Fig. 6.1
Kidney cancers are a diverse group of malignancies with distinct genetic and molecular alterations and disparate histologic features (Permission granted from Elsevier) (Linehan et al.)
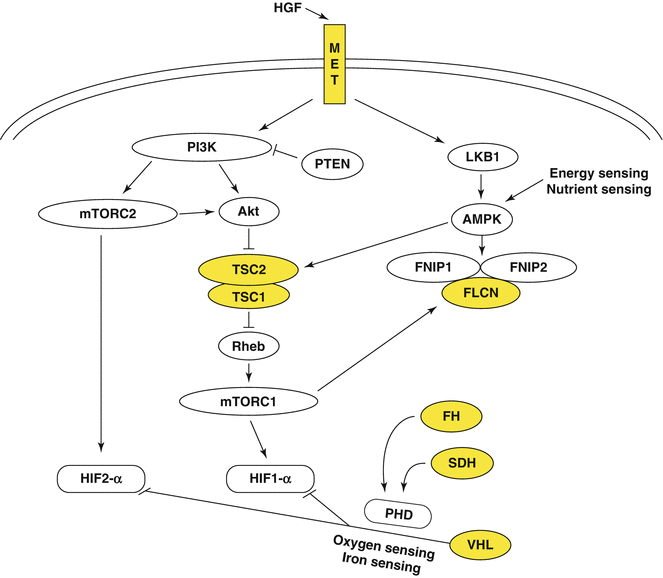
Fig. 6.2
The genes known to cause kidney cancer, VHL, MET, FLCN, FH, SDH, TSC1, and TSC2 share the common feature in that each is involved in various pathways that regulate cell growth, proliferation, or nutrient metabolism pathways (Permission granted from Elsevier) (Linehan et al.)
6.2 Overview of Metabolism in Cancer
Proliferating cells require a steady source of nutrients to meet their demand for energy (ATP) and to serve as building blocks for macromolecules (lipids, proteins, nucleic acids, etc.) that are essential components of newly formed cells. Cancer cells are particularly adept at diverting available nutrients toward pathways conducive to their agenda of dysregulated proliferation [5]. This is accomplished, at least in part, by extensive reorganization of cellular metabolism to ensure (1) a constant supply of critical intermediates required for biosynthesis and (2) the generation of sufficient energy to fuel this process [6, 7]. While our understanding of the metabolic alterations in cancer cells is still evolving, considerable progress has been made toward this end in the past decade.
6.2.1 Glycolysis, the Krebs Cycle, and the Warburg Effect
Glucose is both the primary source of cellular ATP and a major contributor of carbon for macromolecule synthesis. Mammalian cells convert glucose into pyruvate in a multistep process referred to as glycolysis. In normal cells, in the presence of adequate oxygen, pyruvate is diverted to the mitochondrial Krebs cycle (tricarboxylic acid cycle, TCA), leading to the generation of ATP via oxidative phosphorylation. However, under anaerobic conditions, ATP is generated by a process that results in conversion of pyruvate to lactate in the cytoplasm. While the latter process is able to meet cellular bioenergetic requirements as long as the availability of glucose is not limiting, it is a far less efficient means of generating ATP than is oxidative phosphorylation. In addition to its essential role in ATP synthesis, the Krebs cycle also plays an important role in macromolecule synthesis with intermediates such as citrate, oxaloacetate, and α-ketoglutarate serving as critical components of biosynthetic pathways leading to generation of lipids, nucleotides, and proteins.
