Primary central nervous system (CNS) lymphoma is a challenging subtypes of aggressive non-Hodgkin lymphoma. Emerging clinical data suggest that optimized outcomes are achieved with dose-intensive CNS-penetrant chemotherapy and avoiding whole brain radiotherapy. Anti-CD20 antibody-based immunotherapy as a component of high-dose methotrexate-based induction programs may contribute to improved outcomes. An accumulation of insights into the molecular and cellular basis of disease pathogenesis is providing a foundation for the generation of molecular tools to facilitate diagnosis as well as a roadmap for integration of targeted therapy within the developing therapeutic armamentarium for this challenging brain tumor.
Key points
- •
Long-term survival and cure is feasible in primary central nervous system lymphoma (PCNSL) without whole brain radiotherapy (WBRT).
- •
WBRT consolidation is associated with severe neurotoxicity, particularly in patients older than 60.
- •
High-dose chemotherapy is currently under investigation as first-line consolidation.
- •
There is a need for novel therapies that target key survival pathways in PCNSL, including activation of nuclear factor-κB survival signaling.
Introduction
Although primary central nervous system lymphoma (PCNSL) remains a rare neoplasm, representing only 2% to 3% of all cases of non-Hodgkin’s lymphoma (NHL), the incidence of PCNSL among immunocompetent patients seems to be increasing, particularly among persons age 65 years and older. The characteristic pathobiology of PCNSL is that of an aggressive lymphoma, localized within the central nervous system (CNS) and often disseminated within brain, cranial nerves, leptomeninges, cerebrospinal fluid (CSF), intraocular structures and spinal cord, without overt systemic disease.
PCNSL has long been recognized to be an aggressive brain tumor associated with a poor prognosis. Historically known as reticulum cell sarcoma or microglioma, management principles for this disease have emerged slowly. Beginning in the 1960s, in the absence of prospective data, whole brain radiotherapy (WBRT) was used as the first-line intervention as a means to elicit immediate responses in patients faced with a rapidly deteriorating course; WBRT alone typically resulted in median survival of 12 months. To date, the most significant advance in PCNSL has been the recognition, in the 1970s, of the efficacy of high-dose methotrexate (HD-MTX). Several recent prospective trials have demonstrated markedly improved outcomes in PCNSL. Our goal is to highlight significant advances in our understanding of disease biology, diagnosis, staging, and therapeutic management.
Introduction
Although primary central nervous system lymphoma (PCNSL) remains a rare neoplasm, representing only 2% to 3% of all cases of non-Hodgkin’s lymphoma (NHL), the incidence of PCNSL among immunocompetent patients seems to be increasing, particularly among persons age 65 years and older. The characteristic pathobiology of PCNSL is that of an aggressive lymphoma, localized within the central nervous system (CNS) and often disseminated within brain, cranial nerves, leptomeninges, cerebrospinal fluid (CSF), intraocular structures and spinal cord, without overt systemic disease.
PCNSL has long been recognized to be an aggressive brain tumor associated with a poor prognosis. Historically known as reticulum cell sarcoma or microglioma, management principles for this disease have emerged slowly. Beginning in the 1960s, in the absence of prospective data, whole brain radiotherapy (WBRT) was used as the first-line intervention as a means to elicit immediate responses in patients faced with a rapidly deteriorating course; WBRT alone typically resulted in median survival of 12 months. To date, the most significant advance in PCNSL has been the recognition, in the 1970s, of the efficacy of high-dose methotrexate (HD-MTX). Several recent prospective trials have demonstrated markedly improved outcomes in PCNSL. Our goal is to highlight significant advances in our understanding of disease biology, diagnosis, staging, and therapeutic management.
Etiology
Risk factors for PCNSL include acquired and/or congenital immunodeficiency states. PCNSL is an AIDS-defining illness associated with a low CD4 cell count (<50 cells/mL) and Epstein–Barr virus (EBV) infection. In systemic AIDS-related lymphomas, EBV infection of the lymphoma may be predictive of secondary CNS involvement. Congenital immunodeficiency states such as Wiskott-Aldrich syndrome, severe combined or common variable immunodeficiency, or ataxia-telangiectasia carry an approximately 4% risk of PCNSL. Posttransplant lymphoproliferative disorder (PTLD) involving CNS develops in 1% to 2% of renal transplant recipients and 2% to 7% recipients of liver, cardiac, and lung transplant recipients. CNS PTLD is associated with EBV in the setting of iatrogenic T-cell immunodeficiency induced by immunosuppressive agents such as mycophenolate mofetil (CellCept, Genentech, San Francisco, CA). Among patients with PCNSL without clinically overt immunosuppression, EBV infection of lymphoma is rare.
