postulate that GRP78 can be used as a marker to distinguish between quiescent and active HSCs. Advanced technologies able to accurately measure oxygen tension within the BM microenvironment at high-resolution overlaid with imaging assessing HSC localization are likely to be highly informative and perhaps will resolve some discrepancies and unanswered questions as to the specifications of the HSC niche.
Oxygen tension is also important in regulating osteolineage cell function. BM MSCs cultured in hypoxic conditions demonstrated increased self-renewal division and increased integrin expression allowing for more robust migration compared to their normoxic-cultured controls , consistent with reports that hypoxic conditioning can increase osteogenic, adipogenic, and chondrogenic differentiation potential , though other reports propose opposite effects suggesting that regulation of SSCs by hypoxia may be highly dynamic. A recent report has also demonstrated that VEGF production by osteolineage cells is cooperatively regulated by osterix and Hif-1α , suggesting hematopoietic and angiogenic support by osteolineage cells is also regulated by oxygen tension. Further exploration into these mechanisms is likely to lead to advancements in therapies, particularly in terms of bioengineering scaffolds and ex vivo culture conditions for cellular therapeutics of bone degeneration .
4.6.6
Nervous system mediators
While it is clear that cellular and molecular constituents within the microenvironment interact and coregulate each other, the question arises as to whether there is a global regulator of the entire bone system able to rapidly alter numerous components of the niche to respond as needed. The recently described Nestin + MSCs are highly intriguing, as Nestin, an intermediate filament protein, is normally restricted to nerve cells. Intriguingly, work by the same laboratory demonstrated that HSC and HPC mobilization by G-CSF required peripheral β 2 -adrenergic signals in studies utilizing chemically sympathectomized mice treated with 6-hydroxydopamine; mice treated with the β-blocker propranolol; or mice genetically deficient in the gene for dopamine β-hydroxylase ( Dbh ), an enzyme that converts dopamine into norepinephrine. They also showed that treatment with the β 2 -adrenergic agonist clenbuterol reversed the phenotype of Dbh knockout mice . The Frenette laboratory also demonstrated that the nervous system regulates HSC niche retention via circadian rhythms . Signaling via β 3 -adrenergic stimulations causes rhythmic oscillations controlling norepinephrine release, CXCR4 expression, and CXCL12 production, causing regular transient trafficking from the BM niche. Nervous system signaling has also been demonstrated to have direct effects on HSC, as human CD34 + hematopoietic cells express β 2 -adrenergic and dopamine receptors and neurotransmitters, such as norepinephrine, serve as direct chemoattractants for HSC , demonstrating nervous system global control over multiple cellular components of the hematopoietic ecologic system. A recent report showed that β 2 -adrenergic signaling causes upregulation of the vitamin D receptor (VDR) on osteolineage cells, and that VDR signaling was necessary for osteolineage responses to G-CSF treatment . VDR expression is also regulated by circadian rhythms demonstrating further interconnectivity of the nervous system control over the bone microenvironmental system.
A population of nonmyelinating Schwann cells that ensheath sympathetic nerves in the BM has been described . These cells are a primary producer of TGF-β that has been demonstrated to be a quiescence inducing signal for HSC . When the bone was denervated, TGF-β signaling was reduced leading to a reduction in the HSC pool . Given that TGF-β is also capable of attracting SSCs , this may suggest that the nervous system can coordinate SSC trafficking and migration to sites of need, though further exploration is required.
4.7
Conclusion
The relationship of bone and BM remains a highly intriguing biologic paradigm for how heterologous cells interrelate to accomplish complex and highly divergent physiologic processes. While the hematopoietic system has been more accessible and more extensively studied in terms of cellular hierarchies and stem cell function, applying approaches and concepts learned from it have again found marked connectedness between bone and BM. The bone, much as the blood, appears to be organized in a hierarchical manner where stem and progenitor cells are required for replenishment of short-lived mature cells. Indeed, the turnover times for mature bone cells, osteoblasts, and mature red cells are similar in both mouse and man. In addition, both skeletal and HSC compartments appear to be heterogeneous, and both stem cell populations appear capable of migration, particularly through the vascular tree. Further exploration of the similarities and differences between these systems promises to both teach important cell and organismal biology and to offer the potential of defining novel therapeutic strategies for diseases in each system.
References
Related posts:
 The bone organ system: form and function
The bone organ system: form and function
 Impact of physical characteristics and lifestyle factors on bone density and fractures
Impact of physical characteristics and lifestyle factors on bone density and fractures
 Clinical and epidemiological studies: skeletal changes across menopause
Diabetes, diabetic medications, and risk of fracture
Clinical and epidemiological studies: skeletal changes across menopause
Diabetes, diabetic medications, and risk of fracture
 Relationship between periodontal disease, tooth loss, and osteoporosis
Relationship between periodontal disease, tooth loss, and osteoporosis
 On the evolution and contemporary roles of bone remodeling
On the evolution and contemporary roles of bone remodeling
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


