FIGURE 1.1 Schematic representation of the genomic and histopathological steps associated to tumor progression: from the occurrence of the initiating mutation in the founder cell to metastasis formation. It has been convincingly shown that the genomic landscape of solid tumors such as that of pancreatic and colorectal requires the accumulation of many genetic events, a process which requires decades to complete This timeline offers an incredible window of opportunity for the early detection (often associated to excellent prognosis) of this disease.

FIGURE 1.2 Timeline of seminal hypotheses, research discoveries, and research initiatives that have led to an improved understanding of the genetic etiology of human tumorigenesis within the past century. The consensus cancer gene data were obtained from the Wellcome Trust Sanger Institute Cancer Genome Project website (http://www.sanger.ac.uk/genetics/CGP). Redrawn from ref. 80.
CANCER GENOME INVESTIGATION: TOOLS AND QUALITY CONTROLS
In order to perform mutational analysis of cancer genomes it is imperative to acquire high-quality reagents and to perform several quality controls to verify that the derived data are reliable. To detect somatic (i.e., tumor-specific) mutations in cancer both the tumor DNA and the germline DNA from the same individual are required, especially because knowledge of the variations in the normal human genome is as yet incomplete. Normal genomic DNA from the same individual may be derived either from blood or from tumor neighboring tissue in cases where solid tumors are investigated.
A cancer sample (either from bioptic or surgical origin) typically contains both malignant and nonmalignant (stromal) cells. Most genomic analyses require that samples are highly enriched for tumor tissue. These can either be generated by deriving early passage tumor cell lines, mouse xenografts, or through a pathologist-guided selective macro- or microdissection of neoplastic tissue. This allows the isolation of tumor-derived genomic DNA and sensitive detection of somatic mutations that would otherwise be masked by contamination of normal tissue. Importantly, the quality of the derived genomic DNA may be affected by its source. Surgical resection specimens are usually large and therefore appropriate for these studies. However, biopsies from patients usually contain few cells, thus reducing the quantity of genomic DNA available. Although whole-genome amplification may be a possibility when low genomic DNA amounts are available, this method can give rise to artifactual genetic alterations.9 Another reason that negatively affects the quality of genomic DNA is that cancer samples (for example, liver metastases) often contain significant numbers of necrotic or apoptotic cells. These issues might also be resolved by increased genetic coverage utilizing second-generation sequencing approaches,10 as detailed below.
Prior to genomic analysis multiple key quality controls should be applied to the tumor and normal tissues. These include verification that the tumor sample contains at least 75% cancer cells, a threshold that allows the identification of homozygous and hemizygous deletions, copy-neutral loss of heterozygosity, duplication, and amplification.11–13 To unequivocally assess the somatic tumor-specific nature of sequence changes, genotyping of SNPs in the tumor and normal tissue is also required to prove that both are derived from the same individual.
Cancer Gene Discovery by Sequencing Candidate Gene Families
The availability of the human genome sequence provides new opportunities to comprehensively search for somatic mutations in cancer on a larger scale than previously possible. Progress in the field has been closely linked to improvements in the throughput of DNA analysis and the continuous reduction in sequencing costs. Below some of the achievements in this research area are described, as well as how they affected knowledge of the cancer genome.
A seminal work in the field was the systematic mutational profiling of the genes involved in the RAF-RAS pathway in multiple tumors. This candidate gene approach led to the discovery that BRAF is frequently mutated in melanomas and is mutated at a lower frequency in other tumor types.14 Follow-up studies quickly revealed that mutations in BRAF are mutually exclusive with alterations in KRAS,14,15 genetically emphasizing that these genes function in the same pathway, a concept that had been previously demonstrated in lower organisms such as Caenorhabditis elegans and Drosophila melanogaster.16,17
In 2003, identification of cancer genes shifted from a candidate gene approach to the mutational analyses of gene families. The first gene families to be completely sequenced were those that involved protein18,19 and lipid phosphorylation.20 The rationale for focusing initially on these gene families was threefold:
1. The corresponding proteins were already known at that time to play a pivotal role in signaling and proliferation of normal and cancerous cells.
2. Multiple members of the protein kinases family had already been linked to tumorigenesis.
3. Kinases are clearly amenable to pharmacological inhibition, making them attractive drug targets.
The mutational analysis of all the tyrosine kinase domains in colorectal cancers revealed that 30% of cases had a mutation in at least one tyrosine kinase gene, and overall mutations were identified in eight different kinases, most of which had not previously been linked to cancer.18 An additional mutational analysis of the coding exons of 518 protein kinase genes in 210 diverse human cancers, including breast, lung, gastric, ovarian, renal, and acute lymphoblastic leukemia, identified approximately 120 mutated genes that probably contribute to oncogenesis.19 A recent somatic mutations interrogation of the protein tyrosine kinases in cutaneous melanoma identified ERBB4 to be mutated in 19% of cases, making it the most highly mutated protein tyrosine kinase in melanoma.21 ERBB4 is a member of the ERBB/HER family of receptor tyrosine kinases. Other family members, including ERBB1 (EGFR) and ERBB2 (HER-2), have been implicated by mutations or amplifications in a number of cancers, including lung, colon, and breast cancers. The high mutation frequency as well as the nonsynonymous (NS) to synonymous (S) ratio, which was 24:3, significantly higher than the NS:S ratio predicted for non-selected mutations (P <.01)22 indicated that ERBB4 mutations are selected for during tumorigenesis and therefore contribute to melanoma tumorigenesis.
As kinase activity is attenuated by enzymes that remove phosphate groups called phosphatases, the rational next step in these studies was to perform a mutation analysis of the protein tyrosine phosphatases. Mutational investigation of this family in colorectal cancer identified that 25% of cases had mutations in six different phosphatase genes (PTPRF, PTPRG, PTPRT, PTPN3, PTPN13, or PTPN14).23 Combined analysis of the protein tyrosine kinases and the protein tyrosine phosphatases showed that 50% of colorectal cancers had mutations in a tyrosine kinase gene, a protein tyrosine phosphatase gene, or both, further emphasizing the pivotal role of protein phosphorylation in neoplastic progression. Many of the identified genes had previously been linked to human cancer, thus validating the unbiased comprehensive mutation profiling. These landmark studies led to additional gene family surveys.
