Molecular Evolution of the Vertebrate Blood Coagulation System
Marianne A. Grant
William C. Aird
Most multicellular organisms have evolved hemostatic mechanisms to prevent the loss of fluid and promote blood vessel repair following injury. In invertebrates, hemostasis is mediated by blood cell (hemocyte) aggregation and/or gelatinization of hemolymph. In vertebrates, blood clotting involves both cells (thrombocytes in nonmammalian vertebrates and platelets in mammals) and a highly conserved network of protease reactions that ultimately lead to thrombin-catalyzed conversion of a soluble plasma protein, fibrinogen, into an insoluble, polymeric fibrin clot. Hemostasis in vertebrates is balanced by a number of important natural anticoagulant pathways.
An interesting question is when did the clotting cascade evolve? Previous studies have demonstrated that thrombincatalyzed conversion of fibrinogen to fibrin occurs in all extant vertebrates, from jawless fish to mammals, but is conspicuously absent in invertebrates. These findings indicate that the clotting cascade evolved during an extremely narrow window of time (a mere 50 to 100 million years) in the ancestral vertebrate following the divergence of the protochordates (urochordates and cephalochordates) some 500 to 600 million years ago. A related question is how did the clotting system evolve? The answer, of course, is through natural selection of heritable traits that conferred a survival advantage. However, any such explanation must take into account not only the short time period during which the vertebrate clotting mechanism appeared, but also the fact that virtually every component of the clotting cascade is important and that some factors depend on others for their activity. As we will discuss in this chapter, the explanation appears to involve a combination of gene duplications, exon shuffling, and functional diversification. Finally, we must ask why did the clotting cascade evolve in the first place? At first glance, the response seems straightforward enough: to stem the loss of blood at sites of injury in animals with a highly pressurized closed circulation (i.e., vertebrates). However, there is now increasing evidence that coagulation may have played a critical early role in innate immunity.
The goal of this chapter is to review various evolutionary questions related to the clotting cascade. We begin by introducing basic principles of evolutionary biology. Next, we briefly review the mammalian clotting cascade and reframe the various components in terms of their protein domains or modules, which serve as a substrate for natural selection. After a brief discussion of comparative hemostasis, we provide a summary of what is known about the clotting cascade in extant vertebrates and invertebrates. Finally, we draw on these data to propose a scheme that explains the evolutionary origins of the clotting system.
PRINCIPLES IN EVOLUTIONARY BIOLOGY
Terminology
An important challenge in uncovering evolutionary histories is to reconstruct the past based on evidence from the present. Unfortunately, there is little in the fossil record that sheds light on the origins of hemostasis. Rather, we are left with comparative analyses of morphology, function, and molecular sequences. A central assumption in comparative anatomy and physiology is that two structures that look or act alike are closely related. However, that is not always the case. Structures may look similar simply because they have evolved similar adaptations. This is termed convergent evolution. An example is seen in the case of the horseshoe crab (Tachypleus tridentatus), which contains a clotting system comprised of a protease cascade of three serine protease (SP) zymogens and a clottable protein (coagulogen). However, these components arose independently of the vertebrate clotting proteins. Among other things, they lack γ-carboxylated glutamic acid (GLA) domains, epidermal growth factor (EGF)-like domains, and kringle domains, which promote essential interactions of vertebrate clotting factors. Conversely, divergent evolution describes dissimilar phenotypes that share a common ancestor. The interpretation of molecular phylogenies for gene sequence divergence is similarly wrought with limitations. It is widely believed that the vertebrate genome has undergone two whole-genome duplication (WGD) events in addition to many local duplications, translocations, gene conversions, and gene loss. Thus, it is important to distinguish between orthologous genes (orthologs) and paralogous genes (paralogs). Orthologous genes in different species have evolved directly from a common ancestral gene, whereas paralogs have evolved from a common ancestral gene through duplication and have diverged in sequence and function from the parent gene through mutations and selection or drift. Stated another way, orthology is the relationship where sequence divergence follows speciation, whereas paralogy is defined as that condition where sequence divergence follows gene duplication.
