The Factor XII-Driven Plasma Contact System
Thomas RennÉ
SUMMARY
The contact system is a protease cascade that is initiated by coagulation factor XII activation and operates on cardiovascular cells. The plasma proteins trigger procoagulant and proinflammatory reactions, via the intrinsic pathway of coagulation and the kallikrein-kinin system, respectively. The biochemistry of the contact system is well understood; however, its in vivo functions are just beginning to emerge. Recent data in animal models have shown that deficiency in contact system proteins largely abolishes thrombus formation but does not interfere with hemostatic mechanisms. Here we present an overview about the biochemistry and cell biology of the plasma contact system and its contribution to thromboembolic and inflammatory diseases.
PROTEINS OF THE PLASMA CONTACT SYSTEM
In the original waterfall cascade model, fibrin formation may be initiated by two distinct pathways, triggered by exposure of blood to a damaged vessel wall (extrinsic) or to blood-borne (intrinsic) factors.1,2 The intrinsic pathway of coagulation is initiated by factor XII (FXII, Hageman factor), in a reaction involving high molecular weight kininogen (HK) and plasma kallikrein (PK), collectively referred to as plasma contact system.3,4,5,6,7,8,9 Contact with negatively charged surfaces induces a conformational change in zymogen FXII resulting in a small amount of active FXII (FXIIa).10 FXIIa cleaves PK to generate active kallikrein, which in turn reciprocally activates FXII zymogen.11 FXIIa initiates fibrin formation through its principal substrate of the intrinsic coagulation pathway, factor XI (FXI), and also triggers liberation of the inflammatory mediator bradykinin (BK) by PK-mediated HK cleavage.5 Binding of BK to the kinin B2 receptor (B2R) activates various proinflammatory signaling pathways that dilate vessels, induce chemotaxis of neutrophils, and increase vascular permeability and fluid efflux12 (FIGURE 15B.1). The serpin C1 esterase inhibitor (C1INH) is the major plasma inhibitor of FXII and PK and controls proteolytic activity of the system.13
Factor XII
Factor XII (FXII, Hageman factor, EC = 3.4.21.38) is the 596-amino acid single-chain zymogen14 of the plasma serine protease α-factor XIIa.15 The single-chain glycoprotein migrates with an apparent molecular weight of 80 kDa in SDS-polyacrylamide gel electrophoresis (PAGE). FXII has a plasma concentration of 375 nM (30 µg/mL; normal range 15 to 45 µg/mL) and circulates in plasma with halftime of 50 to 70 hours.16 The human FXII gene spans approximately 12 kb on chromosome 5 (q33-qter) and has 14 exons.17,18 The protein is primarily expressed by hepatocytes; however, low levels of extrahepatic FXII mRNA synthesis have been detected in other tissues such as the placenta.19 Hepatocyte-specific expression of FXII in mice depends on the transcription factor HNF4α.20 Whether HNFa accounts for liver-specific expression of the coagulation factor in humans is not entirely clear; however, HNF4α antagonizes estrogen-induced upregulation of FXII gene expression in humans.21 Estrogens increase expression of FXII, and elevated FXII plasma levels are found in woman on hormonal contraception and during pregnancy. Indeed, the FXII gene promotor harbors a consensus motive (5′-G-43GGCANNNTGACC-31-3′) for an estrogen-responsive element.22
Protein Biochemistry of Factor XII
FXII is a multidomain plasma protein. The clotting factor is composed of a N-terminal fibronectin type II domain (encoded by exons 3 and 4), an epidermal growth factor (EGF) domain (exon 5), a fibronectin type 1 domain (exon 6), a second EGF domain (exon 7), a kringle domain (exons 8 and 9), a 55-amino acid long “proline-rich” hinge region (exon 9), and a trypsin-like protease domain (exons 10 to 14).18 The signal peptide that mediated secretion of the protein is coded by exon 2, whereas the first exon is noncoding. FXII binds to negatively charged surfaces via its heavy chain (N-terminal 353 residues) with implications for contact-mediated autoactivation of the zymogen (see below).