The phosphatidylinositol 3-kinase (PI3K) gene family, which also plays a role in proliferation, adhesion, survival, and motility, was also comprehensively investigated.24 Sequencing of the exons encoding the kinase domain of all 16 members belonging to this family pinpointed PIK3CA as the only gene to harbor somatic mutations. When the entire coding region was analyzed, PIK3CA was found somatically mutated in 32% of colorectal cancers. At that time, the PIK3CA gene was certainly not a newcomer in the cancer arena, as it had previously been shown to be involved in cell transformation and metastasis.24 Strikingly, its staggering high mutation frequency was discovered only through systematic sequencing of the corresponding gene family.20 Subsequent analysis of PIK3CA in other tumor types identified somatic mutations in this gene in additional cancer types, including 36% of hepatocellular carcinomas, 36% of endometrial carcinomas, 25% of breast carcinomas, 15% of anaplastic oligodendrogliomas, 5% of medulloblastomas and anaplastic astrocytomas, and 27% of glioblastomas.25–29 It is known that PIK3CA is one of the two (the other being KRAS) most commonly mutated oncogenes in human cancers. Further investigation of the PI3K pathway in colorectal cancer showed that 40% of tumors had genetic alterations in one of the PI3K pathway genes, emphasizing the central role of this pathway in colorectal cancer pathogenesis.30 The relevance and the functional role of the PI3K pathway in tumorigenesis is further described in Chapter 5.
Although most cancer genome studies of large gene families have focused on the kinome, recent analyses have revealed that members of other families highly represented in the human genome are also a target of mutational events in cancer. This is the case of proteases, a complex group of enzymes consisting of at least 569 components that constitute the so-called human degradome.31 Proteases exhibit an elaborate interplay with kinases and have traditionally been associated with cancer progression because of their ability to degrade extracellular matrices, thus facilitating tumor invasion and metastasis.32,33 However, recent studies have shown that these enzymes hydrolyze a wide variety of substrates and influence many different steps of cancer, including early stages of tumor evolution.34 These functional studies have also revealed that beyond their initial recognition as prometastatic enzymes, they play dual roles in cancer, as assessed by the identification of a growing number of tumor-suppressive proteases.35
These findings emphasized the possibility that mutational activation or inactivation of protease genes occurs in cancer. The first clear evidence of this is derived from systematic analysis of genetic alterations in breast and colorectal cancers, which revealed that proteases from different catalytic classes were candidate cancer genes that had somatically mutated in cancer.36 These results have prompted the mutational analysis of entire protease families such as MMPs (matrix metalloproteinases), ADAMs (a disintegrin and metalloproteinase) and ADAMTSs (ADAMs with thromsbospondin domains) in different tumors. These studies led to identification of protease genes frequently mutated in cancer, such as MMP8, which is mutated and functionally inactivated in 6.3% of human melanomas.37,38 Other MMP genes, including MMP2, MMP9, MMP14, and MMP27, are also somatically mutated in melanomas and other malignant tumors, albeit at low frequency.37,39 Systematic mutational analysis of all members of the ADAM family of membrane-bound metalloproteases has shown that ADAM7 and ADAM29 are also often mutated in melanoma, whereas parallel studies of the ADAMTS family have revealed that ADAMTS15 is mutated in colorectal carcinomas and ADAMTS18 and ADAMTS20 in melanomas.40,41 Functional analyses have indicated that ADAM7, ADAM29, and ADAMTS18 mutations affect adhesion of melanoma cells to specific extracellular matrix proteins and in some cases increase their migrating and invasive properties, suggesting that these mutated genes play a role in melanoma progression.41,42 In contrast, functional studies of ADAMTS15 mutations in colorectal cancer cells have revealed that this metalloprotease restrains tumor growth and invasion, further validating the concept that secreted proteases may have tumor-suppressor properties.40
The mutational status of caspases has also been extensively analyzed in different tumors as these proteases play a fundamental role in execution of apoptosis, one of the hallmarks of cancer.43 These studies demonstrated that CASP8 is deleted in neuroblastomas and inactivated by somatic mutations in a variety of human malignancies, including head and neck, colorectal, lung, and gastric carcinomas.44–46 Likewise CASP3, CASP4, CASP5, CASP6, CASP7, CASP10, and CASP14 are occasionally inactivated by mutation in different human cancers.47–54 Other large protease families whose components are often mutated in cancer are the deubiquitylating enzymes (DUBs), which catalyze the removal of ubiquitin and ubiquitinlike modifiers of their target proteins.55 Some DUBs were initially identified as oncogenic proteins, but recent work has shown that other deubiquitylases such as CYLD, A20, and BAP1 are tumor suppressors inactivated in cancer. CYLD is mutated in patients with familial cylindromatosis, a disease characterized by the formation of multiple tumors of skin appendages.56 A20 is a DUB family member encoded by the TNFAIP3 gene, which is mutated in a large number of Hodgkin’s lymphomas and primary mediastinal B-cell lymphomas.57–60 Finally, the BAP1 gene, encoding an ubiquitin C-terminal hydrolase, has been found to be somatically mutated in 86% metastasizing uveal melanomas of the eye.61
Mutational Analysis of Exomes Using Sanger Sequencing
Although the gene family approach for the identification of cancer genes has proven extremely valuable, it still is a candidate approach and thus biased in its nature. The next step forward in the mutational profiling of cancer has been the sequencing of exomes, which is the entire coding portion of the human genome (18,000 protein-encoding genes). As of today the exomes of breast, colorectal, pancreatic, and ovarian clear cell carcinomas, glioblastoma multiforme, and medulloblastoma have been analyzed using Sanger sequencing. These large-scale analyses for the first time allowed researchers to describe and understand the genetic complexity of human cancers.22,36,62–65 The declared goals of these exome studies were to provide for the first time methods for exome-wide mutational analyses in human tumors, to characterize their spectrum and quantity of somatic mutations, and, finally, to discover new genes involved in tumorigenesis as well as novel pathways that have a role in these tumors. In these studies, sequencing data were complemented with gene expression and copy number analyses, thus providing for the first time a comprehensive view of the genetic complexity of human tumors.62–65 A number of conclusions can be drawn from these analyses:
1. Cancer genomes have an average of 30 to 100 somatic alterations per tumor, which was a higher number than previously thought. Although the alterations included point mutations, small insertions, deletions, or amplifications, the great majority of the mutations observed were single-base substitutions.62,63
2. Even within a single cancer type, there is a significant intertumor heterogeneity. This means that multiple mutational patterns (encompassing different mutant genes) are present in tumors that cannot be distinguished based on histological analysis. The concept that individual tumors have a unique genetic milieu is highly relevant for personalized medicine, a concept that will be discussed below.
