The Field of Hemostasis and Thrombosis: Selected Translational Achievements
Victor J. Marder
William C. Aird
Joel S. Bennett
Sam Schulman
Gilbert C. White II
INTRODUCTION
As with any field of active scientific inquiry, the field of “Hemostasis and Thrombosis” has experienced its own evolution of thoughts and concepts, and the ever refining of the details of existing information. Given the enormous worldwide impact of hemostatic and thrombotic disorders, the intense focus of scientists on the field is justified, even essential. The modern era of coagulation could arguably be dated from 1905, when Morawitz proposed that four ingredients are involved in clot formation.1 By this formula, fibrinogen (factor I) is converted to fibrin by thrombin, produced from prothrombin (factor II) by the action of “thrombokinase” (factor III) and calcium (factor IV). The designations factors I, II, III, and IV were preserved in the nomenclature consensus agreement of 1959,2 which brought some semblance of order to the plethora of synonyms used for each real or implied clotting participant reported by that time. Observations regarding thrombotic disorders in the venous circulation were reported independently, and one could date the origin of this discipline to the late 19th century with the reports of Virchow (1856)3 and Trousseau (1865).4 Virchow’s many and dramatic breakthrough observations include the now-obvious connection between venous thrombosis of the lower extremity with pulmonary embolism,3 although the often-cited “Virchow’s triad” of causes for venous thrombosis may actually not belong to his credit alone.5 Trousseau brought to our attention the association of carpal spasm with tetany, but more memorably, the association of malignancy with venous thrombosis.4,6
RECOGNITION OF THROMBOSIS AND HEMOSTASIS AS A UNIFIED FIELD OF SCIENTIFIC INQUIRY
Despite the slow pace of discovery during the first half of the 20th century, there were a number of important and remarkable observations. Three separate discoveries of potential therapeutic agents are representative. First is the serendipitous purification of an anticoagulant from liver in 1916 by Jay McLean, a Johns Hopkins medical student,7,8 named “heparin” by his faculty mentor William Howell.9 Second is the serendipitous finding10 by Tillett and Garner in 1933 to 1934 of a clot-lysing ingredient in Streptococcal broth, termed fibrinolysin.11,12 Third is the detective work that solved the mystery of hemorrhagic disease in cattle caused by spoiled (moldy) sweet clover hay13, leading ultimately to the purification of dicoumarol by Campbell and Link in 1941.14,15 Translation of these observations to the clinic was likewise a slow process; it took 20 years until heparin was tested in a relevant animal model,16 14 years for a coumarin agent to be applied as a rodenticide,17 and 12 years for fibrinolysin (Streptokinase) to be used in animals18 and 16 in humans but for the lysis of pleural adhesions, not vascular thrombosis.19,20 Concurrent with these discoveries, inquiry into the components of blood coagulation began to reveal the complexity of this process. Historical reviews detail these events,21,22,23,24,25 but suffice it to say that patients with distinct deficiencies were identified in a flurry of discovery in the 1940s and 1950s, following work on antihemophilic factor (factor VIII) in 1937 by Patek and Taylor.26 The full cast of clotting factors was then identified, mostly through clinical comparisons and the differential correction of abnormal blood coagulation in the laboratory. In this way, patients were found who were deficient in “proaccelerin” (factor V) by Owren,27 “serum prothrombin conversion accelerator” (factor VII) by Alexander,28 “Christmas factor” (factor IX) by groups led by Aggeler,29 Biggs,30 and Schulmann,31 “plasma thromboplastin antecedent” (factor XI) by Rosenthal et al.,32 “Hageman factor” (factor XII) by Ratnoff,33 “Stuart-Prower factor” (factor X) by Hougie et al.,34 and “fibrin stabilizing factor” (factor XIII) by Duckert,35 some 17 years after its biochemical identification by Robbins36 and by Laki and Lorand.37
These observations were facilitated by the development of simple, yet incredibly important, bench technology for measuring blood coagulation, namely, the prothrombin time devised by Quick in 1935 (the “Quick time”)38 and the partial thromboplastin time by Langdell, Wagner, and Brinkhous in 1953.39,40 With the development of platelet aggregometry by Born and by O’Brien in 1962,41,42 the field of platelet physiology and pathology took a major leap forward21 beyond the 19th century description of a role for platelets in clot and thrombus structure,43,44,45 and the early 20th century descriptions of Glanzmann thrombasthenia,46 von Willebrand disease,47 and Bernard-Soulier syndrome.48
The expanding number of hereditary bleeding disorders and the vexing problem of competing terminology for the same clotting component contributed to a complexity that even to this day, frightens the medical student and humbles the young scientist upon entering the field. The nomenclature issue was resolved by the committee,2 providing for simplified nomenclature for the bewildering array of blood clotting components, using Roman numerals for each factor. More importantly, clarity to the question of how the factors interact to initiate clotting was provided in 1964 via the brilliant syntheses of the science by Ratnoff and Davie49 of the “waterfall” and by Macfarlane50 of the
“cascade” of biochemical events. Admittedly, these explanations were not without need for modification.22,23 For example, it was necessary to account for thrombin activation of factors V and VIII,51 for their function as cofactors and not enzymes,52 and for the activation of factor IX by the “extrinsic” system factor VII: tissue factor complex.53 Still, the schemes devised by Ratoff and Davie and by Macfarlane had a profound impact on our concepts of blood coagulation and provided a framework for a remarkable, ever-accelerating application of new scientific technology to the “field.”