Methods
To establish the evolutionary history of the coagulation cascade, we must turn to modern-day animals (especially early diverging vertebrates) and examine how their blood clots and/or determine whether their genome contains sequences for the various clotting factors. Functional studies of nonmammalian vertebrates have routinely employed the same clotting time assays that are used in humans, including whole-blood clotting time, prothrombin (PT) time, and activated partial thromboplastin
time (aPTT). However, the absence of factor-deficient plasma from many of these species has precluded the use of mixing studies to measure individual clotting factor levels. Functional studies of blood clotting in early vertebrates are further hampered by inter- and intraspecies variations in clotting times, small volumes of available blood (e.g., in small fish such as the zebrafish), and the nonuniform plasma composition and body temperatures of poikilotherms.
time (aPTT). However, the absence of factor-deficient plasma from many of these species has precluded the use of mixing studies to measure individual clotting factor levels. Functional studies of blood clotting in early vertebrates are further hampered by inter- and intraspecies variations in clotting times, small volumes of available blood (e.g., in small fish such as the zebrafish), and the nonuniform plasma composition and body temperatures of poikilotherms.
Protein purification has been used to isolate fibrinogen in different species. However, such a strategy is not feasible for many of the SPs, which circulate at trace levels. With the advent of polymerase chain reaction (PCR), it has been possible to use degenerate primers (based on multiple sequence alignment of available gene sequences) against specific domains of clotting factors to clone the cDNA for SP clotting factors. More recently, searching for DNA sequences (orthologs) has been facilitated by the availability of the whole-genome sequence of the sea squirt (Ciona intestinalis), amphioxus (Branchiostoma floridae), puffer fish (Fugu rubripes), and other higher vertebrates. Complete genome sequence is not available for lamprey or hagfish. However there is a lamprey trace database maintained by the NCBI and EBO. Once DNA sequences are available from multiple species, it is possible to align sequences and construct phylogenic trees, which are diagrams that depict the lines of evolutionary descent of different species, organisms, or genes from a common ancestor. Sequence similarities and phylogenic trees based on aligned sequences provide important information about when clotting factors arose during evolution. In addition to analyzing DNA sequences, it is valuable to examine gene organization. For example, the occurrence and arrangement of catalytic and peripheral domains yields clues about gene duplication and exon shuffling. Similarities in intron-exon structures are characteristic of orthologous genes. The presence of two similar genes (paralogs) on the same chromosome is a clue that they originated from local duplication, rather than genome-wide duplication.
Taxonomy
Studies on the evolution of the clotting cascade have focused on the vertebrate-invertebrate divide, namely the chordates. Chordates form a phylum of animals that includes vertebrates and closely related invertebrates. There are three subphyla: urochordates or tunicata (composed of more than 3,000 species and represented by the sea squirt), cephalochordates or lancelets (composed of ˜29 species and represented by amphioxus), and the extant vertebrates including jawless fish or cyclostomes (hagfishes and lampreys) and jawed vertebrates or gnathostomes (composed of ˜53,000 species) (figure 2.1). Urochordates and cephalochordates are referred to as basal chordates, or protochordates. Among other features, they have a notochord that persists throughout life but do not have backbones. Traditionally, cephalochordates were considered to represent the closest living relatives of vertebrates, with tunicates representing the earliest chordate lineage. However, recent evidence indicates that urochordates are more closely related to vertebrates than are cephalochordates.1,2 Jawless fish are the earliest appearing vertebrates. The two extant genera are hagfish and lamprey. Although morphologic and molecular phylogenic studies have yielded conflicting results, it is believed that hagfish and lamprey are not monophyletic, but rather represent separate divergences, with hagfish being more primitive.3 The jawed vertebrates include cartilaginous fish (also termed elasmobranchs), the bony fishes (also termed teleosts), amphibians, reptiles, birds, and mammals.