3. The spectrum and nucleotide contexts of mutations differ between different tumor types. For example, over 50% of mutations in colorectal cancer were C:G to T:A transitions, and 10% were C:G to G:C transversions. In contrast, in breast cancers, only 35% of the mutations were C:G to T:A transitions, and 29% were C:G to G:C transversions. Knowledge of mutation spectra is vital as it allows insight into the mechanisms underlying mutagenesis and repair in the various cancers investigated.
4. A considerably larger number of genes that had not been previously reported to be involved in cancer were found to play a role in the disease.
5. Solid tumors arising in children, such as medulloblastoma, harbor on average five to ten times less gene alterations compared to a typical adult solid tumor. These pediatric tumors also harbor fewer amplifications and homozygous deletions within coding genes compared to adult solid tumors.
Importantly, to deal with the large amount of data generated in these genomic projects, it was necessary to develop new statistical and bioinformatic tools. Furthermore, examination of the overall distribution of the identified mutations allowed the development of a novel view of cancer genome landscapes and a novel definition of cancer genes. These new concepts in the understanding of cancer genetics are further discussed below. The compiled conclusions derived from these analyses have led to a paradigm shift in the understanding of cancer genetics.
A clear indication of the power of the unbiased nature of the whole exome surveys was revealed by the discovery of recurrent mutations in the active site of IDH1, a gene with no known link to gliomas, in 12% of tumors analyzed.63 As malignant gliomas are the most common and lethal tumors of the central nervous system, and glioblastoma multiforme (GBM; World Health Organization grade IV astrocytoma) is the most biologically aggressive subtype, the unveiling of IDH1 as a novel GBM gene is extremely significant. Importantly, mutations of IDH1 predominantly occurred in younger patients (median age of 34 versus 56 years for anaplastic astrocytomas and 32 versus 59 years for GBMs) and were associated with a better prognosis, as patients with IDH mutations have a median overall survival of 31 months, and patients with wild type IDH1 and IDH2 have a median 15-month survival.66 Follow-up studies showed that mutations of IDH1 occur early in glioma progression, the R132 somatic mutation is harbored by the majority (greater than 70%) of grades II and III astrocytomas and oligodendrogliomas, as well as in secondary GBMs that develop from these lower grade lesions.66–72 In contrast, less than 10% of primary GBMs harbor these alterations. Furthermore, analysis of the associated IDH2 revealed recurrent somatic mutations in the R172 residue, which is the exact analog of the frequently mutated R132 residue of IDH1. These mutations occur mostly in a mutually exclusive manner with IDH1 mutations,66,68 suggesting that they have equivalent phenotypic effects. Subsequently, IDH1 mutations have been reported in additional cancer types such as myeloid leukemia samples,73–75 a single case of colorectal cancer, two prostate carcinomas,71 one melanoma case,76 and a few cases of adult supratentorial primitive neuroectodermal tumors.69 Further description of the function of IDH1 and IDH2 mutations in cancer is found in Chapter 8.
Next-Generation Sequencing and Cancer Genome Analysis
The introduction in 1977 of the Sanger method for DNA sequencing with chain-terminating inhibitors has transformed biomedical research.8 Over the past 30 years, this first-generation technology has been universally used for elucidating the nucleotide sequence of DNA molecules. However, the launching of new large-scale projects, including those implicating whole-genome sequencing of cancer samples, has made necessary the development of new methods that are widely known as next-generation sequencing technologies.77–79 These approaches have significantly lowered the cost and the time required to determine the sequence of the 3 × 109 nucleotides present in the human genome. Moreover, they have a series of advantages over Sanger sequencing, which are of special interest for the analysis of cancer genomes.80 First, next-generation sequencing approaches are more sensitive than Sanger methods and can detect somatic mutations even when they are present only in a subset of tumor cells.81 Moreover, these new sequencing strategies are quantitative and can be used to simultaneously determine both nucleotide sequence and copy number variations.82 They can also be coupled to other procedures such as those involving paired-end reads, allowing the identification of multiple structural alterations, such as insertions, deletions, and rearrangements, commonly occurring in cancer genomes.81 Nonetheless, next-generation sequencing still presents some limitations mainly derived from the relatively high error rate in the short reads generated during the sequencing process. In addition, these short reads make the task of de novo assembly of the generated sequences and the mapping of the reads to a reference genome extremely complex. To overcome some of these current limitations, deep coverage of each analyzed genome is required and a careful validation of the identified variants must be performed, typically using Sanger sequencing. As a consequence, there is a substantial increase in both cost of the process and time of analysis. Therefore, it can be concluded that whole-genome sequencing of cancer samples is already a feasible task but not yet a routine process. Further technical improvements will be required before the task of decoding the entire genome of any malignant tumor of any cancer patient can be applied to clinical practice.