“cascade” of biochemical events. Admittedly, these explanations were not without need for modification.22,23 For example, it was necessary to account for thrombin activation of factors V and VIII,51 for their function as cofactors and not enzymes,52 and for the activation of factor IX by the “extrinsic” system factor VII: tissue factor complex.53 Still, the schemes devised by Ratoff and Davie and by Macfarlane had a profound impact on our concepts of blood coagulation and provided a framework for a remarkable, ever-accelerating application of new scientific technology to the “field.”
One might assume that the “field” of hemostasis and thrombosis existed from the start. However, a formal designation of a discipline for the study of both bleeding and clotting disorders was glaringly absent until the mid-1900s, and many basic and clinical research groups focused uniquely on the diagnosis and treatment of patients with hereditary bleeding disorders caused by clotting factor or platelet disorder, or on the pathology, treatment, and prevention of thrombosis, mostly in the venous circulation. Arguably, the field of Hemostasis and Thrombosis began with three organizational events, the first being the founding of the journal Thrombosis et Diathesis Haemorrhagica (TDH) in 1957, formally inviting scientific submissions in both thrombosis and bleeding. Second, the International Committee on Clotting Factors, upon completing its nomenclature goals, changed its focus and name in 1964 to the International Committee on Thrombosis and Hemostasis (ICTH). Third, the International Society on Thrombosis and Haemostasis (ISTH) was established in 1969, incorporating the ICTH as the Scientific and Standardization Committee and collaborating with the journal TDH as a major outlet for publishing reports and scientific studies. Now, many former Hemophilia Centers are termed Hemophilia and Thrombosis Centers, reflecting a broader scope of interest and cementing the collaboration of basic scientists working in the areas of vascular and cell biology, biochemistry, genetics, physiology, immunology, and pathology with clinical scientists with expertise in diagnosis, pharmacology, biostatistics, epidemiology, and public health. A major goal of the field is to apply basic principles to clinical practice, and notable achievements have been accomplished in the field of Hemostasis and Thrombosis.
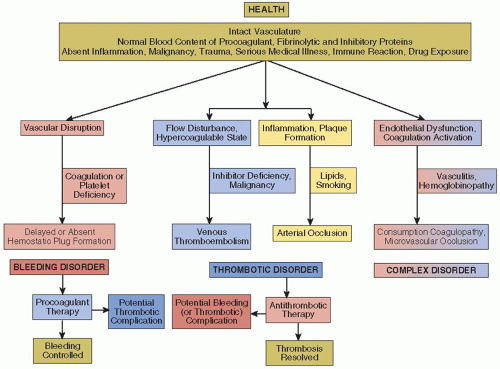 FIGURE 1.1 Schematic of bleeding and thrombotic disorders, contributing factors, and complications of therapy. |
OVERVIEW OF HEMOSTASIS AND THROMBOSIS
In a healthy individual with an intact vascular system, blood flow is unimpeded and the soluble and cellular components of the blood are balanced to maintain blood fluidity yet are poised for a rapid and effective response to vascular trauma (FIGURE 1.1). This balance is complex and multilayered but can be affected by confounding influences such as hereditary polymorphisms, inflammatory processes, malignancy, vascular injury, superimposed medical illness or surgical intervention, or drug exposure yet be primed for effective protection against excessive bleeding after vascular trauma.