Clinical features and pathogenesis
PCNSL is typically a highly infiltrative neoplasm that has been characterized as a “whole brain disease,” particularly at relapse. For this reason, its radiographic appearance typically underestimates disease extent, and like malignant gliomas, PCNSL is not amenable to curative resection. One of the archetypical histologic features of PCNSL is angiotropism; the accumulation of lymphoma cells around tumor vessels, a phenotype that likely disrupts the blood–brain barrier and enables radiographic detection of lesions via pathologic contrast enhancement. PCNSL commonly is diagnosed as a solitary mass, typically with vasogenic edema and mass effect. The frequency of multiple lesions is increased among the immunosuppressed ( Fig. 1 ).
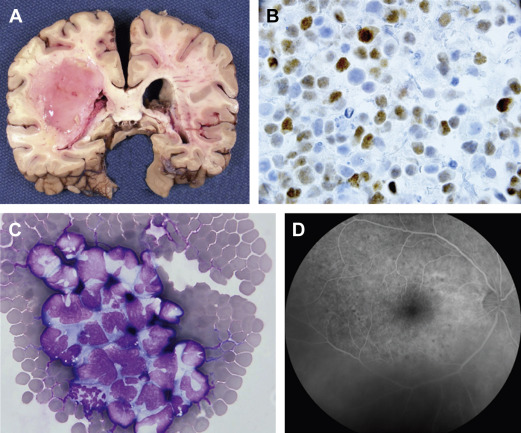
Although NHL presenting in the brain is typically classified as PCNSL, subclinical tumor-related clones are often detectable in the blood and bone marrow of PCNSL patients, suggesting that the brain microenvironment might promote malignant progression.
Intraocular disease is a common manifestation: 20% of PCNSL patients present with involvement of the retina, uvea, and vitreous. An important principle is that apparently localized intraocular lymphoma (IOL) will disseminate within brain in greater than 80% of cases; therefore, detection of IOL mandates staging of the neuroaxis. Therapies for IOL that address this risk should be strongly considered.
Approximately 95% of PCNSL are large B-cell lymphoma; other include T-cell (2%), Burkitt, lymphoblastic, and marginal zone lymphomas. PCNSL is distinguished from dural-based marginal zone lymphomas because these rarely invade brain parenchyma and typically share overlapping radiographic features on MRI with meningioma.
Molecular pathogenesis of primary central nervous system lymphoma
Determination of the unique genetic features of PCNSL poses a greater challenge than for systemic diffuse, large B-cell lymphoma, both because of the rarity of this neoplasm and the paucity of material available for investigational studies. Most diagnostic specimens are obtained by stereotactic biopsy or via analysis of the CSF. Most investigations have focused on the elucidation of PCNSL, large cell type. Between 50% and 80% of PCNSL express BCL6 by immunohistochemistry ; 95% stain positive for MUM-1 and, therefore, the majority of PCNSL cases are of a late germinal center or an activated B-cell immunophenotype. Immunohistochemical characterization of tumors from patients that participated in CALGB 50202 demonstrated that high BCL6 correlated with refractory disease and shorter progression-free and overall survival, thus representing a potentially useful molecular prognostic biomarker. Although the adverse prognostic significance of high BCL-6 in PCNSL was confirmed recently in an independent large prospective trial, several small retrospective studies provided a conflicting result, namely that BCL-6 correlates with a better prognosis. This discrepancy raises the possibility that the prognostic significance of BCL-6 may depend on treatment-related factors, such as rituximab or whole-brain radiotherapy. Between 56% and 93% of PCNSL express BCL2.