The number of next-generation sequencing platforms has substantially grown over the past few years and currently includes technologies from Roche/454, Illumina/Solexa, Life/APG’s SOLiD3, Helicos BioSciences/HeliScope, and Pacific Biosciences/PacBio RS.79 Noteworthy also are the recent introduction of the Polonator G.007 instrument, an open source platform with freely available software and protocols, the Ion Torrent’s semiconductor sequencer, as well as those involving self-assembling DNA nanoballs or nanopore technologies.83–85 These new machines are driving the field toward the era of third-generation sequencing, which brings enormous clinical interest as it can substantially increase speed and accuracy of analysis at reduced costs and facilitate the possibility of single-molecule sequencing of human genomes. A comparison of next-generation sequencing platforms is shown in Table 1.1. These various platforms differ in the method utilized for template preparation and in the nucleotide sequencing and imaging strategy, which finally result in their different performance. Ultimately, the most suitable approach depends on the specific genome sequencing projects.79
Current methods of template preparation first involve randomly shearing genomic DNA into smaller fragments from which a library of either fragment templates or mate-pair templates are generated. Then, clonally amplified templates from single DNA molecules are prepared by either emulsion polymerase chain reaction (PCR) or solid-phase amplification.86,87 Alternatively, it is possible to prepare single-molecule templates through methods that require less starting material and do not involve PCR amplification reactions, which can be the source of artifactual mutations.88 Once prepared, templates are attached to a solid surface in spatially separated sites, allowing thousands to billions of nucleotide sequencing reactions to be performed simultaneously.
The sequencing methods currently used by the different next-generation sequencing platforms are diverse and have been classified into four groups: cyclic reversible termination, single-nucleotide addition, real-time sequencing, and sequencing by ligation79,89 (Fig. 1.3). These sequencing strategies are coupled with different imaging methods, including those based on measuring bioluminescent signals or involving four-color imaging of single molecular events. Finally, the extraordinary amount of data released from these nucleotide sequencing platforms is stored, assembled, and analyzed using powerful bioinformatic tools that have been developed in parallel with next-generation sequencing technologies.90
TABLE 1.1
COMPARATIVE ANALYSIS OF NEXT-GENERATION SEQUENCING PLATFORMS
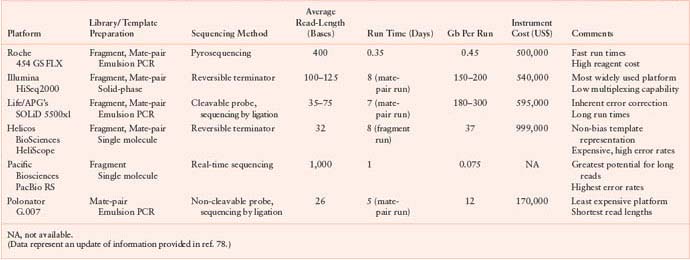
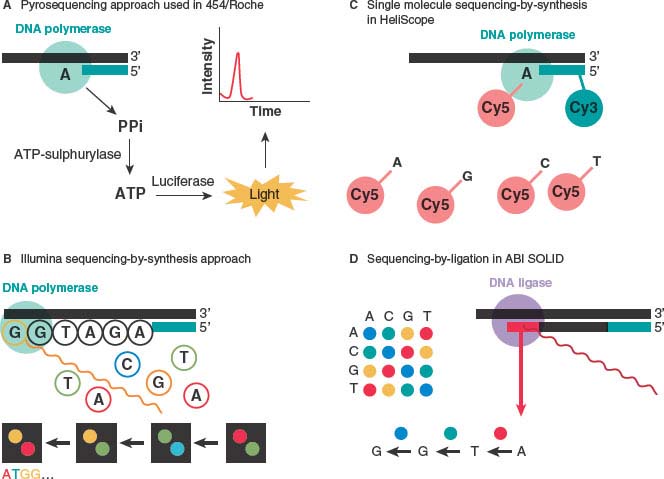
FIGURE 1.3 Advances in sequencing chemistry implemented in next-generation sequencers. A: The pyrosequencing approach implemented in 454/Roche sequencing technology detects incorporated nucleotides by chemiluminescence resulting from PPi release. B: The Illumina method utilizes sequencing-by-synthesis in the presence of fluorescently labeled nucleotide analogs that serve as reversible reaction terminators. C: The single-molecule sequencing-by-synthesis approach detects template extension using Cy3 and Cy5 labels attached to the sequencing primer and the incoming nucleotides, respectively. D: The SOLiD method sequences templates by sequential ligation of labeled degenerate probes. Two-base encoding implemented in the SOLiD instrument allows for probing each nucleotide position twice. (From ref. 88.)
Next-generation sequencing approaches represent the newest entry into the cancer genome decoding arena and have already been applied to cancer analysis. The first research group to apply these methodologies to whole cancer genomes was that of Ley et al.,91 who reported in 2008 the sequencing of the entire genome of a patient with acute myeloid leukemia (AML) and its comparison with the normal tissue from the same patient, using the Illumina/Solexa platform. As further described below, this work has allowed the identification of point mutations and structural alterations of putative oncogenic relevance in AML and represents proof-of-principle of the relevance of next-generation sequencing for cancer research.
Whole-Genome Analysis Utilizing Second-Sequencing
The sequence of the first whole cancer genome was reported in 2008, where an AML and skin from the same patient were described.91 Numerous additional whole-genomes, together with the corresponding normal genomes of patients with a variety of malignant tumors, have been reported since then.73,92–95 The first available whole-genome of a cytogenetically normal AML subtype M1 (AML-M1) revealed eight genes with novel mutations along with another 500 to 1,000 additional mutations found in noncoding regions of the genome. Most of the identified genes were not previously associated with cancer. Validation of the novel mutations identified no novel recurring mutations.91 Concomitantly, with the expansion in the use of next-generation sequencers, other whole-genomes have been evaluated in a similar manner, including malignant melanoma, small cell lung cancer bone metastasis, lung adenocarcinoma, and a second AML.