HEMOSTATIC DISORDERS
Hemostatic disorders may first become apparent after vascular injury such as during surgery or may seem to be “spontaneous” without evident injury, but following otherwise trivial activities such as walking or dental care (FIGURE 1.1, left). While the distinction between normality and a mild bleeding state is often problematic, as in the case of “simple” purpura, fully manifest bleeding disorders, regardless of the etiology, may be dramatic and life threatening. Based on the current state of knowledge and clinical experience, the diagnosis of a hereditary coagulation disorder is usually straightforward. Nonetheless, the relative contribution of coagulation factor deficiencies to bleeding varies, and in some individuals with abnormal in vitro coagulation tests, bleeding may be unusually absent (factor XII deficiency) and there may even be a risk of thrombosis (lupus anticoagulant). The hemorrhagic manifestations of thrombocytopenia and/or platelet dysfunction are distinctive, manifest as mucocutaneous bleeding due to small capillary leaks. Immune disorders such as acquired hemophilia due to autoantibodies against or thrombocytopenia due to the appearance of platelet autoantibodies can appear suddenly in a patient with no prior history of bleeding. Once a diagnosis is made, treatment is usually evident and effective, but the same cellular and protein components that are involved in hemostasis also promote thrombosis. Thus, in some instances, such as with the use of recombinant factor VIIa to arrest hemorrhage, treatment carries a potential complication of thrombosis.51
VENOUS THROMBOSIS
The predispositions for venous or arterial thrombotic disease differ markedly. A triad of conditions, generally attributed to Virchow,5 consisting of the confluence of a systemic hypercoagulable state, disturbance of venous blood flow (stasis), and vascular injury makes for a perfect storm of venous thromboembolic (VTE) disease. For example, in 1959, Sevitt and Gallagher52 highlighted the VTE risk that can complicate hip fracture in which hypercoagulability is induced by the release of bone marrow contents into the circulation, the fracture causes complete immobility, and there is associated venous damage due to the initial injury or surgical repair. However, not all three components of the triad are required to present an increased risk of VTE, as reflected by the 80% prevalence of VTE after total knee arthroplasty in the absence of DVT prophylaxis53 or the smaller risk of VTE that accompanies ankle and even arthroscopic surgery54 In the latter instances, it would not be unreasonable to anticipate that additional factors, such as the absence of a natural anticoagulant inhibitor such as protein C (PC) or the presence of the factor V Leiden mutation, strongly impact on clinical outcome.55,56,57,58 The balance of hemostasis and thrombosis is reflected in their respective hereditary predispositions of absent inhibitors of procoagulant proteins or coagulation inhibitors.
Options of the effective treatment of venous thromboembolism are expanding rapidly with the recent introduction of oral thrombin and factor Xa inhibitors to the standard regimen of coumarins and heparins.59 Nonetheless, in converse to the treatment of bleeding disorders, (FIGURE 1.1, bottom center), treatment of VTE with anticoagulants carries a risk of a bleeding complication. Again, concomitant hereditary predispositions, in this case to bleeding, increase the risk of such a complication.
ARTERIAL THROMBOSIS
Arterial thrombosis is responsible for the most prevalent and lethal pathologic conditions in the world, myocardial infarction and ischemic stroke.60 The pathogenesis of these arterial events is distinct from that in venous thrombosis and usually results from the rupture of an atherosclerotic plaque in epicardial coronary vessels and often, but not always, involves an upstream embolic event in ischemic stroke. Thrombi in the vicinity of a ruptured atherosclerotic plaque have a prominent platelet component, initiated by exposure of blood to tissue factor on the plaque surface and in plaque debris. This results in a “white thrombus,” composed predominantly of platelets and fibrin, although a retrograde red thrombus composed of red cells and fibrin may form when arterial blood flow is blocked. The “white thrombus” is analogous to the primary hemostatic platelet plug that forms after vascular trauma, a clear example of the coalescence of cellular and coagulation responses in physiologic and pathologic conditions. Predispositions to arterial thrombosis are distinct from those that predispose to venous thrombosis, being primarily those influences that promote arthrosclerosis, and less commonly resulting from inflammation (vasculitis) or immune damage as in the antiphospholipid syndrome (APLS). Less often, arterial occlusion can result from the same pathophysiology as VTE, as after embolization of an atrial thrombus in a patient with atrial fibrillation or from paradoxical embolization in a patient with a patent foramen ovale.