Transcriptional analyses of PCNSL identified several candidate mediators of disease pathogenesis, including expression of Pim 1 and MYC . Increased MYC in PCNSL was confirmed in the recent CALGB 50202 study. Upregulation of microRNAs associated with MYC pathway (miR-17-5p, miR-20a, miR-9) has also been demonstrated. Circulating extracellular microRNAs in CSF such as miR-21were also recently described in PCNSL, suggesting usefulness as clinical biomarkers.
Given that aberrant somatic hypermutation contributes to the pathobiology of diffuse, large B-cell lymphoma, Montesinos-Rongen et al evaluated the potential role of this mechanism in PCNSL. Somatic hypermutation of 4 protooncogenes was identified— PAX5 , PIM1 , c-MYC , and RhoH/TTF —genes that regulate B-cell development, proliferation, and apoptosis. Mutational frequencies for these were 2- to 5-fold higher in PCNSL compared with extraneural diffuse, large B-cell lymphoma. Such high mutation frequencies suggest a prolonged interaction of the tumor cell in the germinal center microenvironment. Whole exome sequencing studies of PCNSL identified protein-coding mutations involving mediators of cell signaling, CARD14, CD79A/B, TLR2, TLR6, and TLR10, regulators of cell cycle, CCND3, and CDK20, and chromatin structure CREBBP, MLL2, ARID1A/B, and SMARCA4.
The p16 INK4a gene is commonly inactivated by either homozygous deletion (40%–50%) or 5′-CpG hypermethylation (15%–30%) in PCNSL. Inactivation of p14 ARF and p16 INK4a genes by homozygous deletion or promoter hypermethylation may represent an important step in the molecular pathogenesis of PCNSL. The p14 ARF gene normally induces growth arrest and stabilizes p53 protein in the nucleus. Both p14 ARF and p16 INK4a genes are frequently codeleted; moreover, mice lacking the murine homolog of p14 ARF develop a variety of tumors, including lymphomas, sarcomas, and gliomas. In contrast, mutations in the TP53 gene have been observed in only a small proportion of PCNSL.
Comparative genomic hybridization has identified other genetic lesions in PCNSL. Recurrent gains have been detected on chromosome 12 as well as on the long arms of chromosomes 1, 7, and 18; gain on chromosome 12 seems to be the most common chromosomal alteration, specifically in the 12q region harboring STAT6, MDM2, CDK4, and GLI1.
Another common genomic aberrational hotspot in PCNSL involves losses on chromosome 6p21 that harbor loci for HLA as well as broad deletions involving chromosome 6q. Chromosome 6q deletions, in particular 6q21-23, may be most frequent and occur in 40% to 60% of PCNSL. Candidate tumor suppressors linked to chromosome 6q include PRDM1 , a tumor suppressor and regulator of B-cell differentiation ; PTPRK , a protein tyrosine phosphatase that participates in cell adhesion signaling events ; and A20 ( TNFAIP3 ), a negative regulator of nuclear factor-κB (NF-κB) signaling.
Further evidence for the aberrant activation of NF-κB pathways in PCNSL is supported by a gain in DNA copy number for MALT1 as well as activating mutations of CARD11 and MyD88 . The activating exchange of leucine to proline at position 265 of MyD88 may be enriched in PCNSL and has been demonstrated to occur in between 38% and 50% of cases ( Fig. 2 ).

Several lines of investigation support a role for JAK/STAT signaling pathway as a mediator of prosurvival signals in PCNSL. Interleukin (IL)-4, a B-cell growth factor that mediates intracellular signals via JAK/STAT, is upregulated at the transcript and protein levels within the vascular microenvironment in PCNSL tumors. Increased concentration of IL-10 (another activator of JAK/STAT) is detectable in the vitreous and CSF in PCNSL and, in independent studies, correlated with adverse prognosis. A recent analysis demonstrated upregulated IL-10 transcripts in primary CNS lymphoma tumors compared with secondary CNS lymphomas and nodal lymphomas, with concomitant upregulated IL-10 protein in CSF from cases of PCNSL. In addition, CSF concentration of IL-10 correlated with tumor response and progression in patients treated with rituximab and methotrexate. Finally, intratumoral JAK1 transcripts are upregulated in PCNSL. Importantly, increased IL-10 expression plus activation of JAK/STAT signaling in PCNSL are manifestations of aberrant activation of the MyD88 pathway (see Fig. 2 ; Figs. 3 and 4 ).