In contrast to the first AML whole genome, the second did observe a recurrent mutation in IDH1, encoding isocitrate dehydrogenase.73 Follow-up studies extended this finding and reported that mutations in IDH1 and the related gene IDH2 occur at a 20% to 30% frequency in AML patients and are associated with a poor prognosis in some subgroups of patients.96–98 A good example illustrating the high pace at which second-generation technologies and their accompanying analytical tools are found is demonstrated by the following finding derived from reanalysis of the first AML whole genome. Thus, when improvements in sequencing techniques were available, the first AML whole genome described above, which identified no recurring mutations and had a 91.2% diploid coverage, was re-evaluated by deeper sequence coverage, yielding 99.6% diploid coverage of the genome. This improvement together with more advanced mutation naming algorithms allowed the discovery of several nonsynonymous mutations that had not been identified in the initial sequencing. This included a frameshift mutation in the DNA methyltransferase gene DNMT3A. Validation of DNMT3A in 280 additional de novo AML patients to define recurring mutations led to the significant discovery that a total of 22.1% of AML cases had mutations in DNMT3A that were predicted to affect translation. The median overall survival among patients with DNMT3A mutations was significantly shorter than that among patients without such mutations (12.3 months vs. 41.1 months; P <.001).
Shortly after this study, complete sequences of a series of cancer genomes together with matched normal genomes of the same patients have been reported.73,92,93,99 These works have opened the way to more ambitious initiatives, including those involving large international consortia, aimed at decoding the genome of malignant tumors from thousands of cancer patients. In addition to these direct applications of next-generation sequencing technologies for the mutational analysis of cancer genomes, these methods have an additional range of applications in cancer research. Thus, genome sequencing efforts have begun to elucidate the genomic changes that accompany metastasis evolution through comparative analysis of primary and metastatic lesions from breast and pancreatic cancer patients.94,100–102 Likewise, massively parallel sequencing has been used to analyze the evolution of a tongue adenocarcinoma in response to selection by targeted kinase inhibitors.103 Detailed information of several of these whole genome projects is found below.
The first solid cancer to undergo whole-genome sequencing was a malignant melanoma that was compared to a lymphoblastoid cell line from the same person.92 Impressively, a total of 33,345 somatic base substitutions were identified, with 187 nonsynonymous substitutions in protein-coding sequences, at least one order of magnitude higher than any other cancer type. Most somatic base substitutions were C:G greater than T:A transitions and of the 510 dinucleotide substitutions, 360 were CC.TT/GG.AA changes, which is consistent with ultraviolet light exposure mutation signatures previously reported in melanoma.19 Such results from the most comprehensive catalog of somatic mutations not only provide insight into the DNA damage signature in this cancer type but can also be useful in determining the relative order of some acquired mutations. Indeed, this study shows that a significant correlation exists between the presence of a higher proportion of C.A/G.T transitions in early (82%) compared to late mutations (53%). Another important aspect that the comprehensive nature of this melanoma study provided was that cancer mutations are spread out unevenly throughout the genome, with a lower prevalence in regions of transcribed genes, suggesting that DNA repair occurs mainly in these areas.
An interesting example of the power of whole-genome sequencing in deciphering the mutation evolution in carcinogenesis was seen in a study in which a basal-like breast cancer tumor, a brain metastasis, a tumor xenograft derived from the primary tumor, and the peripheral blood from the same patient were compared (Fig. 1.4).94 This analysis showed a wide range of mutant allele frequencies in the primary tumor, which was narrowed in the metastasis and xenograft samples. This suggested that the primary tumor was significantly more heterogeneous in its cell populations compared to its matched metastasis and xenograft samples as these underwent selection processes whether during metastasis or transplantation. The clear overlap in mutation incidence between the metastatic and xenograft cases suggests that xenografts undergo similar selection as metastatic lesions and are therefore a reliable source for genomic analyses. The main conclusion of this whole-genome study was that although metastatic tumors harbor an increased number of genetic alterations, the majority of the alterations found in the primary tumor are preserved.
Whole-Exome Analysis Utilizing Second-Generation Sequencing
Another application of second-generation sequencing involves utilizing nucleic acid “baits” to capture regions of interest in the total pool of nucleic acids. These could either be DNA, as described above,104,105 or RNA.106 Indeed, most areas of interest in the genome can be targeted, including exons and noncoding RNAs. Despite inefficiencies in the exome targeting process, including the uneven capture efficiency across exons, which results in not all exons being sequenced, and the occurrence of some off-target hybridization events, the higher coverage of the exome makes it highly suitable for mutation discovery in cancer samples.

FIGURE 1.4 Covering all the bases in metastatic assessment. Ding et al.94 performed genome-wide analysis on three tumor samples: a patient’s primary breast tumor; her metastatic brain tumor, which formed despite therapy; and a xenograft tumor in a mouse, originating from the patient’s breast tumor. They find that the primary tumor differs from the metastatic and xenograft tumors mainly in the prevalence of genomic mutations (permission from Gray et al. Nature 2010).
A recent study using exome capture followed by massively parallel sequencing surveyed somatic mutations in metastasizing uveal melanoma,61 which is the most common primary cancer of the eye and is at high risk for fatal metastasis.107 In this impressive study only two class II uveal melanoma tumors and their matching normal DNA were investigated. Although not much is known about the genetic basis of uveal melanoma, class II tumors are strongly associated with monosomy 3.108 The authors therefore chose to specifically survey tumors that were monosomic for chromosome 3 to see whether loss of one copy of chromosome 3 could unmask a mutant gene on the remaining copy that promotes metastasis. This strategy was extremely fruitful as it allowed the identification of inactivating somatic mutations in BAP1, located at chromosome 3p21.1 and encoding a deubiquitylating enzyme. Further functional studies have implicated mutational inactivation of BAP1 as a key event in uveal melanoma metastasis, thus expanding the relevance of DUBs as potential therapeutic targets in cancer.61
Use of Next-Generation Sequencing for Additional Cancer Genome Applications
Next-generation sequencing of RNA extracted from tumor cells can be used for the precise and complete characterization of cancer transcriptomes to sample the expressed part of the genome.109 This approach, called RNA-seq, has higher sensitivity than methods of RNA profiling based on DNA microarrays and can be also useful to find novel genes mutated in cancer, as illustrated by the identification of a recurrent FOXL2 mutation in granulose-cell ovarian tumors.109 An additional example of the power of RNA-seq was a survey in which the whole transcriptome of 18 ovarian clear-cell carcinomas and 1 ovarian clear-cell carcinoma cell line were sequenced, leading to the discovery of somatic mutations in ARID1A in 6 of the samples.110 Validation analyses of ARID1A identified it to be somatically mutated in 46% of ovarian clear-cell carcinomas and 30% of endometrioid carcinomas. The spectrum of the identified mutations suggested that ARID1A, which encodes BAF250a, part of the SWI–SNF chromatin remodeling complex, is a novel tumor suppressor.