Mechanisms
It is widely accepted that the vertebrate genome has undergone multiple rounds of WGD (reviewed in Ref.4) (figure 2.1). Although the timing and the genomic scale of such duplications remain poorly understood, the favored hypothesis is that the first round occurred in a common ancestor of jawed and jawless vertebrates (˜630 to 565 million years ago), with the second round occurring in a common ancestor of the jawed vertebrates (˜565 to 525 million years ago). An additional WGD may have occurred in an ancestor of the majority of ray-finned fish some 350 to 320 million years ago. The idea of genome duplication has been strengthened by the observations that model vertebrates often have multiple duplicate genes (paralogs) corresponding to a set of invertebrate orthologs.5,6 When a gene is duplicated, one of the copies is redundant and escapes purifying selection. Natural selection cannot operate on idle proteins, but only on those that are being used. Thus, most gene duplications from WGDs are rapidly lost (this accounts for why the genome of sea squirt is smaller [330 Mb] than that of amphioxus [540 Mb]). Rarely, the mutations may provide the gene with a new function. Indeed, duplicate functional modules arising from WGDs are likely to have played a critical role in enhancing morphologic and functional complexity during vertebrate evolution.4,7,8
Although it is widely accepted that large regional or WGD events contributed to the structure of vertebrate genomes,9,10,11 local duplication and translocation have certainly contributed to the evolution of multigene families on separate chromosomal regions. Domain shuffling (insertion) is another important mechanism for the evolution of novel genetic material. Since the domains are often correlated with exon boundaries, exon shuffling is considered to be the driving force behind domain shuffling.12 In this way, different domains of modular proteins may have evolved in unrelated protein families, and then been subsequently fused to previously existing domains.13 Other mechanisms of domain shuffling include simple fusion of genes and recruitment of mobile elements.
OVERVIEW OF MAMMALIAN BLOOD COAGULATION
Mammalian blood coagulation is a complex, balanced process initiated and regulated by a networked series of protein reactions and interactions that form positive and negative feedback loops. In humans, blood coagulation involves more than two dozen extracellular proteins, almost half of which are members of the SP family. Blood coagulation in mammals (figure 2.2) is initiated by the exposure and binding of factor VII (FVII) to membrane-bound (cell surfaces in the subendothelial matrix) tissue factor (TF). Bound TF activates FVII to the protease FVIIa generating a TF-FVIIa protease complex that can proteolytically activate FIX and FX, producing proteases FIXa and FXa, respectively. In the absence of its activated cofactor FVa, FXa generates trace amounts of thrombin by the proteolytic processing of PT. This thrombin cleaves FXI to FXIa.
FXIa then cleaves FIX to FIXa, which along with FVIIIa cleaves larger amounts of FX to FXa. Although insufficient to initiate significant fibrin polymerization, trace amounts of thrombin formed in the “initiation” stage of coagulation are sufficient to proteolytically activate FV and FVIII in a positive feedback loop. Greater amounts of activated FVa and FVIIIa function as cofactors for FXa and FIXa, respectively, forming complexes that amplify the generation of thrombin during the “propagation” stage, which leads to the generation of the large amounts of thrombin required to convert fibrinogen to fibrin. These reactions occur on the surface of platelets that are anchored to the subendothelial matrix via collagen and von Willebrand factor, which themselves activate platelets along with the newly generated thrombin.
FXIa then cleaves FIX to FIXa, which along with FVIIIa cleaves larger amounts of FX to FXa. Although insufficient to initiate significant fibrin polymerization, trace amounts of thrombin formed in the “initiation” stage of coagulation are sufficient to proteolytically activate FV and FVIII in a positive feedback loop. Greater amounts of activated FVa and FVIIIa function as cofactors for FXa and FIXa, respectively, forming complexes that amplify the generation of thrombin during the “propagation” stage, which leads to the generation of the large amounts of thrombin required to convert fibrinogen to fibrin. These reactions occur on the surface of platelets that are anchored to the subendothelial matrix via collagen and von Willebrand factor, which themselves activate platelets along with the newly generated thrombin.
