The Role of Platelets in Blood Coagulation
Peter N. Walsh
The hemostatic response to vascular injury consists of a series of proteolytic reactions that occur on the surface of activated platelets and other intravascular cells including monocytes and neutrophils.1,2,3 This localizes coagulation complex assembly to the hemostatic thrombus and protects coagulation enzymes from inactivation by plasma and cellular inhibitors of serine proteases.1,2,3,4,5,6,7,8 The initial hypotheses explaining the normal coagulation mechanism, referred to as the cascade9 and waterfall10 theories, were proposed more than 45 years ago and have since been extensively revised.11,12,13 The goal of this chapter is to describe our current understanding of the normal hemostatic mechanism. The contributions of leukocytes and monocytes to coagulation complex assembly and the role of cell membrane receptors in the assembly of the anticoagulant complexes involving activated protein C (APC), protein S, and thrombomodulin are discussed in Chapter 19. The interactions of thrombin with platelets and other cells is considered in Chapter 33.
SEQUENTIAL COAGULATION COMPLEX FORMATION
The cascade9 and waterfall10 theories of blood coagulation originally postulated a mechanism by which a minute initial stimulus was sequentially amplified through a series of enzymatic reactions to result in the local explosive generation of thrombin. These hypotheses postulated two alternative pathways for the initiation of coagulation, each leading to the activation of factor X (FX). One was termed the intrinsic pathway in which the “contact factors,” FXII, prekallikrein (PK), high molecular weight kininogen (HK), and FXI, are assembled on negatively-charged surfaces,14 and the other was termed the extrinsic pathway, initiated by FVIIa and by the exposure of tissue factor (TF).13 It was subsequently shown that platelets make major contributions to each pathway, including an alternative, platelet-dependent pathway for the activation of FXI.1,15,16,17,18,19 However, these hypotheses required revision because patients with deficiencies of FXII, PK, and HK do not experience abnormal bleeding,14 whereas patients with deficiencies of factors VIII and IX (hemophilia A and B, respectively) experience severe, life-threatening, spontaneous, and posttraumatic bleeding,20,21 and patients with FXI deficiency22,23,24,25,26,27,28,29 and FVII deficiency30 may bleed excessively after surgery or trauma. Nonetheless, because FIX activation can be catalyzed not only by FXIa31,32,33,34,35,36,37,38,39,40 but also by FVIIa-TF,37 it has been suggested that the TF pathway can initiate the intrinsic pathway directly and independently of the contact factor pathway. However, this does not account for the hemostatic defect observed in FXI deficiency,22,23,24,25,26,27,28,29 and in 1991 an alternative mechanism for proteolytic activation of FXI by thrombin11,12 was demonstrated. Thus, the initial event in hemostasis is likely the exposure of TF on cell membranes, resulting in the activation of FX by FVIIa-TF,30 and leading to the generation of trace concentrations of thrombin because the TF pathway is rapidly shut down by tissue factor pathway inhibitor (TFPI).41,42,43,44,45,46 The initial trace amounts of thrombin formed are sufficient to activate platelets, FXI, FVIII, and FV, permitting the intrinsic (or consolidation) pathway of coagulation to generate sufficient quantities of thrombin to promote hemostasis.11,12 The intrinsic pathway is thought to occur on the surface membranes of activated platelets, where there are binding sites for FXI,47,48,49,50,51,52 FXIa,53,54,55,56 FIX,57,58,59,60 FIXa,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73 FVIII,74,75,76,77,78 FVIIIa,74,75,76,77,78 FX,75,79,80 FXa,81,82,83,84,85,86,87,88,89,90,91,92,93 FV,81,82,83,84,85,86,87,88,89,90,91,92,93 FVa,81,82,83,84,85,86,87,88,89,90,91,92,93 prothrombin,79,80 and thrombin.2,94 These binding sites promote the assembly of coagulation enzyme-cofactor-substrate complexes that compared with solution-phase reactions, accelerate the kinetics of FXI activation by FXIIa,95 of FX activation by FIXa,57,58,62,64,65,66 and of prothrombin activation by FXa.81,82,83,84,85,86,87,88,89,90 The assembly of these complexes is presented in schematic form in FIGURE 35.1, providing a point of reference for understanding the role of platelets and other cells in the initiation and consolidation pathways of blood coagulation.