Next-generation sequencing technologies have also been relevant in the identification of noncoding RNAs, including both microRNAs and large noncoding RNAs, which are encoded by a new class of genes of growing importance in cancer.89,111,112 Likewise, RNA-seq data have also proven to be useful for detecting alternative splicing events or novel fusion transcripts in cancer samples.113,114 Finally, several large-scale approaches such as ChIP-seq, which involves chromatin immunoprecipitation coupled with massively parallel sequencing, have facilitated the genome-wide identification of epigenetic alterations in cancer cells.115,116
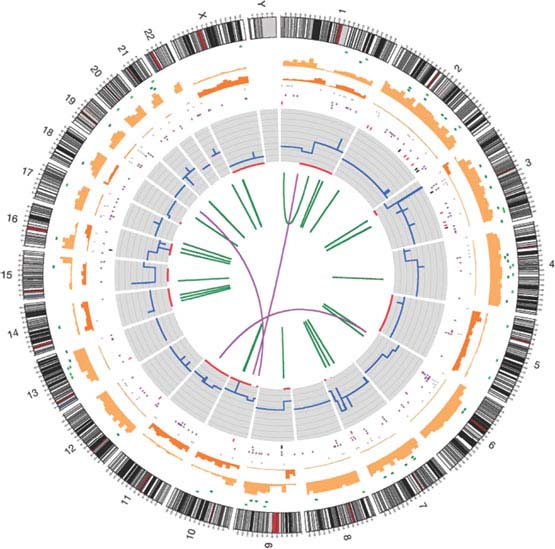
FIGURE 1.5 The catalog of somatic mutations in COLO-829. Chromosome ideograms are shown around the outer ring and are oriented pter–qter in a clockwise direction with centromeres indicated in red. Other tracks contain somatic alterations (from outside to inside): validated insertions (light green rectangles); validated deletions (dark green rectangles); heterozygous (light orange bars), and homozygous (dark orange bars) substitutions shown by density per 10 megabases; coding substitutions (colored squares: silent in gray, missense in purple, nonsense in red, and splice site in black); copy number (blue lines); regions of loss of heterozygosity (LOH) (red lines); validated intrachromosomal rearrangements (green lines); validated interchromosomal rearrangements (purple lines). (From ref. 92.)
SOMATIC ALTERATION CLASSES DETECTED BY CANCER GENOME ANALYSIS
Whole-genome sequencing of cancer genomes has an enormous potential to detect all major types of somatic mutations present in malignant tumors. This large repertoire of genomic abnormalities includes single nucleotide changes, small insertions and deletions, large chromosomal reorganizations, and copy number variations (Fig. 1.5).
Nucleotide substitutions are the most frequent somatic mutations detected in malignant tumors, although there is a substantial variability in the mutational frequency among different cancers.78 On average, human malignancies have one nucleotide change per million bases, but melanomas reach mutational rates tenfold higher and tumors with mutator phenotype caused by DNA mismatch repair deficiencies may accumulate tens of mutations per million nucleotides. By contrast, tumors of hematopoietic origin have less than one base substitution per million. Several bioinformatic tools and pipelines have been developed to efficiently detect somatic nucleotide substitutions through comparison of the genomic information obtained from paired normal and tumor samples from the same patient. Likewise, there are a number of publicly available computational methods to predict the functional relevance of the identified mutations in cancer specimens.78 Most of these bioinformatic tools exclusively deal with nucleotide changes in protein coding regions and evaluate the putative structural or functional effect of an amino acid substitution in a determined protein, thus obviating changes in other genomic regions, which can also be of crucial interest in cancer. In any case, current computational methods used in this regard are far from being optimal, and experimental validation is finally required to assess the functional relevance of nucleotide substitutions found in cancer genomes.
Small insertions and deletions (indels) represent a second category of somatic mutations that can be discovered by whole-genome sequencing of cancer specimens. These mutations are about tenfold less frequent than nucleotide substitutions but may also have an obvious impact in cancer progression. Accordingly, specific bioinformatic tools have been created to detect these indels in the context of the large amount of information generated by whole-genome sequencing projects.117
The systematic identification of large chromosomal rearrangements in cancer genomes represents one of the most successful applications of next-generation sequencing methodologies. Previous strategies in this regard had mainly been based on the utilization of cytogenetic methods for the identification of recurrent translocations in hematopoietic tumors. More recently, a combination of bioinformatics and functional methods has allowed the finding of recurrent translocations in solid epithelial tumors such as TMPRSS2–ERG in prostate cancer and EML4–ALK in non–small cell lung cancer.118,119 Now, by using next-generation sequencing analysis of genomes and transcriptomes, it is possible to systematically search for both intrachromosomal and interchromosomal rearrangements occurring in cancer specimens. These studies have already proven their usefulness for cancer research through the discovery of recurrent translocations involving genes of the RAF kinase pathway in prostate cancer, gastric cancer, and melanoma.120 Likewise, massively parallel paired-end genome and transcriptome sequencing has already been used to detect new gene fusions in cancer and to catalog all major structural rearrangements present in some tumors and cancer cell lines.81,113,121,122 The ongoing cancer genome projects involving thousands of tumor samples will likely lead to the detection of many other chromosomal rearrangements of relevance in specific subsets of cancers. It is also remarkable that whole-genome sequencing may also facilitate the identification of other types of genomic alterations, including rearrangements of repetitive elements, such as active retrotransposons or insertions of foreign gene sequences, such as viral genomes, which can contribute to cancer development. Indeed, next-generation sequencing analysis of the transcriptome of Merkell cell carcinoma samples has revealed the clonal integration within the tumor genome of a previously unknown polyomavirus likely implicated in the pathogenesis of this rare but aggressive skin cancer.123
Finally, next-generation sequencing approaches have also demonstrated their feasibility to analyze the pattern of copy number alterations in cancer, as they allow researchers to count the number of reads in both tumor and normal samples at any given genomic region and then to evaluate the tumor-to-normal copy number ratio at this particular region. These new methods offer some advantages when compared with those based on microarrays, including much better resolution, precise definition of the involved breakpoints, and absence of saturation, which facilitates the accurate estimation of high-copy number levels occurring in some genomic loci of malignant tumors.78
PATHWAY-ORIENTED MODELS OF CANCER GENOME ANALYSIS