The FXII surface-binding site is not precisely known, and several regions of the heavy chain appear to contribute to a discontinuous binding pocket. Studies using recombinant FXII deletion variants or antibodies that interfere with surface-mediated FXII activation identified nonoverlapping portions in the proximal N-terminus (residues 1 to 28), the fibronectin type II domain (residues 39 to 47), the fibronectin type 1 domain (residues 134 to 153), the second EGF domain, the kringle domain, and the proline-rich region as potential FXII surface-binding sites.23,24,25,26 The monoclonal anti-FXII antibody (F1) binds to the kringle domain and enhances proteolytic activation.27 Within the first fibronectin type II domain, two histidine-containing Zn2+ ion-binding sites have been identified (residues 40 to 44 and 78 to 8228). Binding to zinc ions stabilized intermediate structures of the activation process and largely accelerates contact-induced formation of the active serine protease.29 FXII is activated by cleavage of a single peptide bond (Arg353-Val354). Limited proteolysis is either a result of autoactivation30 or mediated by another protease such as α-kallikrein, factor XIa,
factor XIIa, plasmin, or trypsin.13 Among these FXII processing enzymes, α-kallikrein is by far the most potent activator.11 The product of activation, α-FXIIa, consists of a 52 kDa N-terminal heavy chain and 28 kDa C-terminal light chain (protease domain), connected by a disulfide bond between residues Cys340 and Cys367.31 Both binding to negatively charged surfaces and binding to zinc ions largely accelerates FXII activation. Plasma prokallikrein (PPK) and factor XI are the principal substrates of α-FXIIa during contact activation.32,33 FXIIa can activate plasminogen and factor VII; however, the in vivo relevance of these steps remains to be established. α-FXIIa gets further cleavages at Arg334 and Arg343 resulting in β-FXIIa. The latter form lacks most of the heavy chain, binds poorly to surfaces, and does not support surface-mediated activation of PPK or factor XI.34,35 β-FXIIa can activate PPK in the fluid phase and also activates complement factor C1 in plasma.36 FXII and FXIIa stimulate the proliferation of cultured human hepatoma cells (HepG2) through a mitogenic activity that may reside in the EGF-like domains.37 Consistently, FXII stimulates ERK1/2 and Akt through uPAR, integrins, and the EGFR to initiate angiogenesis.38 The major plasma inhibitor of both α- and β-FXIIa is C1INH, with antithrombin III (ATIII) and plasminogen activator inhibitor-1 (PAI-1) having FXIIa-blocking activity.39
factor XIIa, plasmin, or trypsin.13 Among these FXII processing enzymes, α-kallikrein is by far the most potent activator.11 The product of activation, α-FXIIa, consists of a 52 kDa N-terminal heavy chain and 28 kDa C-terminal light chain (protease domain), connected by a disulfide bond between residues Cys340 and Cys367.31 Both binding to negatively charged surfaces and binding to zinc ions largely accelerates FXII activation. Plasma prokallikrein (PPK) and factor XI are the principal substrates of α-FXIIa during contact activation.32,33 FXIIa can activate plasminogen and factor VII; however, the in vivo relevance of these steps remains to be established. α-FXIIa gets further cleavages at Arg334 and Arg343 resulting in β-FXIIa. The latter form lacks most of the heavy chain, binds poorly to surfaces, and does not support surface-mediated activation of PPK or factor XI.34,35 β-FXIIa can activate PPK in the fluid phase and also activates complement factor C1 in plasma.36 FXII and FXIIa stimulate the proliferation of cultured human hepatoma cells (HepG2) through a mitogenic activity that may reside in the EGF-like domains.37 Consistently, FXII stimulates ERK1/2 and Akt through uPAR, integrins, and the EGFR to initiate angiogenesis.38 The major plasma inhibitor of both α- and β-FXIIa is C1INH, with antithrombin III (ATIII) and plasminogen activator inhibitor-1 (PAI-1) having FXIIa-blocking activity.39
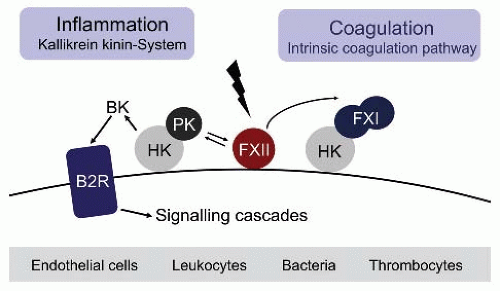 FIGURE 15B.1 The factor XII (FXII)-driven contact system. Contact with negatively charged surfaces activates coagulation factor XII (FXII) on leukocytes, bacteria, endothelial cells, and thrombocytes and initiates procoagulant and proinflammatory protease cascades. FXII triggers fibrin formation through the factor XI (FXI)-driven intrinsic pathway of blood coagulation. Activated FXII initiates the kallikrein-kinin system that generates the proinflammatory peptide hormone BK by PK-mediated cleavage of HK. |
Plasmakallikrein
PPK is the zymogen of the trypsin-like serine protease α-PK.40,41 PPK circulates in plasma at a concentration of approximately 580 nM (50 µg/mL), 75% to 90% in a noncovalent 1:1 complex with HK.42 PPK is primarily expressed by hepatocytes; however, PPK mRNA is found in numerous tissues, and small amounts of PPK are secreted from non-liver cells including pancreatic islets and kidney tubules.43 PK is a paralog of factor XI and both genes have evolved from a common ancestor.44 The human PK gene covers approximately 30 kb on chromosome 4q34-35,45 and is approximately 8 kb distant from the factor XI gene locus, consistent with a gene duplication event as underlying mechanisms for the two distinct proteins.46 Exon 1 of the PK gene is not translated, exon 2 encodes the signal peptide, exons 3 to 10 the four apple domains, and exons 11 to 15 the protease domain.