The concept of altered metabolism in cancer is not new; as early as the 1920s, the German scientist and subsequent Nobel laureate Otto Warburg noted that compared to normal cells, tumor cells (derived from ascitic fluid in his original experiments) consume larger amounts of glucose and generate excessive amounts of lactic acid. Based on these observations, he concluded that cancer cells utilize glycolysis preferentially as a means of ATP synthesis (i.e., aerobic glycolysis or the “Warburg Effect”) [8, 9]. He further posited that this phenomenon was the result of an intrinsic defect in mitochondrial function and oxidative phosphorylation, forcing cells to turn to aerobic glycolysis. Although there is overwhelming evidence of the Warburg effect in a wide range of cancers, Warburg’s suggestion that this results from mitochondrial dysfunction is supported only in a small minority of tumors, including at least two subsets of kidney cancer [10, 11]. Instead, in most cancers where the Warburg effect is evident, the reliance on aerobic glycolysis for ATP generation appears to be an attempt to spare Krebs cycle intermediates for preferential use as substrates for biosynthetic pathways [12]. Regardless, aerobic glycolysis appears to be essential for tumor proliferation and survival in many human cancer models and is a valid and promising target for therapeutic intervention.
6.2.2 Drivers of the Warburg Effect in Human Cancer
The precise mechanisms that promote aerobic glycolysis in cancer cells are a matter of ongoing debate. It is becoming increasingly clear that there is no single unifying mechanism driving the glycolytic shift across all cancers, with distinct processes identified in disparate tumor types. Activation of several oncogenes as well as inactivation of a variety of tumor suppressor genes has been implicated in promoting the glycolytic phenotype. Inactivation of the Krebs cycle enzymes fumarate hydratase or succinate dehydrogenase in some forms of familial kidney cancer offers the best examples of direct interruption of oxidative phosphorylation and obligate use of aerobic glycolysis for ATP generation [10, 11]. The PI3K/Akt/mTOR pathway is an important regulator of cell growth and proliferation and plays a key role in nutrient sensing and cellular responses to growth factors [13]. Activation of this pathway has been described in several cancers and can lead to upregulation of glycolysis by several mechanisms, including increased influx of glucose and other nutrients, transcriptional activation of key glycolytic enzymes, and enhanced translation of a number of proteins essential to the process [14, 15]. Similarly, amplification of the proto-oncogene MYC has been shown to activate glycolysis via upregulation of enzymes regulating this process (including PKM-2, hexokinase, and LDH-A) as well as upregulation of transmembrane glucose transporters such as GLUT-1 [16, 17]. Activation of the NRF2 oxidative stress response pathway is a feature of several cancers including non-small cell lung cancer and some forms of papillary RCC; recent evidence suggests that this pathway promotes metabolic reprogramming for cancer cells by enhancing glycolysis as well as glutaminolysis [18, 19].
Hypoxia-inducible factor-1 (HIF-1) is an important component of the cellular oxygen-sensing apparatus and plays a key role in the regulation of glycolysis in response to hypoxia. In normoxic conditions, key proline residues in the alpha subunit of HIF-1 are hydroxylated by prolyl hydroxylase. The VHL gene encodes the VHL protein which binds to hydroxylated HIF-1α, targeting the latter for ubiquitin-mediated degradation [20, 21]. In hypoxic conditions, prolyl hydroxylase is inhibited, permitting an HIF-1α to accumulate. Stabilization of HIF-1α enables the cell to respond to hypoxic and low-iron conditions by transcriptional upregulation of a number of genes critical for cancer proliferation and activation of aerobic glycolysis including vascular endothelial growth factor (VEGF), epidermal growth factor (EGF), platelet-derived growth factor (PDGF), glucose transporter 1 (GLUT1), phosphofructokinase-2 (PFK2), and pyruvate dehydrogenase (PDH). In many human cancers, HIF-1α is stabilized by a variety of mechanisms independent of ambient oxygen tension. One of the best studied mechanisms mediating this pseudohypoxic HIF response is seen in VHL-deficient tumors. Due to VHL inactivation in these tumors, HIF-1α is no longer appropriately targeted for ubiquitin-mediated degradation even when oxygen is readily available, a phenomenon that is seen in both VHL-associated and sporadic ccRCC [22]. VHL-independent upregulation of HIF-1 has also been described, particularly in RCC variants where disruption of the Krebs cycle leads to accumulation of substrates such as fumarate and succinate, which impede the hydroxylation of the alpha subunits of HIF-1 [11, 23]. Additionally, translational upregulation of HIF-1α has been described with activation of the mammalian target of rapamycin (mTOR) signaling, which can occur by a variety of mechanisms [24, 25].