Genome-wide mutational analyses suggest that the mutational landscape of cancer is made up of a handful of genes that are mutated in a high fraction of tumors, otherwise know as “mountains,” and most mutated genes are altered at relatively low frequencies, otherwise known as “hills”36 (Fig. 1.6). The mountains probably give a high selective advantage to the mutated cell, and the hills might provide a lower advantage, making it hard to distinguish them from passenger mutations. As the hills differ between cancer types, it seems that the cancer genome is more complex and heterogeneous than anticipated. Although highly heterogeneous, bioinformatic studies suggest that the mountains and hills can be grouped into sets of pathways and biologic processes. Some of these pathways are affected by mutations in a few pathway members and others by numerous members. For example, pathway analyses have allowed the stratification of mutated genes in pancreatic adenocarcinomas to 12 core pathways that have at least one member mutated in 67% to 100% of the tumors analyzed62 (Fig. 1.7). These core pathways deviated to some that harbored one single highly mutated gene, such as in KRAS in the G1/S cell cycle transition pathway and pathways where a few mutated genes were found, such as the transforming growth factor (TGF-β) signaling pathway. Finally, there were pathways in which many different genes were mutated, such as invasion regulation molecules, cell adhesion molecules, and integrin signaling. Importantly, independent of how many genes in the same pathway are affected, if they are found to occur in a mutually exclusive fashion in a single tumor, they most likely give the same selective pressure for clonal expansion.

FIGURE 1.6 Cancer genome landscapes. Nonsilent somatic mutations are plotted in two-dimensional space representing chromosomal positions of RefSeq genes. The telomere of the short arm of chromosome 1 is represented in the rear left corner of the green plane and ascending chromosomal positions continue in the direction of the arrow. Chromosomal positions that follow the front edge of the plane are continued at the back edge of the plane of the adjacent row, and chromosomes are appended end to end. Peaks indicate the 60 highest-ranking CAN-genes for each tumor type, with peak heights reflecting CaMP scores (7). The dots represent genes that were somatically mutated in the individual colorectal (Mx38) (A) or breast tumor (B3C) (B) displayed. The dots corresponding to mutated genes that coincided with hills or mountains are black with white rims; the remaining dots are white with red rims. The mountain on the right of both landscapes represents TP53 (chromosome 17), and the other mountain shared by both breast and colorectal cancers is PIK3CA (upper left, chromosome 3). (Redrawn from ref. 36. Reprinted with permission from AAAS.)

FIGURE 1.7 Signaling pathways and processes. A: The 12 pathways and processes whose component genes were genetically altered in most pancreatic cancers. B, C: Two pancreatic cancers (Pa14C and Pa10X) and the specific genes that are mutated in them. The positions around the circles in (B) and (C) correspond to the pathways and processes in (A). Several pathway components overlapped, as illustrated by the BMPR2 mutation that presumably disrupted both the SMAD4 and hedgehog signaling pathways in Pa10X. Additionally, not all 12 processes and pathways were altered in every pancreatic cancer, as exemplified by the fact that no mutations known to affect DNA damage control were observed in Pa10X. NO, not observed. (Redrawn from ref. 61. Reprinted with permission from AAAS.)
The idea of genetically analyzing pathways rather than individual genes has been applied previously, revealing the concept of mutual exclusivity. Mutual exclusivity has been shown elegantly in the case of KRAS and BRAF where a KRAS mutated cancer generally does not also harbor a BRAF mutation, as KRAS is upstream of BRAF in the same pathway.14 A similar concept was applied for PIK3CA and PTEN, where both mutations do not usually occur in the same tumor.30
“Passenger” and “Driver” Mutations
By the time a cancer is diagnosed, it is comprised of billions of cells carrying DNA abnormalities, some of which have a functional role in malignant proliferation but also many genetic lesions acquired along the way that have no functional role in tumorigenesis.19 The emerging landscapes of cancer genomes include thousands of genes that were not previously linked to tumorigenesis but are found to be somatically mutated. Many of these changes are likely to be “passengers” or neutral in that they have no functional effects on the growth of the tumor.19 Only a small fraction of the genetic alterations are expected to drive cancer evolution by giving cells a selective advantage over their neighbors. Passenger mutations occur incidentally in a cell that later or in parallel develops a “driver” mutation, but are not ultimately pathogenic.124 Although neutral, cataloging passengers mutations is important as they incorporate the signatures of previous exposures the cancer cell underwent as well as DNA repair defects the cancer cell has. As in many cases passenger and driver mutations occur at similar frequencies, and identification of drivers versus the passenger is of utmost relevance and remains a pressing challenge in cancer genetics.125–127 This goal will eventually be achieved through a combination of genetic and functional approaches, some of which are listed below.
The most reliable indicator that a gene was selected for and therefore is highly likely to be pathogenic is identification of recurrent mutations, whether at the same exact amino acid position or in neighboring amino acid positions in different patients. Further than that, if somatic alterations in the same gene occur very frequently (mountains in the tumor genome landscape), these can be confidently classified as drivers. For example, cancer alleles that are identified in multiple patients and different tumors types such as those found in KRAS, TP53, PTEN, and PIK3CA are clearly selected for during tumorigenesis.
However, most genes discovered thus far are mutated in a relatively small fraction of tumors (hills), and it has been clearly shown that genes that are mutated in less than 1% of patients can still act as drivers.128 The systematic sequencing of newly identified putative cancer genes in the vast number of specimens from cancer patients will help in this regard. However, even if examination of large numbers of samples can provide helpful information to classify drivers versus passengers, this approach alone is limited by the marked variation in mutation frequency among individual tumors and individual genes. The statistical test utilized in this case calculates the probability that the number of mutations in a given gene reflects a mutation frequency that is greater than expected from the nonfunctional background mutation rate,36,129 which is different between different cancer types. These analyses incorporate the number of somatic alterations observed, the number of tumors studied, and the number of nucleotides that were successfully sequenced and analyzed.