Plasma Kallikrein Functions
The principal mechanism of PK is to cleave the high molecular weight bradykinin precursor (HK) that liberates BK.47,48 Despite similar names, PK is not related to the large group of kininogenases called tissue-type (urinary or glandular) kallikreins. Mature human PK is a 619-amino acid single-chain polypeptide. Similarly to factor XI, PK is composed of four 90- to 91 amino acid apple domains (Apple 1 to 4) that form the heavy chain of the molecule and a C-terminal protease domain.49 The high degree of homology between PK and factor XI is supported by the findings that apple domains of PK and factor XI can be exchanged without compromising the capacity of the protein to be synthesized and secreted.50 In contrast to factor XI, PK is a monomer. PK migrates as a doublet with 86 and 88 kDa on nonreducing SDS-polyacrylamide gels. The two bands represent differentially glycosylated proteins. The residue Cys321 that forms the interchain disulfide bond in factor XI (connecting the two polypeptides of the dimeric protein) forms an intrachain bond with Cys326 in Apple 4 domain. PPK is a substrate of α-FXIIa, β-FXIIa, and trypsin.51 PPK is converted by these proteases to active α-kallikrein by proteolytic cleavage of the Arg371-Ile372 bond.52 Limited proteolysis of the single peptide bond results in heavy and light chain fragments (50 and 35 kDa) that remain connected by a disulfide bond connecting Cys364 in the Apple 4 domain and Cys484 within the protease domain.41 Similarly to FXII, a surface-dependent PPK autoactivation has been shown, but the physiologic importance of that mechanism is disputed. Following long-term incubation in buffer, α-kallikrein undergoes further conversion to β-kallikrein. Cleavage of the Lys140-Ala141 bond within Apple 2 domain abolished β-kallikrein binding to HK.50,53 So far the β-kallikrein form has not been identified in vivo. PPK may also be activated independent of FXII activity involving the prolylcarboxypeptidase (PRCP).54 PRCP has been originally recognized as a lysosomal exopeptidase exopeptidase. The physiologic relevance of PRCP as potential PPK activator in vivo and the molecular mechanisms by an exopeptidase that could function as an endopeptidase mediating limited proteolysis of PPK are subject to ongoing research. Gene targeting of PRCP accelerates thrombosis in mice.55 In contrast, targeting PPK expression interferers with thrombosis,56 arguing against PRCP-mediated PPK activation in thrombotic diseases. Additionally, extracellular carbonic anhydrase may activate PPK with implications for control of retinal permeability.57 PK functions as a kininogenase and cleaves HK resulting in release of BK. In a positive feedback loop, α-kallikrein also activates surface-bound FXII to α- and β-XIIa.58,59 Furthermore, PK has the capacity to convert prourokinase to urokinase60 and plasminogen to plasmin61 and for triggering the renin-angiotensin system by conversion of prorenin to renin.62 Due to its BK forming capacity, α-kallikrein has potent in vitro effects on leukocyte migration, stimulates monocytes, and induces neutrophils aggregation,63 chemotaxis, and elastase release.64 α-Kallikrein bound to mast cell surfaces has been identified as an activator of plasminogen during epithelial cell apoptosis, adipocyte differentiation, and stromal remodeling during mammary gland involution. Recent studies have suggested a role of PK for control of intracerebral hemorrhage in hyperglycemia.65 In contrast, hereditary deficiency in PK (Fletcher trait) and its substrate HK
(Fitzgerald trait) is not associated with increased bleeding risk or hemostatic abnormalities.66 The enzymatic activity of PK is primarily regulated by C1INH and α2-macroglobulin, with some contribution of protein C inhibitor, PAI-1, and ATIII.67,68
(Fitzgerald trait) is not associated with increased bleeding risk or hemostatic abnormalities.66 The enzymatic activity of PK is primarily regulated by C1INH and α2-macroglobulin, with some contribution of protein C inhibitor, PAI-1, and ATIII.67,68
High Molecular Weight Kininogen
HK is a 120-kDa β-globulin glycoprotein that circulates in plasma at a concentration of 670 nM (80 µg/mL) and is a member of the cystatin family of cysteine protease inhibitors. The human kininogen gene that is 27 kb in size contains 11 exons, and is located on chromosome 3 (3q-26qter). It is part of a cluster of related genes including α2-HS glycoprotein (α2-HS; also termed fetuin-A) and histidine-rich glycoprotein (HRG).8,69,70 Through alternative mRNA splicing, the kininogen gene produces transcripts for HK (3.5 kb) and its splice variant low molecular weight kininogen (LK, 1.7 kb), a 68 kDa glycoprotein with a plasma concentration of 1.3 µM (90 µg/mL).71 Alternative splicing of the primary transcript is controlled by the direct interaction of U1 small nuclear ribonucleoprotein (“snurp”) and the complementary sequences of the kininogen hnRNA. Exons 1 to 9 and a portion of exon 10 that codes for BK and its C-terminal 12 amino acids are common to the mRNAs for HK and LK, and the proteins share identical heavy chain regions (domains D1 to D3) and a BK-containing domain (D4) coded by these exons. The 5′-terminal portion of exon 10 encodes the unique 56-kDa C-terminal light chain of HK. This part of exon 10 and its flanking 90-nucleotide sequence are spliced out in the LK mRNA. The 4 kDa light chain of LK is coded by exon 11.72