Because platelets are the predominant component of arterial thrombi, antiplatelet drugs are the agents of choice in treating these disorders. As with treatment for VTE disease, there is a risk of antithrombotic treatment inducing a hemorrhagic complication, with the added burden that the hemorrhage may be intracranial in a patient with an ischemic stroke. Adding complexity to treatment, we now appreciate that treatment can result in thrombosis as well as hemorrhage. Thus, the heparin-induced thrombocytopenia (HIT) that results from immune complexes containing antibodies against platelet factor 4 (PF4) can produce arterial and venous thrombi, and vitamin K antagonists (coumarins) can induce cutaneous microvascular thrombosis producing skin necrosis in a patient with inherited PC deficiency.62,63
COMPLEX DISORDERS
These disorders manifest both bleeding and thrombosis and emphasize the disparate properties of some coagulation proteins. Thrombin, the central player in the coagulation cascade, mediates feedback amplification of the “intrinsic” system by activating factor XI; activates platelets to expose phosphatidylserine, thereby accelerating the formation of factor Xa; activates factors V, VIII, and XI; activates factor XIII and the thrombinactivated fibrinolysis inhibitor making clots more resistant to plasmin lysis; and cleaves fibrinopeptides A and B from fibrinogen to induce fibrin formation. Thrombin also binds to thrombomodulin (TM) on microvascular endothelial cells and changes its character to activate PC. When bound to the endothelial cell activated protein C (APC) receptor and in concert with cofactor protein S (PS), APC proteolytically inactivates factors Va and VIIIa, thus effectively turning off the coagulation cascade. Accordingly, thrombin is not only the quintessential procoagulant enzyme but also the quintessential anticoagulant molecule.
The prototypical example of a complex disorder (FIGURE 1.1, right) is disseminated intravascular coagulation (DIC), most
commonly associated with sepsis and endotoxemia, in which there is activation of both platelets and coagulation.64 This results in loss of endothelial antithrombotic functions, microclot formation, consumption of platelets and coagulation factors, microvascular occlusion causing organ failure, and bleeding. The difficulty in recognizing and managing DIC derives in part from the variable multi-organ dysfunction that can be mimicked by other pathologic syndromes. For example, thrombotic thrombocytopenic purpura, initially described by Moschowitz in 1924, results from the failure to cleave ultra-large high molecular weight von Willebrand factor (vWF) multimers because of an inherited or acquired ADAMTS 13 deficiency. This results in platelet thrombi in the microcirculation of the brain, kidney and even heart, lungs and liver.65 Thus, at one extreme are the abnormal or absent vWF proteins that cause the hemorrhagic syndrome of von Willebrand disease,66 while at the other extreme is the persistence of ultra-large multimers on the endothelial surface67 that are decorated with platelets and lead to microvascular occlusion.
commonly associated with sepsis and endotoxemia, in which there is activation of both platelets and coagulation.64 This results in loss of endothelial antithrombotic functions, microclot formation, consumption of platelets and coagulation factors, microvascular occlusion causing organ failure, and bleeding. The difficulty in recognizing and managing DIC derives in part from the variable multi-organ dysfunction that can be mimicked by other pathologic syndromes. For example, thrombotic thrombocytopenic purpura, initially described by Moschowitz in 1924, results from the failure to cleave ultra-large high molecular weight von Willebrand factor (vWF) multimers because of an inherited or acquired ADAMTS 13 deficiency. This results in platelet thrombi in the microcirculation of the brain, kidney and even heart, lungs and liver.65 Thus, at one extreme are the abnormal or absent vWF proteins that cause the hemorrhagic syndrome of von Willebrand disease,66 while at the other extreme is the persistence of ultra-large multimers on the endothelial surface67 that are decorated with platelets and lead to microvascular occlusion.