INITIATION PATHWAYS
Assembly and Regulation of the Tissue Factor Pathway
As discussed in Chapter 11, the contact phase proteins can initiate intrinsic coagulation when foreign surfaces are introduced into the blood stream,14 the initiating event in hemostasis is the expression of the transmembrane protein TF on cell membranes.30 A number of cell types, including monocytes and leukocytes and those underlying the endothelium, can synthesize and expose TF to plasma proteins, but the specific cell type and mechanism of TF presentation has not been definitively identified.30,96 Unactivated platelets do not constitutively express TF. However, TF-bearing microparticles,97,98,99,100,101,102,103 derived from specialized membrane microdomains (i.e., lipid rafts) in monocytes, can fuse with platelet membranes, perhaps utilizing adhesive receptors such as P-selectin on activated platelets and PSGL-1 on microparticles to transfer TF to platelets by membrane fusion.
Platelets and the Contact Activation Pathway
The reciprocal proteolytic activation of FXII and PK on negatively charged surfaces in the presence of high molecular weight kininogen (HK) can lead to the limited proteolytic activation of FXI14,104,105,106,107,108,109,110,111,112 and activation of the intrinsic coagulation pathway. However, the physiologic role of these reactions is uncertain
since patients deficient in contact activation pathway proteins do not bleed.4,7,8,22,23,24,25,26,27,28,29,94,113,114 In fact, HK may downregulate platelet activation by impairing thrombin binding to the platelet glycoprotein (GP) Ib/IX/V.115 Nonetheless, the contact activation pathway may contribute to the formation of pathologic thrombi, especially in patients with intravascular prosthetic devices.14
since patients deficient in contact activation pathway proteins do not bleed.4,7,8,22,23,24,25,26,27,28,29,94,113,114 In fact, HK may downregulate platelet activation by impairing thrombin binding to the platelet glycoprotein (GP) Ib/IX/V.115 Nonetheless, the contact activation pathway may contribute to the formation of pathologic thrombi, especially in patients with intravascular prosthetic devices.14
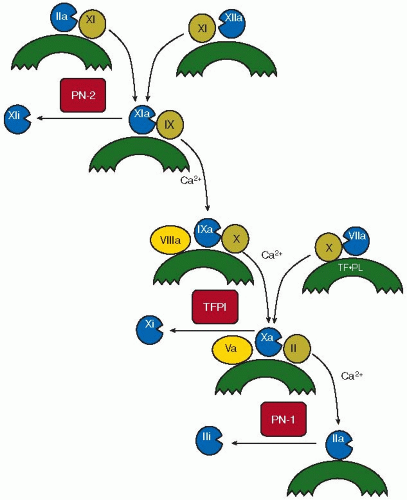 FIGURE 35.1 Schematic diagram representing a current hypothesis of the contribution of platelets to blood coagulation, and the postulated sequence of coagulation reactions that comprises the normal hemostatic mechanism. The crescentic forms represent the activated platelet membrane, except in the case of the form labeled TF·PL, which represents tissue factor expressed on a phospholipid membrane surface. The roman numerals represent the coagulation proteins including zymogens or substrates (in circles), cofactors (in ellipses), and enzymes (in circles with segmental excisions). TF, tissue factor; TFPI, tissue factor pathway inhibitor; PN2, protease nexin-2; PN1, protease nexin-1. The curved arrows represent conversions from zymogens (e.g., prothrombin, shown as II) to active enzymes (e.g., thrombin, shown as IIa), whereas the leftward pointing arrows represent the inhibition of these active enzymes (e.g., FXa, shown as Xa) to their inactivated forms (e.g., inactivated FXa, shown as Xi). Shown in boxes are the Kunitz-type inhibitors, PN-2 and TFPI, and the serpin, PN-1. The coagulation mechanism is initiated as the consequence of the assembly of the FVIIa/FX/TF/PL complex and is regulated by TFPI, whereas the consolidation phase of coagulation, regulated by PN-2 and PN-1, is activated by the resulting generation of low concentrations of thrombin sufficient to activate platelets, FXI, FVIII, and FV to produce thrombin in sufficient quantities to form a fibrin clot. (From Walsh PN. Platelets: yin and yang. Blood 2010;115:1-2, with permission.) |