In rats a pseudogene resembling the “canonical” kininogen gene as well as two other functional kininogen (“T-kininogen”) genes have been identified that are each transcribed as a single mRNA species. The T-kininogen mRNAs produce two closely related isoforms of T-kininogen (TK-I and TK-II), the precursors of T-kinin (TK, ISRPPGFSPFR).73 In contrast, mice have two functional highly homologous genes for kininogens (Kngl, Kng2) that result from a gene duplication event. Each Kng gene account for about half of the concentration of H-kininogen 1 and 2 and L-kininogen 1 and 2, respectively, in mouse plasma.74
In humans and rodents, HK and LK are predominantly expressed in the liver but can also be found in endothelial cells, granulocytes, renal tubular cells, and α-granules of platelets.75,76,77 It is not clear whether these cells synthesize kininogens or whether the proteins are taken up from plasma or cell culture supernatant containing serum components. Kininogen secretion into the plasma is mediated by amino-terminal signal peptides of 18 residues. The mature forms of LK (409 amino acids) and HK (626 amino acids) contain multiple posttranslational modifications such as hydroxylation and glycosylation.78 The detailed functions of the various carbohydrates are unknown, but they most likely affect solubility, half-life, and secretion of the kininogens. LK and HK contain 18 cysteine residues each, which form 9 disulfide bridges.79 Eight loops are present in the N-terminal portions, while a single disulfide bridge spans the extreme amino- and carboxy-terminal portions of LK and HK. The kinin-free form of HK (HKa) migrates with largely different apparent molecular weight than the 120 kDa single-chain form. The discrepancy indicates that HK undergoes major structural changes upon BK liberation. Under reducing conditions on SDS-PAGE, HKa has a heavy chain of 63 kDa and a light chain of 58 kDa, the latter of which is rapidly degraded by α-kallikrein and FXIa to smaller-sized fragments. HK is a typical multidomain plasma protein. HK domains D1, D2, and D3 of the heavy chain are homologous to cystatins, with D2 and D3 retaining cysteine protease inhibitory activity.80 The D4 domain harbors the kinin peptide that is releasable from both HK and LK through limited proteolysis mediated by kininogenases. HK is the preferred substrate for α-kallikrein, and its cleavage at residues Lys362 and Arg371 generates the nonapeptide BK (NH4-Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe-Arg).81 In contrast, LK is cleaved predominantly by tissue kallikreins resulting in formation of Lys-BK (kallidin).82,83 Kininogens are also cleaved by factor XIIa, factor XIa, and plasmin in reconstituted systems, but the physiologic relevance of these reactions is not clear. Release of BK and kallidin differs in kinetics and possible mechanism. BK is liberated locally and in response to stimuli on cell surfaces by the contact activation mechanism. There is a constant turnover of LK and basal static kallidin release.84 The two-chain form of LK that is the result of kallidin release is composed of a heavy chain (residues 1 to 362) and a light chain (residues 372 to 409). A single disulfide bridge spanning Cys10 with Cys389 connects both fragments. BK-free HK contains the same heavy chain as LK (domains D1 to D3, residues 1 to 362), and a much larger light chain composed of domains D5H and D6H (residues 372 to 626) is connected to the heavy chain by a single disulfide bond (Cys10-Cys596).8,85
A total deficiency of kininogens is rarely found in humans. In the case of Williams trait, a single C to T base transition at position 587 changes a CGA codon (Arg) to a TGA codon (stop) in exon 5, affecting the synthesis of both LK and HK.86 HK and LK deficiency in Brown-Norway Katholiek rats results from a single base pair substitution leading to a Tyr for Ala exchange at amino acid 163. This subtle change in kininogen sequence does not affect biosynthesis but leads to defective posttranslational modification and/or secretion of the kininogens such that their plasma levels are <1% of normal.87 T-Kininogen genes and, therefore, T-kininogen biosynthesis are unaffected in these rats.