6.2.3 Glutaminolysis and Reductive Carboxylation
Glutamine is an additional important nutrient substrate in tumor cells, contributing to the generation of citrate and acetyl coenzyme A for lipid synthesis as well as acting as a nitrogen donor for amino acid/protein synthesis. A key step in glutamine metabolism is its deamidation by glutaminases to generate glutamate. While this reaction is bidirectional in normal cells, in tumor cells, deamidation of glutamine to glutamate is favored, partly a result of the overexpression of glutaminases and suppression of glutamine synthetase [26, 27]. Glutamate is converted to α-ketoglutarate which enters the Krebs cycle and can be converted to oxaloacetate in one of two ways. In tumors with an intact Krebs cycle and electron transport chain, mitochondrial oxidative metabolism is the predominant pathway used. However, in instances where oxidative metabolism is impaired (e.g., mutations in FH or SDH), an alternative pathway of reductive carboxylation is employed to generate both oxaloacetate and acetyl CoA [28]. In the latter instances, glutamine is the major carbon source for fatty acid synthesis [28]. Glutaminolysis also allows tumor cells to meet the NADPH demands of growth and supports enhanced fatty acid synthesis, nucleotide biosynthesis, and maintenance of the glutathione pool [12].
6.3 Metabolic Alterations in Kidney Cancer
Although metabolic reprogramming is a common theme in the different subtypes of kidney cancer, there are significant differences in the nature of the metabolic alterations and the genetic and molecular mechanisms driving these alterations between individual RCC variants. Elucidating the precise changes underlying each entity is crucial for developing specific targeting strategies against a given subtype.
6.3.1 Kidney Cancers Harboring Mutations in Krebs Cycle Enzyme Genes
Inactivating germ line mutations or deletions of fumarate hydratase and subunits of succinate dehydrogenase are associated with distinct forms of RCC that offer classic examples of the Warburg effect in human cancer. In both cases, the basic underlying defect is an enzymatic deficiency that impairs the Krebs cycle and oxidative phosphorylation, with a consequent glycolytic shift.
6.3.1.1 Hereditary Leiomyomatosis and Renal Cell Carcinoma (HLRCC)
HLRCC is an autosomal dominant hereditary cancer syndrome characterized by the presence of potentially painful cutaneous leiomyomas occurring on the arms or trunk and development of early-onset uterine fibroids in affected women (often necessitating hysterectomy in the third or fourth decade of life) [29]. In addition, affected patients are at risk for developing a particularly aggressive form of renal cancer with potential for rapid growth and early metastasis [30]. HLRCC-associated kidney cancer presents as early-onset, unifocal or bilateral, multifocal renal cysts or type 2 papillary tumors that have a characteristic histologic appearance that differentiates it from other forms of RCC [31]. Localized renal tumors are managed surgically, with even small tumors being removed due to a heightened risk of metastases. Once metastatic, this form of kidney cancer is almost always fatal.