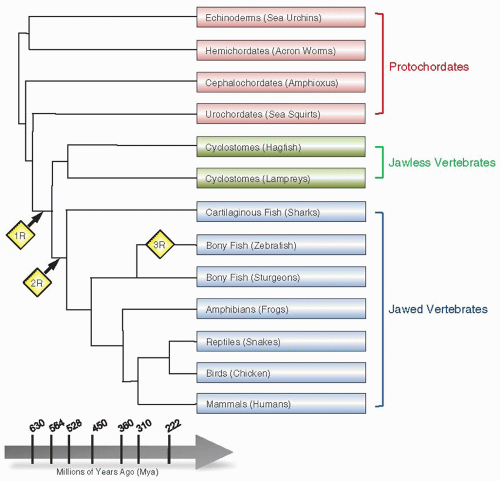 FIGURE 2.1 A phylogenetic tree representation of WGD during evolution. The points in evolutionary time where WGD events have been hypothesized to occur are indicated by yellow diamonds. The first round of WGD (1R) is thought to have occurred at the stage of divergence between urochordates and cyclostomes. The second WGD round (2R) is estimated to have occurred after the divergence of jawless fish (cyclostomes) and jawed vertebrates. A third round of WGD (3R) that is hypothesized to be a lineage-specific or fish-specific genome duplication is shown during divergence in the bony fish lineages. |
Thrombin also proteolytically activates the anticoagulant pathway (see figure 2.2). Thrombin forms a complex with the endothelial thrombomodulin (TM) receptor, and by an allosteric mechanism its substrate specificity is altered. The procoagulant substrates of thrombin, including FV, FVIII, and fibrinogen, are no longer efficiently proteolyzed. Instead, the thrombin-TM complex activates protein C (PC), generating activated PC (APC), which then associates with its protein S (PS) cofactor to rapidly inactivate procoagulant cofactors FVa and FVIIIa. Thrombin-TM-mediated activation of PC is further augmented 10- to 20-fold if PC is bound to another receptor, termed endothelial protein C receptor (EPCR).14 The procoagulant process of “propagation” described above occurs independently of the TF-VIIa complex, which is rapidly inactivated
by the tissue factor pathway inhibitor (TFPI). Thrombin, FIXa, and FXa are specifically inhibited by antithrombin (AT), heparin cofactor II (HC II), and Z-dependent protein inhibitor/protein Z (PZ) complex. The fibrin clot is lysed in fibrinolysis (see figure 2.2) by plasmin, which is generated from plasminogen by cleavage with tissue-type plasminogen activator (t-PA) and urokinase-type plasminogen activator (u-PA). This reaction is subsequently inhibited by two plasminogen activator inhibitors, namely plasminogen activator inhibitor (PAI)-1 and PAI-2.
by the tissue factor pathway inhibitor (TFPI). Thrombin, FIXa, and FXa are specifically inhibited by antithrombin (AT), heparin cofactor II (HC II), and Z-dependent protein inhibitor/protein Z (PZ) complex. The fibrin clot is lysed in fibrinolysis (see figure 2.2) by plasmin, which is generated from plasminogen by cleavage with tissue-type plasminogen activator (t-PA) and urokinase-type plasminogen activator (u-PA). This reaction is subsequently inhibited by two plasminogen activator inhibitors, namely plasminogen activator inhibitor (PAI)-1 and PAI-2.