Bradykinin
BK and kallidin are ligands of kinin B2 receptors and bind to the G-protein-coupled receptor with binding affinities in the lower nanomolar range.88,89 Kinins in plasma have short half-lives of <20 seconds and are rapidly degraded to inactive peptides by angiotensin converting enzyme, neutral endopeptidases, and aminopeptidases. BK and kallidin are also cleaved by carboxypeptidases, collectively referred to as kininase I (carboxypeptidase N, EC 3.4.17.3), to yield des-Arg9-BK and des-Arg10-Kallidin, which have longer half-lives and activate the kinin B1 receptor. Kininase II, identical to the angiotensin-converting enzyme (ACE), and neutral endopeptidase (NEP24.11, EC 3.4.24.11) inactivate BK or kallidin by removing a dipeptide, Phe-Arg, from the C-terminus.90,91 Kinins are potent proinflammatory mediators. The peptide hormones initiate an increase in intracellular Ca2+ that activates endothelial nitric oxide synthase-mediated nitric oxide formation.92 Endothelial denudation of large arteries abrogates the vasodilatory response to kinins. Kinins also stimulate the production and release of autacoids such as prostaglandin PGI2 and prostacyclin PGE2.93 Kinins induce vasodilatation, regulate local tissue perfusion, and importantly increased vascular permeability.94 Excessive BK production is the cause for a life-threatening swelling disease, hereditary angioedema (HAE) (see below). The importance of
the blood pressure lowering effects of kinins is highlighted in the clinical use of ACE inhibitors such as captopril and ramiprilate for the treatment of hypertension and cardiac failure.95,96
the blood pressure lowering effects of kinins is highlighted in the clinical use of ACE inhibitors such as captopril and ramiprilate for the treatment of hypertension and cardiac failure.95,96
Kinin-Independent HK Functions
In addition to the kinin sequence, other portions of HK have been associated with biologic function. The histidine/glycinerich region in the HK D5H domain mediates binding of the protein to anionic surfaces such as heparin.97 A peptide spanning the region in D5H between His479 and His498 of HK (HKH20) has been identified as the major cell-binding site of HK. Residues in the D3 domain contribute to HK binding to a broad variety of cells with 1,000 to 107 binding sites per cell.98 Heparan and chondroitin sulfate type proteoglycans have been identified as major HK cell surface binding site.99,100 The glycosaminoglycan chains of proteoglycans such as syndecans and glypicans bind the plasma protein to cardiovascular cells. Consistent with the interaction of the histidine-rich polypeptide in D5H with negatively charged glycosaminoglycans, zinc ions critically promote HK cell binding—presumably by an intercalating mechanism. Binding to glycosaminoglycans protects HK from cleavage, and detaching the hormone precursor seems to be a prerequisite for BK formation.101 Alternatively, HK has been shown to interact with p33/gC1qr (a mitochondrial protein102), urokinase receptor, and the cytoskeleton compound cytokeratin-1.103,104 The physiologic relevance of the proposed ternary complex for providing cell-surface HK-docking sites on intact cells is currently not precisely known.