The underlying genetic alteration is a germ line mutation/deletion of the FH gene; a “second hit,” i.e., loss of the normal somatic allele, precedes development of kidney cancer. Inactivation of FH has several consequences. Loss of fumarate hydratase activity leads to accumulation of its substrate, fumarate, interrupting a key oxidative metabolic pathway critical for meeting cellular bioenergetic and biosynthetic needs. Impairment of the Krebs cycle imposes a metabolic shift characterized by a reliance on aerobic glycolysis for generating ATP and by the use of glutamine as the major source of carbon for fatty acid synthesis. The metabolic shift in FH−/− cells is initiated and sustained by a number of factors. First, excess fumarate inhibits hydroxylation of HIF-1α, leading to the accumulation of this key driver of aerobic glycolysis. Additionally, FH–/− cells are characterized by decreased activity of activated protein kinase adenosine monophosphate kinase (AMPK), the master energy sensor of the cell, with consequent upregulation of mTOR activity [32]. Decreased AMPK activity also leads to decreased cytosolic iron levels by downregulating iron transporter activity (DMT1), which further diminishes the catalytic activity of prolyl hydroxylases, complementing the competitive inhibition of these enzymes imposed by fumarate accumulation. Lastly, excess fumarate leads to activation of an oxidative stress response mediated by nuclear factor E2-related factor 2 (Nrf2) [33]. Nrf2 activity is normally regulated tightly by an E3 ubiquitin ligase complex composed of Kelch-like ECH-associated protein 1(KEAP1) and cullin 3 (cul3). In FH−/− cells, KEAP1 undergoes a posttranslational modification (succination at cysteine residues) disrupting its interaction with Nrf2, thus allowing the latter to accumulate in the nucleus. Nrf2 is an essential transcriptional regulator mediating the cellular antioxidant response which is critical for neutralizing the effect of reactive oxygen species during oxidative stress. Activation of Nrf2 has been shown to redirect glucose and glutamine into anabolic pathways including glycolysis and the pentose phosphate pathway. The Nrf2 pathway is also activated in some forms of sporadic papillary RCC; mutations in KEAP1, Nrf2, and cul3 leading to disruption of protein–protein interactions have been described and are thought to mediate the oxidative stress response in these tumors [34]. The reliance of FH−/− cells on glycolysis and glutamine catabolism offers rational therapeutic targets. Approaches directed against these pathways are subjects of ongoing preclinical and clinical investigation.
6.3.1.2 Succinate Dehydrogenase Deficiency Kidney Cancer
SDH is the only enzyme that participates in both the TCA cycle and the electron transport chain; SDH is composed of four subunits that are bound to the inner mitochondrial membrane. Germ line mutations in genes encoding the SDH-C, SDH-B, and SDH-D subunits are associated with hereditary paraganglioma of the head and neck in addition to hereditary pheochromocytoma [35, 36]. Alteration of these subunits has been found to be associated with familial kidney cancer that can present with or without paragangliomas or pheochromocytoma [37, 38]. Loss of SDH activity leads to disruption of the TCA cycle and accumulation of succinate, with many biochemical and metabolic consequences similar to those seen with FH inactivation. Elevated levels of succinate inhibit the activity of prolyl hydroxylases, preventing the hydroxylation of HIF with resultant accumulation [11]. SDH-RCC presents with an aggressive phenotype associated with early onset and presentation with local symptoms or systemic manifestations associated with metastatic disease [39]. Treatment strategies similar to those employed in HLRCC are under investigation.
6.3.2 Von Hippel–Lindau Syndrome and Clear Cell Renal Cell Carcinoma
Patients with VHL syndrome present with a constellation of manifestations including central nervous system hemangioblastomas (cerebellum and spine), retinal angiomas, endolymphatic sac tumors of the inner ear, pheochromocytoma, pancreatic serous cystadenomas, neuroendocrine tumors, and epididymal cystadenomas, as well as renal cysts and clear cell kidney cancer. VHL patients often develop numerous, bilateral renal cysts, solid tumors, and mixed lesions. Renal tumors in VHL patients rarely metastasize when less than 3 cm in greatest dimension. Active surveillance of these cancers until they reach this size threshold has allowed for renal preservation while minimizing the number of surgical interventions needed to maintain oncologic outcomes [40, 41].