Multiple small and medium-sized vessels can be occluded as part of specific disorders, such as sickle cell disease, polyarteritis, and the APLS. APLS in particular may present with complex features, including non-bacterial thrombotic endocarditis, systemic activation of coagulation, embolic phenomena, placental ischemia, as well as the lupus inhibitor that prolongs the PTT and prothrombin time (INR). While patients with APLS may also have a specific coagulation factor inhibitor that causes bleeding,68 the inhibitor actually represents a risk for the thrombotic, not hemorrhagic, manifestations.69 Thus, patients with APLS can present a dilemma for clinicians as patients could simultaneously manifest both bleeding and thrombosis. Additional examples could include patients with the Budd-Chiari syndrome (hepatic vein thrombosis) portal hypertension and bowel ischemia who also have active bleeding from esophageal varices or patients with postoperative hip or knee replacement patient and significant bleeding into joint prosthesis, but also having a proximal DVT and PE.
SELECTED TRANSLATIONAL ACHIEVEMENTS IN THE FIELD OF HEMOSTASIS AND THROMBOSIS
Translational advances made by the “clotters” have had an immense impact in medicine. A limited number of highly selected translational achievements are described below and reflect the broad application of their insights on medical practice beyond the field of hemostasis and thrombosis.
NEW ORAL ANTICOAGULANTS
Heparin and coumarin-based vitamin K antagonists have been the mainstays of thrombosis treatment and prevention for more than half a century, and efforts to improve on their safety, efficacy, and especially their pharmacologic limitations, have led to development of some very inventive agents. Examples of new directions in anti-coagulant therapy include the strict anti-thrombin leech salivary gland extract, hirudin,70 and its recombinant or synthetic derivatives,71 and short peptide inhibitors of thrombin such as argatroban,72 which remain in clinical use as intravenous antithrombotic agents for patients with HIT. In 1992, Bode et al.73 at the Max-Planck Institute for Biochemistry reported the crystallographic solution of the 3-dimensional configuration of thrombin with a resolution of 1.9 Å and revealed the active site pocket in complex with D-Phe-Pro-Arg CH2C1 and other non-peptidic molecules. This observation paved the way for the design of highly-specific thrombin inhibitors with perfect fit into the serine protease pocket. Drug development at the Swedish company Hässle (subsequently Astra and then Astra-Zeneca) led to the first orally available thrombin inhibitor melagatran (and its prodrug ximelagatran).74 The concept applied to the design of these synthetic agents was to mimic the structure of fibrinopeptide A and, as opposed the slow-tight binding of another prototype (efegatran), melagatran demonstrated a high association rate constant indicating that it would rapidly inhibit thrombin.75 Gustafsson et al. pioneered the preclinical development and clinical trials with melagatran,76,77 showing that the drug had similar efficacy and bleeding risk as low molecular weight heparins and warfarin for prophylaxis against VTE,78,79 prophylaxis against stroke in atrial fibrillation,80,81 and treatment of VTE.82,83 Notwithstanding the liver toxicity that appeared during the phase III clinical trials and led to the withdrawal of the drug in 2006, the concept of an oral anticoagulant that avoided the multiple drug interactions and which did not require the careful laboratory monitoring used for coumarin agents, was correct. Direct-acting thrombin inhibition, not requiring activation of the serpin antithrombin (AT), is effective and with a bleeding risk similar or less than with conventional anticoagulants. A favorable risk/benefit-ratio was confirmed with the successor to melagatran, dabigatran, in a large trial in patients with atrial fibrillation, showing that reduction of ischemic stroke can be achieved simultaneously with reduction of intracranial bleeding.84 Currently, more than 500,000 patients are receiving long-term dabigatran for stroke prophylaxis. New small molecule anticoagulants that bind in the factor Xa serine protease pocket have also been developed, including rivaroxaban, apixaban and edoxaban and advanced clinical trials show impressive efficacy and safety results in stroke prevention in atrial fibrillation,84,85,86 in DVT prophylaxis and treatment,87,88,89,90,91 and in acute coronary syndromes.92 It now appears that we have entered a new age of anticoagulant pharmacotherapeutics that will decrease our reliance on parenteral heparins or the oral coumarins.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