Peptides from the D5H domain have antimicrobial activity and potently kill pathogens such as Escherichia coli, Pseudomonas aeruginosa, and Enterococcus faecalis by disrupting bacterial membranes. Similarly, an antibacterial fragment of the HK D3 domain has antibacterial activity versus Streptococcus pyogenes, Staphylococcus aureus, and Salmonella.105 A synthetic 26-amino acid peptide had similar properties. A protease from P. aeruginosa generates antimicrobial peptides from the D5H domain, demonstrating that products of HK processing other than BK provide part of the first line of defense against microbial invasion in vivo. D5H fragments have also been shown to have antiangiogenetic106 and leukocyte-recruiting107 activities with potential therapeutic implication.108 Cleaved HK or peptides from HK’s domain D5H blocks FXII-induced ERK1/2 and Akt phosphorylation.38 The D6H domain in the HK light chain region contains binding sites for factor XI and PK, and most of these proteins circulate in plasma bound in a complex with HK. Both PK and factor XI bind with high affinity (9 nM) to HK. The factor XI and PK binding sites on HK overlap, therefore, one molecule of HK can bind either a molecule of factor XI or a molecule of PK. Consistent with a high degree of homology PK and factor XI, apple domains A1, A2, and A4 form a discontinuous binding site for HK. Domain swapping experiments between PK and factor XI support a similarity in binding site among the zymogens and indicate that the Apple 2 domain contains the core region of the discontinuous HK-binding site.50 The contact induced procoagulant activity of HK as measured in the activated partial thrombin time (PTT) assay requires both D5H-mediated association of HK with artificial anionic surfaces and D6H-mediated binding of factor XI and PK to HK. Since LK is not binding to PK or factor XI, the protein lacks procoagulant activity in the PTT assay (FIGURE 15B.2A,B).
CONTACT ACTIVATION
Mechanism of Contact Activation
Contact activation is the mechanism of contact-induced FXII zymogen activation. Contact activation is initiated by activation of FXII on a surface, and activated FXII (α-FXIIa) catalyzes activation of additional FXII. Alternatively, α-FXIIa catalyzes conversion of PK to α-kallikrein, which in turn amplifies the process through reciprocal activation of FXII.11 The relative importance of FXII auto activation on surfaces versus reciprocal activation of FXII and PK remains to be analyzed and may depend on the nature of the contact system activating surface. Contact activation can propagate from the activating surface into the plasma fluid phase. α-Kallikrein cleaves HK and liberates BK, activates the renin-angiotensin system by converting prorenin to renin, and initiates the alternative complement pathway by activation of C3. Surface-bound α-FXIIa is concerted to β-FXIIa, which does not bind to surfaces but potently activates PK, and the classical pathway of complement via its substrate C1. The importance of contact system-driven activation of the complement system for host-defense reactions in vivo is not precisely known; however, some individuals with severe deficiency in contact system proteins appear to suffer from increased susceptibility for bacterial infections.109
Contact activation provides the mechanistic basis for one of the most commonly used diagnostic clotting test, the activated partial thromboplastin time (aPTT). The contact surface used in aPTT assays is typically a negatively charged nonphysiologic substance such as kaolin (aluminum silicate), celite (diatomaceous earth), or ellagic acid. The aPTT is widely used in clinical practice for preoperative screening and monitoring of anticoagulation therapy. Despite its indispensible role in fibrin formation in the aPTT assay, FXII initiated coagulation was believed to have no important function in vivo. This notion is based on the clinical observation that FXII deficiency in humans and animals is not associated with abnormal bleeding despite causing a marked prolongation of the aPTT.110,111 In contrast, deficiency of the extrinsic pathway trigger factor VII results in severe bleeding in humans, whereas complete tissue factor (TF) deficiency in mice is not compatible with life due to intrauterine bleeding.112 Similar to FXII-deficient individuals, humans lacking the other contact proteins, PK or HK, do not have impaired hemostasis and do not show signs of any symptoms of pathologic spontaneous or injury-related bleeding. Contact factors can assemble and activate ex vivo when blood is exposed to bioincompatible surfaces, contributing to the systemic inflammatory response that occurs when blood is passed through an extracorporeal circulation system, such as during cardiopulmonary bypass surgery.
Contact activation-independent pathways for FXII activation have been suggested. Several proteinases including elastase, trypsin, bacterial proteases, and plasmin, convert FXII to FXIIa (aFXIIa form). Activation of the contact proteases by bacterial surface proteases is important for tissue invasion by the organism. The pathogens utilize the host contact system to form BK on their surfaces. Local BK activity increases vascular permeability and facilitates the invasion of bacteria from the blood into the surrounding tissues.113
Contact Activators
FXII can be activated by macromolecular constituents of the subendothelial matrix such as glycosaminoglycans and collagens, nucleotides, sulfatides, soluble polyanions, or nonphysiologic
materials such as glass or polymers (reviewed by9), but the physiologically relevant mechanism for FXII activation has remained enigmatic for decades. Just recently, physiologic FXII contact activators have been identified (see below).
materials such as glass or polymers (reviewed by9), but the physiologically relevant mechanism for FXII activation has remained enigmatic for decades. Just recently, physiologic FXII contact activators have been identified (see below).