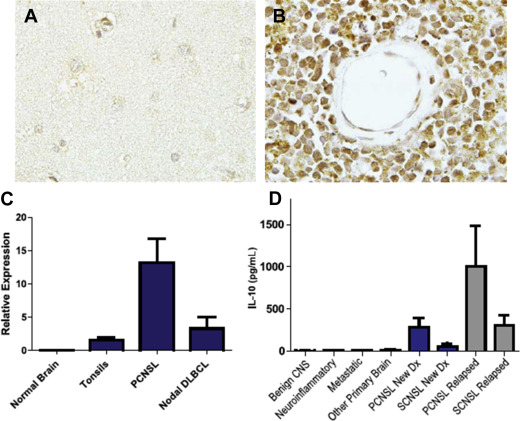
In addition, CD79B , a component of the B-cell receptor signaling pathway, is mutated in approximately 20% of cases, providing further data that dysregulation of the B-cell receptor and the NF-κB signaling pathway contribute to pathogenesis of PCNSL (see Fig. 2 ). Silencing of CDKN2A , a cell cycle regulator, occurs in 50% of cases and is linked with inferior outcomes.
Tumor microenvironment in primary central nervous system lymphoma
The molecular basis for tropism and selective dissemination of lymphoma within the brain are problems that are fundamental to the pathogenesis of PCNSL. In vitro chemotactic responses by large B-cell lymphoma cells isolated from brain lesions have been demonstrated in response to chemokines CXCL12 (SDF-1) and CXCL-13 (B-lymphocyte chemoattractant), providing evidence for their role as neurotropic factors. Moreover, high CXCL-13 concentration in CSF from CNS lymphoma patients correlates with adverse prognosis, supporting its role as a prosurvival factor in PCNSL. In addition, determination of the CSF concentration of CXCL-13, as well as IL-10, facilitate diagnosis of CNS lymphoma in that bivariate expression of each molecule has diagnostic sensitivity at least 2-fold greater than cytology or flow cytometry. In a multicenter investigation, the positive predictive value of bivariate increases in IL-10 plus CXCL-13 in CSF was 95% in the identification of untreated PCNSL.
Given that expression of B-cell chemokines CXCL13 and SDF-1 by retinal pigment epithelium has also been demonstrated in primary IOLs, these chemokines may also contribute to lymphoma cell homing to the retinal pigment epithelium from choroidal circulation.
While under physiologic conditions, the brain is believed to be immunologically quiescent, diagnostic specimens of PCNSL often reveal a robust inflammatory response, with infiltrating reactive T cells and activated macrophages. Notably, perivascular T-cell infiltrates in PCNSL may predict a favorable outcome, suggesting that immunotherapeutic interventions that potentiate T-cell–mediated immune surveillance may be effective.
Diagnostic Evaluation of the Patient with a Focal Brain Lesion
Because patients with PCNSL/IOL commonly present with a variety of nonspecific and/or subtle neurologic symptoms, the diagnostic process may be protracted and extend for months to years ( Fig. 5 ). The cornerstone of diagnostic testing in suspected PCNSL is MRI of the brain, with gadolinium contrast. In 95% of PCNSL, pathologic enhancement localizes homogeneously to dominant lesions. Among the immunocompetent, lesions are solitary in 65% of PCNSL patients and multifocal in 35%. Involvement of the cerebral hemispheres is most common (38%) followed by the thalamus and basal ganglia (16%), corpus callosum (14%) periventricular loci (12%), and cerebellum (9%).
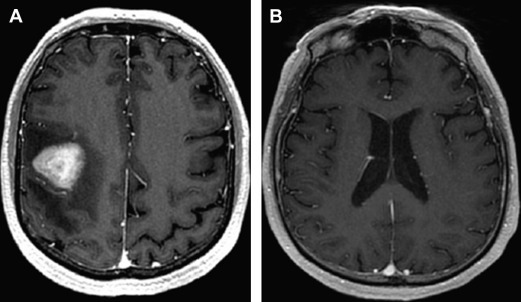
Although there are insufficient data to recommend newer techniques such as quantitative diffusion-weighted and perfusion MRI as “standard of care,” an accumulation of recent data demonstrates the usefulness of these metrics in prognostication as well as diagnosis of CNS lymphomas and related inflammatory and malignant conditions. Given the quantitative nature of these techniques, and their universal availability in standard MR neuroimaging packages, it is possible that these will be incorporated in future studies.