Genetic linkage analysis in VHL families led to the identification of the VHL tumor suppressor gene, located on the short arm of chromosome 3 [42, 43]. As a well-studied hereditary kidney cancer syndrome, it has provided profound insight into the behavior of both VHL-associated tumors and of nonfamilial clear cell kidney cancer. Indeed, somatic VHL gene alterations have been identified in up to 90 % of patients with sporadic ccRCC [44]. The VHL protein product is a component of an E3 ligase that targets several proteins for ubiquitin-mediated degradation [20]. The most widely studied consequence of VHL inactivation is its effect on the alpha subunits of the hypoxia-inducible factors. Loss of functional VHL protein prevents the ubiquitin-mediated degradation of HIF-α, leading to intracellular accumulation of HIFs [21, 22]. The stabilization of HIFs independent of ambient oxygen tension leads to the transcriptional activation of a number of genes including those encoding proangiogenic factors (VEGF, PDGF), glucose transporters (GLUT1), and growth factors (TGF-α/EGFR) which are necessary for tumor growth and survival. Elucidation of the critical role played by dysregulated VHL/HIF activity in the genesis of ccRCC has been instrumental in the development of clinically effective targeted strategies in this disease; antagonists of the VEGF pathway are currently a mainstay of therapy for patients with metastatic ccRCC.
The metabolic alterations underlying ccRCC are not as well characterized as those in tumors with impaired Krebs cycle activity. It is, however, clear that ccRCC is characterized by several features that promote a glycolytic phenotype. HIF-1 is often upregulated in VHL-deficient ccRCC, usually in conjunction with an activated Akt/mTOR pathway. The Cancer Genome Atlas recently conducted a comprehensive molecular characterization of over 400 primary ccRCC tumors [45]. In addition to VHL mutations, activation of the PI3K/Akt pathway was a recurrent theme, as was amplification of MYC. This analysis also uncovered evidence of metabolic reprogramming reminiscent of the Warburg effect, particularly in high-grade/high-stage tumors associated with poor survival; Krebs cycle enzymes and AMPK were downregulated, while glycolytic enzymes and enzymes catalyzing lipid synthesis were upregulated in these tumors. While attempts to target metabolic aberrations in ccRCC are still in the early stages of development, it is notable that patients with poor prognosis appear to derive clinical benefit from temsirolimus, an inhibitor of mTOR activity. Given the central role played by mTOR in regulating cellular metabolism, it is tempting to speculate that the activity of mTOR inhibitors in this context might relate to their ability to reverse the altered metabolic phenotype.
6.3.3 Hereditary Papillary Renal Cell Carcinoma and Type 1 Papillary Renal Cell Carcinoma
HPRC is an autosomal dominant condition resulting from activating mutations of the MET proto-oncogene located on 7q31. Affected individuals are at risk for developing bilateral, multifocal type 1 papillary RCC. Somatic MET mutations are seen in approximately 10–15 % of papillary kidney cancers; additionally, chromosome 7, on which both MET and its ligand HGF are located, is duplicated in a significant percentage of papillary RCC, and it has been suggested that this may represent an alternative means of activating the HGF/MET pathway [46].
MET encodes the hepatocyte growth factor receptor which exhibits tyrosine kinase activity [47]. In normal cells, MET is activated by binding to HGF; however, mutations in the tyrosine kinase domain, as seen in HPRC, render MET constitutionally active. Activation of MET engages multiple signal transduction pathways including the phosphatidylinositol 3-kinase pathway (PI3K). PI3K-RAS and PI3K-AKT activation play a role in increased expression of nutrient transporters resulting in additional uptake of glucose and amino acids. Mutations leading to constitutive activation of HGF/MET can also overcome the negative regulation of AMPK in response to nutrient starvation and/or low cellular ATP (which in normal conditions will promote fatty acid oxidation and ketogenesis).
Clinical management of HPRC is similar to that of ccRCC in VHL syndrome, with active surveillance of lesions until growth of at least one tumor to 3 cm triggers nephron-sparing surgical intervention. A small molecule kinase inhibitor of MET and VEGF receptors (Foretinib) demonstrated an overall response rate of 13.5 %, with the presence of a germ line MET mutation being highly predictive of response (50 % response rate in those with activating germ line MET mutations) [48].