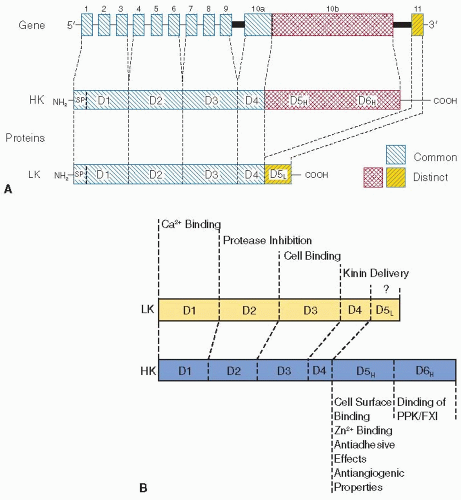 FIGURE 15B.2 Kininogen gene and structure-function relationships. Left: LK and HK are products of a single gene of 11 exons (E1 to E11). E1 to E9 encode domains D1 to D3 preceded by a signal peptide. Domain D4 contains the kinin moiety and is encoded by the 5′-terminal portion of exon 10 (E10a). The mRNAs for LK and HK are identical in their 5′-parts including exons 1 to 10a. They differ in their 3′-portions. The 3′-segment of exon 10 (E10b) encodes HK domains D5H and D6H, and exon 11 codes for LK domain D5L. Abbreviations: SP, signal peptide; LK consists of five domains (D1 to D5L), and HK contains six domains (D1 to D6H). Right: The domain structures of human kininogens are shown (not drawn to scale). Their common domains D1 to D4 bind Ca2+ (D1), inhibit cysteine proteinases (D2, D3), mediate cell binding (D3), and contain the kinin segment (D4). The biologic function, if any, of LKspecific domain D5L is unknown: HK-specific domain D5H contains a major cell binding site, Zn2+ binding site(s), and mediates antiadhesive effects of kininogens: Domain D6H exposes overlapping binding sites for FXI and PPK. |
Glass was one of the first substances identified to activate FXII. The white clay minerals kaolin and celite, two other silicarich compounds, are the most commonly used FXII activators for coagulation diagnostics. Kaolin is also used to terminate blood loss after serious injury in combat victims.114 Although the idea of employing the contact system to seal injuries is interesting, exposing flowing blood to kaolin triggers thromboembolic events115 and produces excessive hydratization energy causing burn necrosis at the wound site. Another classical FXII activator is ellagic acid (2,3,7,8-tetrahydroxy-chromeno[5,4,3-cde] chromene-5,10-dione). Ellagic acid triggers FXII-driven coagulation in plasma,116 and the substance is commonly used as the starter for the aPTT assay. Infusion of ellagic acid into humans is prothrombotic.117 However, it remains unclear whether the hypercoagulant response is a direct result of FXII activation or ellagic acid effects on platelet aggregation, or a combination of both. Ellagic acid also activates the fibrinolytic system possibly involving PK activity118 and when administered in vivo reduces blood pressure presumably involving BK activity.119
A well-characterized FXII contact activator is high molecular weight dextran sulfate (DX500.000). Dextran sulfate is a polysulfated polysaccharide of glucose moieties. DX-mediated FXII activation is critically dependent on the chain length and degree of sulfation of the polyanion.120 High molecular weight DXS (500 kDa) is a potent stimulator of FXII, whereas shorter DXS polymers fail to activate FXII. When injected in large animals, long-chain DXS induces BK-mediated hypotension,121 but does not trigger intravascular coagulation. Polysaccharide-stimulated BK generation may have fatal effects. Starting November 2007, there was a dramatic increase in heparin-induced adverse reactions such as lethal acute hypersensitivity reactions in the United States and Germany in patients intravenously receiving commercial heparin of specific lots from a single manufacturer (http://www.fda.gov/cder/drug/infopage/heparin/adverse_events.htm). Greater than 150 patients deceased from anaphylactic hypotension, associated with intravenous heparin treatment. Comprehensive analyses identified a nonnatural contaminant occurring in suspect preparations of heparin that was characterized as oversulfated chondroitin sulfate (OSCS).122 OSCS-contaminated heparin has greatly increased potency for activating FXII and triggering PK-mediated BK formation in human plasma and in a model of
experimental hypotonic shock in vivo.123 Additionally, infusions of OSCS-contaminated heparin were associated with complement activation indicated by elevated plasma levels of C3a and C5a. These catastrophic reactions in patients are reminiscent of experimental shock models induced by DX-stimulated BK formation in pigs. Infusion of DX500.000120 induced transient systemic hypotension, and icatibant (a B2R antagonist) blocked the drop in blood pressure.121 BK-forming activity seems to be dependent on negative charge density of the polysaccharide rather than on a defined structure. OSCS activates the kallikrein-kinin and complement systems as potently as does DXS.