Activated platelets can participate in the activation of both FXII and FXI.1,15,16,17,18,19 Thus, ADP- or collagen-stimulated platelets can promote the proteolytic activation of FXII by kallikrein and the subsequent proteolytic activation of FXI by FXIIa95 by exposing high-affinity FXI binding sites that require the presence of either HK and zinc ions or prothrombin and calcium ions (Table 35.1).47,48,49,50,51,52,53,54,55 Since there is evidence that activation of FXI on the platelet surface by thrombin can occur in the absence of FXII,116 it is possible that under certain physiologic or pathologic conditions, assembly of contact pathway proteins on activated platelets can trigger intrinsic coagulation. For example, polyphosphates secreted from platelet dense granules117,118,119 and RNA released from damaged vessels120 can activate FXII. A recent systems biology approach, examining the kinetics of thrombin generation in blood treated with corn trypsin inhibitor (CTI) to inhibit contact activation, suggests that activated platelets support the initiation of coagulation via the combined activity of FXIIa and FXIa in the absence of any evidence for kinetically significant blood-borne TF.121
CONSOLIDATION PATHWAY
Interactions of Activated Platelets with FXI
FXI is a unique, homodimeric coagulation protein that circulates in plasma in a complex with HK. Although FXI can be activated by FXIIa, the physiologic relevance of this reaction is uncertain. On the other hand, thrombin has been implicated in feedback activation of FXI on negatively charged surfaces, either in the presence of HK and zinc ions or prothrombin and calcium ions.11,12 Moreover, platelet activation appears to result in FXI binding to GPIb/IX/V, thereby colocalizing FXI with GPIb/IX/V bound in membrane microdomains or lipid rafts.122,123 Although it was originally reported that platelet stimulation by thrombin promotes FXI activation,124,125 these observations could not be confirmed.126 On the other hand, activated platelets can enhance FXI activation when thrombin is generated by low concentrations of TF-FVIIa116 and thrombin propagates further thrombin generation in CTI-treated plasma when FXI and platelets are present.127 Nonetheless, the role of thrombin in FXI activation remains controversial. Although Pedicord et al.128
found no evidence that FXI activation by thrombin occurs in plasma or on the surface of activated platelets, others found that FXI activation by low concentrations of thrombin makes major contributions to the sustained generation of thrombin in plasma in the absence of FXII.116,127,129,130,131,132 The latter observations are supported by evidence that activated platelets, but not endothelial cells, participate in the intrinsic pathway of blood coagulation by promoting the activation of FXI by thrombin and generating FXIa to promote the activation of FIX.53,133
found no evidence that FXI activation by thrombin occurs in plasma or on the surface of activated platelets, others found that FXI activation by low concentrations of thrombin makes major contributions to the sustained generation of thrombin in plasma in the absence of FXII.116,127,129,130,131,132 The latter observations are supported by evidence that activated platelets, but not endothelial cells, participate in the intrinsic pathway of blood coagulation by promoting the activation of FXI by thrombin and generating FXIa to promote the activation of FIX.53,133
Table 35.1 Binding of coagulation zymogens, enzymes, and cofactors to activated platelets | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
 >108-fold)
>108-fold) >108-fold)
>108-fold) >108-fold)
>108-fold) >105-fold)
>105-fold) >105-fold)
>105-fold)Related posts:
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Integrín αIIbβ3 and Platelet Aggregation
Integrín αIIbβ3 and Platelet Aggregation
 Acquired Nonimmune Thrombocytopenia
Acquired Nonimmune Thrombocytopenia
 Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