6.3.4 Birt–Hogg–Dubé Syndrome and Chromophobe Renal Cell Carcinoma
Germ line mutations of the tumor suppressor FLCN, located on the short arm of chromosome 17 at position 11.2, have been noted in a large proportion of Birt–Hogg–Dubé (BHD) families with up to 70 % of BHD-associated tumors demonstrating loss of heterozygosity or sequence alterations in the somatic copy of FLCN [49, 50]. BHD is inherited in an autosomal dominant fashion and can be associated with pulmonary cysts predisposing to spontaneous pneumothorax, fibrofolliculomas, and renal tumors including chromophobe, oncocytic, and clear cell RCC in addition to oncocytomas and hybrid chromophobe/oncocytic tumors.
Folliculin, the protein product of FLCN, interacts with two other cellular proteins, folliculin interacting proteins 1 and 2 (FNIP1 and FNIP2), which aid in folliculin localization to lysosomes during amino acid starvation [51]. FNIP1 and FNIP2 are both phosphorylated by AMPK and bind to the γ-subunit of AMPK which responds to low levels of ATP and nutrients by inhibiting mTOR activity and thereby downregulating cellular metabolism, protein synthesis, and growth [52]. Both in vitro studies and animal models of FLCN inactivation suggest that loss of folliculin is associated with activation of both mTORC1 and mTORC2 activity [26]. The metabolic changes resulting from FLCN loss are yet to be fully characterized. Despite the presence of increased mTOR activity, the preponderance of evidence suggests that oxidative phosphorylation is active in BHD-associated tumors. In a mouse model where FLCN was selectively knocked out in skeletal muscle, FLCN-null cells appeared to have increased mitochondrial activity and demonstrated a shift toward oxidative phosphorylation [53]. The metabolic environment in BHD-associated tumors is likely to be more complex than simply a preference for oxidative phosphorylation and is the subject of ongoing investigation. A sporadic counterpart of BHD-associated tumors has not yet been identified. However, sporadic forms of chromophobe RCC (one of the histologic subtypes seen in BHD patients) appear to be characterized by genetic alterations that lead to increased mTOR activity (including PTEN loss, inactivating mutations in TSC1/2, and activating mTOR mutations) [54]. Additionally, mutations in mitochondrial genes that may promote a glycolytic phenotype have also been identified in these tumors, although their precise role in tumorigenesis and metabolic regulation remains to be determined [44].
6.3.5 Tuberous Sclerosis Complex
Tuberous sclerosis complex (TSC) is an autosomal dominant disorder resulting from germ line mutations in the TSC1 or TSC2 genes which act as tumor suppressor genes. Affected individuals are at risk for involvement of multiple organs, and CNS, skin, pulmonary, cardiac, ocular, and renal manifestations have been described. Although the most common renal manifestation is the presence of angiomyolipomas, clear cell RCC is not uncommon [55].
Tuberin and hamartin, the protein products of TSC1 and TSC2, respectively, form a complex with a GTPase-activating protein complex which acts to inhibit mTOR activity [56]. TSC loss is associated with increase in mTORC1 activity and elevated HIF-1α levels; in vitro, treatment of TSC2-deficient cells with rapamycin has been shown to restore normal levels of HIF, supporting the role of TSC2 in regulation of HIF translation by mTOR inhibition [57].
6.3.6 TFE-Fusion Renal Cell Carcinoma
Transcription factor E (TFE)3 and TFEB are transcription factors that belong to the microphthalmia-associated (MiT) family of transcription factors. Gene translocations involving both TFE3 and TFEB have been described in a variant of kidney cancer commonly referred to as “translocation RCC” characterized histologically by the presence of papillary and clear cell features and comprising around 1–5 % of all kidney cancers. TFE3 and TFEB are located on chromosome Xp11.2 and 6p21.2, respectively. A number of fusion partners for these transcription factors have been identified in kidney cancer (PRCC–TFE3, ASPSCR1–TFE3, SFPQ–TFE3, NONO–TFE3, CLTC–TFE3, and MALAT1–TFEB). Unique characteristics of this group include its predilection for presenting earlier in life, with up to 15 % of those with kidney tumors less than 45 years of age possessing a TFE gene fusion [58, 59]. A recent study suggests that these tumors may have identifiable imaging characteristics although these findings should be validated in a larger study [60–62].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