experimental hypotonic shock in vivo.123 Additionally, infusions of OSCS-contaminated heparin were associated with complement activation indicated by elevated plasma levels of C3a and C5a. These catastrophic reactions in patients are reminiscent of experimental shock models induced by DX-stimulated BK formation in pigs. Infusion of DX500.000120 induced transient systemic hypotension, and icatibant (a B2R antagonist) blocked the drop in blood pressure.121 BK-forming activity seems to be dependent on negative charge density of the polysaccharide rather than on a defined structure. OSCS activates the kallikrein-kinin and complement systems as potently as does DXS.
Charge-dependent contact system activation is not restricted to polysaccharides. It is known for decades that FXII binds to insoluble collagen, enhancing coagulation.124 The interaction is dependent on a repetitious presentation of negative charges in the collagen fiber.124 However, later reports argued against a direct activation of FXII by collagen. Recent studies have reevaluated the importance of collagen for FXII activation.125 FXII was shown to bind to collagen fibrils of various origins. When added to plasma (equine), type I collagen promoted thrombin formation and plasma clotting in an FXII-dependent manner. FXII activity is critical for collagen-stimulated thrombus formation in flow chambers.126 Additionally, collagens may stimulate the contact system indirectly.127
Dextran sulfate may have physiologic analogues. It was believed that mast cell-triggered leakage is predominantly if not exclusively mediated by histamine release, and targeting histamine signaling is widely used therapeutically to treat edema formation associated with aberrant mast cell activity.128 Mast cell secretory granules also contain highly sulfated polysaccharides, with heparin as a major constituent. Addition of mast cell-derived heparin initiated BK formation in plasma in an FXII-dependent manner and triggered the FXII/PK cascade resulting in HK procession. Consistent with ex vivo data,129 topical heparin application increased vascular permeability in an FXII-dependent manner in mice. Intravital microscopy and tracer extravasation experiments identified BK as the active mediator for increased leakage in heparin-driven plasma extravasation in mice.130 Under these conditions the FXII substrate FXI was not activated. The detailed mechanism for selective activation of the FXIIa substrate PK over FXI is not entirely clear but may reflect a higher plasma concentration of PK versus FXI and higher specificities of heparin-dependent plasma inhibitors such as AT III for activated FXI versus PK. Similarly mast cell-derived chondroitin sulfates have been proposed as activators of the kallikrein-kinin system in vitro.129 However, the physiologic relevance of this mechanism has not been elaborated so far.
Based on the experimental data, targeting heparin-initiated BK formation may represent a promising strategy to protect against defective barrier function attributed to mast cell activation in a broad variety of diseases. Indeed, FXII gets activated on particulate matter that is a component of air pollution.131 BK formed by the FXII-driven contact pathway may contribute to intestinal pulmonary edema in allergic responses to air pollution. A similar mechanism contributes to bacterial infections. Contact system proteins assembled on the surface of E. coli and Salmonella through their specific interactions with fibrous bacterial surface proteins, curli and fimbriae, induce proinflammatory reactions via BK release.113 Pharmacologic inhibitors of contact system-driven BK formation interfere with Salmonella-mediated pneumonia and pulmonary leakage in rodent models.132 BK liberation from HK contributes to the host defense response to infection; however, high BK levels can contribute to hypotension and vascular leak associated with septic shock. Furthermore, BK generated on the bacterial membrane contributes to tissue invasion of the pathogen by opening endothelial cell-cell contacts. Plasma levels of contact factors are reduced in septic shock, and low factor XII and HK levels are predictors of fatal outcome during the systemic inflammatory response syndrome that is a feature of sepsis. Contact factor activation has been noted during meningococcal sepsis and in toxic shock syndrome. The importance of contact activation to the sepsis syndrome is shown by studies of E. coli sepsis in baboons, where FXII inhibition prevented hypotension (Table 15B.1).
Related posts:
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Molecular Basis for Platelet Secretion
Molecular Basis for Platelet Secretion
 Acquired Nonimmune Thrombocytopenia
Acquired Nonimmune Thrombocytopenia
 Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