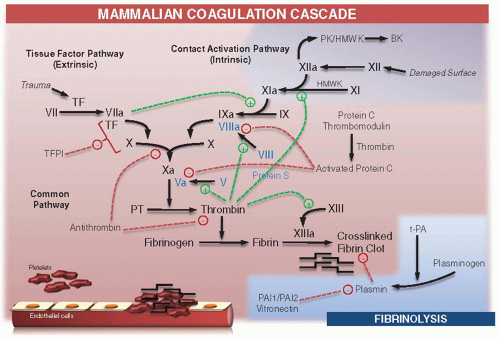 FIGURE 2.2 Schematic representation of coagulation in mammals. Solid black arrows represent the cascade of reactions of zymogen activation to active enzyme (suffix a) and the active complex formations driven by protein-protein interactions of factors in the system. Stimulatory actions are indicated with green dashed lines (+). Inhibitory actions are indicated with red dashed lines (-). PT, prothrombin; TF, tissue factor; TFPI, tissue factor pathway inhibitor; PAI 1 and PAI 2, plasminogen activator inhibitors 1 and 2 ; t-PA, tissue-type plasminogen activator; PK, prekallikrein; HMWK, high molecular weight kininogen; BK, bradykinin. |
The contact system is composed of three proteins: FXII, prekallikrein (PK), and high molecular weight kininogen (HMWK) (reviewed in Ref.15). The intrinsic or contact pathway is initiated when FXII is autoactivated upon exposure to negatively charged surfaces (including kaolin, silicone rubber, polyethylene, celite, and glass), forming FXIIa. FXIIa converts FXI to FXIa, which in turn converts FIX to FIXa. FXII also activates PK, forming plasma kallikrein that can reciprocally activate more FXII and liberate kinins (e.g., bradykinin) from HMWK.15 Kinins are small peptides that bind to receptors on many cell types, leading to an inflammatory response, including vasodilatation and vascular permeability. Although FXII promotes surface-activated clotting in vitro, deficiencies of the contact system proteins (if one excludes FXI) are not associated with clinical bleeding. Under in vivo conditions, FXI is activated by thrombin and not by FXIIa.
CLOTTING FACTORS, CELL SIGNALING, AND INFLAMMATION
In addition to their role in coagulation, several components of the clotting cascade mediate cell signaling. For example, the structural protein fibrinogen has been shown to activate inflammatory cells via distinct ligand-receptor interactions (reviewed in Ref.16). SPs bind to protease-activated receptors (PARs). PARs belong to the family of 7-transmembrane G-protein-coupled receptors. There are four known members (PAR-1, -2, -3, and -4), each of which is activated by proteolytic cleavage of its N-terminal domain. Thrombin binds and activates PAR-1 (and to a lesser extent PAR-3 and PAR-4). FVIIa-TF and FXa signal through PAR-2. In this way, initiation of coagulation is mechanistically coupled with cell-signaling events, which typically contribute to a proinflammatory phenotype (reviewed in Ref.17). In contrast, APC activates PAR-1 and when bound to EPCR is cytoprotective. APC exerts pleiotropic effects not only on endothelial cells, but also on neuronal cells and innate immune cell populations. Other natural anticoagulants, including AT and TFPI, also inhibit inflammation via cell-signaling-dependent mechanisms.18,19
GENE ORGANIZATION AND PROTEIN DOMAIN STRUCTURE
The clotting proteins contain genetically autonomous units (domains or modules), which are linked together to form unique domain architectures.13 In many cases, the domains are encoded by separate exons. Among these domains are SP catalytic domains and subsidiary (noncatalytic, regulatory) domains that mediate biologic specificity of the enzymes through protein-protein interactions and localization of the clot to sites of injury. For example, GLA domains and discoidin domains serve to anchor proteins to platelets at the sites of wounds. Kringle, EGF, and plasminogen-apple-nematode (PAN) domains mediate protein-protein interactions necessary for the ordered assembly of proteins in the clotting cascade. It has been proposed that these various nonregulatory domains represent genetically autonomous units (miniproteins) that have undergone shuffling (e.g., fusion to proteases) during evolution.13 The relationship between SP paralogs can be inferred by the kinds and arrangements of secondary domains associated with the catalytic protease domains. Indeed, the similarities in gene organization and protein domain structure among the major mammalian blood procoagulant, anticoagulant, and clotting cofactors suggest that that they evolved through the duplication and functional diversification of different gene families (figure 2.3).13 Three of these families are discussed in turn.