Although glucocorticoids typically induce radiographic regressions in greater than 40% of patients, steroid-induced responses also increase risk of a nondiagnostic vitreal or brain biopsy. Steroid-induced diagnostic delays may extend for months and, on occasion, steroid-induced regressions of sentinel lesions may delay diagnosis of PCNSL or IOL for years. It is therefore important to emphasize that empiric glucocorticoids such as dexamethasone be tapered rapidly or not administered until a diagnosis is established.
The standard diagnostic approach for PCNSL is stereotactic brain biopsy; in selected cases, a subtotal resection may be appropriate if deemed to be safe. Flow cytometric or cytologic analysis of meningeal lymphoma cells isolated from CSF or vitrectomy may also yield the diagnosis. Notably, although flow cytometry has increased diagnostic yield relative to cytology, CSF needs to be efficiently processed for studies designed to identify, in most cases, a kappa or lambda-restricted B-cell lymphoma. Repeat CSF cytologic or flow cytometric studies infrequently improve diagnostic yield, supporting the development of innovative diagnostic methods based on detection of genomic aberrations, such as detection of oncogenic alleles of MYD88.
Given that approximately 80% of patients with IOL will exhibit CNS dissemination, MRI of the brain with gadolinium may be indicated in the workup of idiopathic uveitis in which lymphoma is a diagnostic consideration. Additional staging tests for IOL include fluorescence angiography and optical coherence tomography, as well as evaluation of mutant L265P MYD88 ; intraocular concentrations of the cytokines IL-10 and IL-6 may also be useful adjuncts to diagnosis.
Staging evaluation for the patient with presumptive PCNSL includes complete ophthalmologic examination plus systemic staging via computed tomography of chest, abdomen, and pelvis plus bone marrow biopsy; the value of PET in staging of PCNSL is not established, but it may be useful in possible concomitant testicular involvement in men older than 60 ( Fig. 6 ).

Clinical prognostic determinants in primary central nervous system lymphoma
The International Extranodal Lymphoma Study Group identified 5 clinical variables that correlate with prognosis in PCNSL; 3 are shared with systemic NHL: increased lactate dehydrogenase, age greater than 60 years, and performance status of greater than 1; parameters specific to PCNSL include elevated CSF protein as well as tumor location within the deep regions of the brain (periventricular, basal ganglia, brainstem, and/or cerebellum). The presence of 0 to 1, 2 to 3, or 4 to 5 adverse risk factors respectively correlates with 2-year survival rates of 80%, 48%, and 15%, respectively. Historically, age has been the most reliable clinical prognostic factor, however there is disagreement regarding the age cutpoint at which prognosis declines. Although the International Extranodal Lymphoma Study Group considers age 60 years to be the cutpoint above which prognosis declines, the Memorial Sloan-Kettering prognostic index uses a cutpoint of age 50. Notably, in CALGB 50202, which evaluated intensive immunochemotherapy followed by high-dose consolidation, without WBRT, outcomes for PCNSL patients older than 60 was similar to younger patients, a result that suggests that the optimal cutpoint for age as a prognostic variable is strongly linked to the effects of delayed neurotoxicity.
Principles of management in primary central nervous system lymphoma
Surgery: Biopsy Versus Resection
Many authorities recommend against neurosurgical resection of PCNSL, given the scant evidence that surgical cytoreduction provides no survival benefit and increases risk of postoperative neurologic deficit. On the other hand, data extracted from the German PCNSL SG-1 Trial provided evidence that aggressive resection of PCNSL correlated with improved progression-free survival (PFS). In many cases, maximum safe resection of lesions provides immediate relief of mass effect, facilitates glucocorticoid taper, and theoretically eliminates drug-resistant tumor clones, without contributing to neurologic deficits, particularly when performed using modern neurosurgical mapping techniques.