The GLA-EGF1-EGF2-Serine Protease Family
Vitamin K-dependent SPs are comprised of a GLA-EGF1-EGF2-SP domain structure common to FVII, FIX, FX, PC, and PZ. In contrast to these latter proteases, PT has two kringle (K) domains in place of the EGF domains. The EGF-like domain is a short sequence containing six cysteine residues, which have been shown to be involved in disulfide bonds. The main structure is a two-stranded beta-sheet followed by a loop to a C-terminal short two-stranded sheet. Similarly, the kringle domain has a unique characteristic triple-loop fold structure, whose conformation is defined by three pairs of cysteine disulfide bonds, a number of hydrogen bonds, and small pieces of antiparallel beta-sheet. (Kringle domains are also found in hepatocyte growth factor, apolipoprotein a, u-PA, t-PA, and plasminogen.) Analysis of amino acid sequence and codon usage at highly conserved residues associated with the active site function in the SP domain distinguishes PT from FVII, FIX, FX, and PC and suggests that thrombin was the ancestral blood coagulation enzyme.20,21,22 With the exception of PT, the genes encoding vitamin K-dependent factors of coagulation have virtually identical exon/intron distributions.
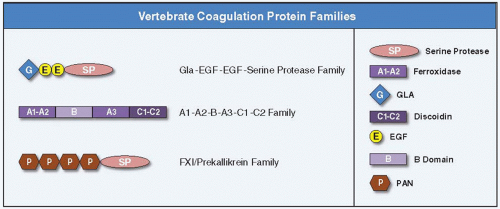 FIGURE 2.3 Schematic representation of the three major vertebrate coagulation protein families. The GLA-EGF-EGF-SP family comprises FVII, FX, FIX, PC, and PZ. The A1-A2-B-A3-C1-C2 protein family includes cofactors FV and FVIII. The PAN-SP family consists of FXI and PK. Domain representations are illustrated on the right. Epidermal growth factor (EGF)-like, discoidin (C), ferroxidase (A), γ-carboxy glutamic acid (GLA), plasminogen-apple-nematode (PAN), serine protease (SP), and B (B) domains. |
The A1-A2-B-A3-C1-C2 Cofactor Family
A1-A2-B-A3-C1-C2 domain structure is common to cofactors FV and FVIII, which are members of the ferroxidase family of proteins. FV and FVIII share sequence identity with the copperbinding plasma protein ceruloplasmin (CP) and the enterocyte transmembrane protein hephastin (HP), which have a common A1-A2-A3 domain structure. FV and FVIII differ from CP and HP in having two discoidin domains (C1 and C2) at their C-terminal ends. The discoidin domains of FV and FVIII bind to thrombocyte surfaces to orient the cofactors normal to the surface where they serve as binding cofactors for FX or FIX, respectively, and allow for optimal conversion of PT to thrombin. In FV and FVIII, there is an additional domain between domains A2 and A3, designated the B domain, which is released following thrombin activation. The B domain has no known structural homologs and is dispensable for procoagulant activity. The primary sequences of human FV and FVIII are 40% identical except in the B domain where there is no homology.
The FXI/Prekallikrein Family
FXI and PK are closely related SP proteins. The most notable feature of FXI and PK is a series of four disulfide-constrained approximately 90-amino acid repeats initially called apple domains. More recently, these domains have been assigned to the larger domain superfamily known as the PAN module superfamily.23 FXI is a disulfide-linked dimer that circulates in blood as a complex with HMWK.24 PK, the zymogen of the protease α-kallikrein, is a monomeric homolog of FXI with the same domain structure that also circulates in complex with HMWK.
COMPARATIVE FUNCTIONAL STUDIES OF BLOOD COAGULATION
The horseshoe crab has a hemolymph coagulation system. However, it uses different enzymes and structural proteins to form a clot compared with vertebrates. In other words, arthropod hemolymph clotting factors are not orthologs of vertebrate blood clotting factors. Indeed, fibrin clots have not been observed in any protochordate (the closest relatives of the vertebrates). In the sea quirt, hemostasis is mediated by circulating cells called hemocytes, which become clumped at sites of injury.3,25 Amphioxus does not have circulating blood cells and appears to lack clotting.3,25
The earliest animals to exhibit fibrin clots are the fishes. The first systematic study of fish coagulation was published in 1962 by Russell Doolittle.26 Blood and plasma were collected from lamprey, dogfish shark (an elasmobranch), blackfish (a teleost), and human. Whole-blood clotting time (blood collected directly into glass tubes without anticoagulant) was fast in blackfish (less than a minute), very slow in dogfish, and weak/poorly organized in lamprey even after 24-hour incubation at room temperature. Addition of lamprey tissue extract to the sample resulted in rapid clot formation within 1 minute. These findings led to the conclusion that lamprey lack an efficient intrinsic pathway. Addition of high concentrations of calcium resulted in accelerated clotting in dogfish and blackfish, but reduced clotting efficiency in human plasma. Plasma clotting in all fish was greatly accelerated by addition of aqueous extracts of homologous tissue (TF source). Although the effect demonstrated relative species specificity, even lamprey skin extract accelerated the clotting of human plasma and bovine brain extracts accelerated the clotting of lamprey plasma. Taken together, this early study suggested that blood coagulation in fish is fundamentally similar to clotting in mammals, with the major difference lying in the intrinsic pathway.
A subsequent report confirmed poor whole-blood clotting of lamprey blood.27 Consistent with an absence of an intrinsic pathway, the aPTT was >120 seconds. Surprisingly, the PT time with bovine thromboplastin (tissue extract) was shorter than it was with lamprey skin extracts. Elasmobranch blood also revealed long whole-blood clotting times, although these were highly variable between specimens. As with lamprey, the aPTT was more than 120 seconds. Comprehensive analyses of multiple species of bony fish revealed highly variable clotting times. Most of the fish clotted in times close to the human range. Homologous tissue extracts had variable thromboplastic effects.
A previous study of blood from multiple elasmobranch species showed very long clotting times in glass tubes (in some cases exceeding 48 hours).28 These data were used to conclude that cartilaginous fish lack an intrinsic clotting mechanism. Additional calcium (up to a certain concentration) resulted in a marked shortening of the clotting time. Beyond that concentration, the clotting time became prolonged again. The clots that formed lysed rapidly. Clot lysis was prevented by the addition of epsilon-amino caproic acid. In assays of the extrinsic pathway, tissue extract from skate accelerated skate clotting time (but not that of human plasma). Human brain extract accelerated human but not skate clotting times.
More recently, hemostatic parameters have been measured in zebrafish.29 Zebrafish thromboplastin was found to clot zebrafish but not human plasma, and rabbit thromboplastin resulted in only partial clotting of zebrafish plasma. Thus, these findings confirmed the species-specific nature of the TF-VII interaction. In addition, the aPTT was measurable (using a rabbit partial thromboplastin reagent), indicating the presence of an intrinsic cascade. This latter result is surprising based on the clear absence of contact factors in zebrafish. Warfarin dissolved directly into the tank water resulted in dose-dependent prolongation of PT time and aPTT. Addition of unfractionated heparin to zebrafish plasma resulted in dose-dependent prolonged aPTT.
Blood clotting in fish is highly dependent on environmental variables. For example, when fish that live in estuarine waters were exposed to normal fluctuations in temperatures and salinity, their whole-blood clotting varied widely.30 Similarly, in reptiles, which also lack an intrinsic system, clotting times are influenced by changes in temperature and pH that reflect variations associated with normal life activities.31
In summary, these and other studies suggest that all groups of fish (cyclostomes, elasmobranchs, and teleosts) generate thrombin through pathways involving vitamin K-dependent factors, exhibit FXIII-dependent fibrin crosslinking, and mediate fibrinolysis that is inhibited by the same factors that inhibit fibrinolysis in mammals.26,32,33
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