Whole Brain Irradiation in Primary Central Nervous System Lymphoma
The positive impact of WBRT in PCNSL is compromised by at least 3 important shortcomings: (1) inadequate local control of lymphoma, (2) subclinical dissemination of radiographically occult lymphoma cells outside of the radiation field, and (3) deleterious delayed effects of radiation on normal brain function. In 1 study, the use of WBRT as the sole intervention in PCNSL yielded a median survival of only 11.6 months, and greater than 60% of patients experienced lymphoma progression within the irradiated field. The archetypical features of the delayed neurotoxicity of WBRT in PCNSL include incontinence, gait, and memory disturbances, toxicities that are most evident in patients older than 60; PCNSL survivors that experience late-delayed neurotoxicity of WBRT may ultimately require custodial care. Although lower doses of WBRT were associated with neurotoxicity that is barely discernable in preliminary studies, additional validation of these results are necessary, and need to be reconciled with the established deleterious neurocognitive effects of prophylactic cranial irradiation at 30 Gy. It seems plausible that the neurotoxicity of WBRT is a continuous variable in terms of its relationship to dose. Importantly, it was recently noted that PCNSL patients older than 60 years of age who received consolidative low-dose WBRT (23.4 Gy) experienced inferior outcome in terms of PFS compared with patients younger than 60 years of age. Given the increasing incidence in PCNSL in older patients, these results substantiate the need for innovative strategies that defer or eliminate WBRT as therapy in PCNSL.
Induction Chemotherapeutic Strategies in Primary Central Nervous System Lymphoma
The feasibility and efficacy of HD-MTX in CNS lymphomas was established in the 1970s and led to its incorporation more broadly in induction and salvage regimens. HD-MTX has been identified in multivariate analysis as the most important treatment-related prognostic variable related to survival in CNS lymphomas.
Although the optimal dose of methotrexate has not been defined, systemic doses of 1 g/m 2 or greater mediate lymphocytotoxic effects within brain parenchyma. In a landmark study, Glantz and colleagues demonstrated that intravenous methotrexate administered at 8 g/m 2 over 4 hours yielded higher cytotoxic levels of methotrexate in serum and CSF compared with intrathecal methotrexate (12 mg) at 48 and 72 hours. Also, investigators at Memorial Sloan-Kettering Cancer Center demonstrated that elimination of intrathecal methotrexate during initial therapy for PCNSL did not affect outcome in patients receiving HD-MTX at doses of at least 3.5 g/m 2 . In summary, these studies indicate that high-dose intravenous methotrexate, administered every 2 weeks for a minimum of 6 cycles, can be used to treat large cell lymphoma within brain and leptomeningeal compartments, without intrathecal therapy.
Prevention and management of high-dose methotrexate toxicity
The principal toxicity of HD-MTX is nephropathy, caused by precipitation of methotrexate and its metabolite 7-OH methotrexate within renal tubules. Measures to prevent this life-threatening complication include hydration, urine alkalinization, and avoidance of drugs that interact with MTX such as penicillins, as well as drainage of third space effusions. Additional interventions for delayed methotrexate clearance include carboxypeptidase–G2 (CPDG2, glucarpidase) a recombinant enzyme that rapidly reduces toxic serum methotrexate, via direct hydrolysis of methotrexate ; glucarpidase was approved by the US Food and Drug Administration in 2012.
Combined modality regimens
Combined modality therapy was pioneered at Memorial Sloan-Kettering Cancer Center and consisted of HD-MTX plus procarbazine and vincristine, followed by WBRT and high-dose cytarabine. Evaluation of this approach in a multicenter Radiation Therapy Oncology Group trial demonstrated a median PFS of 24 months. Because of this encouraging efficacy, combined modality therapy became a widely adopted approach for PCNSL. In a multicenter randomized phase II study, Ferreri and colleagues evaluated HD-MTX–based induction, with or without high-dose cytarabine followed by consolidative WBRT; the median failure-free survival in patients receiving HD-Ara-C in combination with HD-MTX was 8 months; in contrast, median failure-free survival for patients treated with HD-MTX without cytarabine was only 4 months. In the SG-1 randomized trial involving 551 PCNSL patients in which one-half received WBRT as first-line consolidation, Thiel and colleagues provided evidence that omission of WBRT from first-line treatment did not impact overall survival. Although the study revealed a modest effect of WBRT on PFS after methotrexate-based induction, this did not translate into improved overall survival, possibly attributable to the neurotoxicity detected in the radiotherapy arm.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