Another approach often used to distinguish driver from passenger mutations exploits statistical analysis of synonymous versus nonsynonymous changes.130 In contrast to nonsynonymous mutations, synonymous mutations do not alter the protein sequence. Therefore, they do not usually apply a growth advantage and would not be expected to be selected during tumorigenesis. This strategy works by comparing the observed-to-expected ratio of synonymous with that of nonsynonymous mutation. An increased proportion of nonsynonymous mutations from the expected two-to-one ratio implies selection pressure during tumorigenesis.
Other approaches are based on the concept that driver mutations may have characteristics similar to those causing Mendelian disease when inherited in the germ line and may be identifiable by constraints on tolerated amino acid residues at the mutated positions. In contrast, passenger mutations may have characteristics more similar to those of nonsynonymous SNPs with high minor allele frequencies. Based on these premises, supervised machine learning methods have been used to predict which missense mutations are drivers.131 Additional approaches to decipher drivers from passengers include identification of mutations that affect locations that have previously been shown to be cancer causing in protein members of the same gene family. Enrichment for mutations in evolutionarily conserved residues and algorithms, such as SIFT (sorting intolerant from tolerant),132 estimate the effects of the different mutations identified.
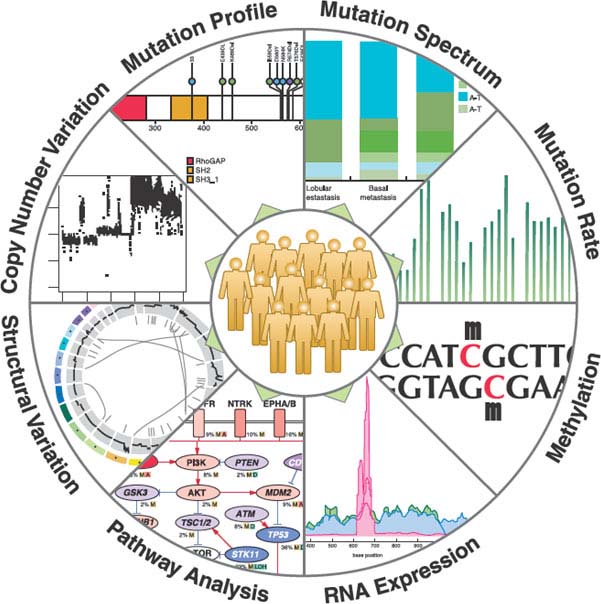
FIGURE 1.8 Landscape of cancer genomics analyses. NGS data will be generated for hundreds of tumors from all major cancer types in the near future. The integrated analysis of DNA, RNA and methylation sequencing data will help elucidate all relevant genetic changes in cancers. (Permission from ref. 94.)
Probably the most conclusive methods to identify driver mutations will be rigorous functional studies using biochemical assays as well as model organisms or cultured cells, using knock-out and knock-in of individual cancer alleles.133 Unfortunately, these methods are not well suited to the analysis of the hundreds of gene candidates that arise from every large-scale cancer genome project. In conclusion, it is fair to say that sequencing cancer genomes is only the beginning of a journey that will ultimately be completed when the thousands of the newly discovered alleles are annotated as being the drivers of this disease. A summary of the various next-generation applications and approaches for their analysis is summarized in Figure 1.8. and Table 1.2.
TABLE 1.2
COMPUTATIONAL TOOLS AND DATABASES USEFUL FOR CANCER GENOME ANALYSIS

NETWORKS OF CANCER GENOME PROJECTS
The first large-scale studies of genes mutated in malignant tumors have allowed the identification of new cancer genes that represent potential targets for therapy in different types of cancer. However, these analyses have also demonstrated that the repertoire of oncogenic mutations is extremely heterogeneous, suggesting that it would be difficult for independent cancer genome initiatives to address the generation of comprehensive catalogs of mutations in the wide spectrum of human malignancies. Accordingly, there have been different efforts to coordinate the cancer genome sequencing projects being carried out around the world. The first initiative in this regard was the Cancer Genome Project (CGP) of the Wellcome Trust Sanger Institute launched in the United Kingdom, which has been followed by two large and ambitious projects called the Cancer Genome Atlas (TCGA) and the International Cancer Genome Consortium (ICGC). Besides these three large cancer genome projects, there are other initiatives that are more focused on specific tumors, such as that lead by scientists at St. Jude Children’s Research Hospital in Memphis, and Washington University, which aims at sequencing 600 pediatric-cancer genomes.
The CGP initially focused on the systematic search for somatic alterations in human tumors and cancer cell lines, analyzing large sets of candidate cancer genes as well as whole genomes. This project has already completed the whole-genome sequencing of several cancer patients and tumor-derived cell lines, including lung carcinomas and melanomas,92,93 and intends to extend these studies to a total of 2,000 to 3,000 cases over the next 5 years.
TCGA began in 2006 at the United States as a comprehensive program in cancer genomics supported by the U.S. National Institutes of Health (NIH). The initial project focused on three tumors: glioblastoma multiforme, serous cystadenocarcinoma of the ovary, and lung squamous carcinoma. These studies have already generated novel and interesting information regarding genes mutated in these malignancies.134 On the basis of these positive results, the NIH has recently announced an expansion of the TCGA program with the aim to produce genomic data sets for at least 10 additional cancers by the end of 2011 and 20 to 25 cancers over the next 5 years.
The ICGC was formed in 2008 to coordinate the generation of comprehensive catalogs of genomic abnormalities in tumors from 50 different cancer types or subtypes that are of clinical and societal importance across the world.135 The project aims to perform systematic studies of over 25,000 cancer genomes at the genomic level and integrate this information with epigenomic and transcriptomic studies of the same cases as well as with clinical features of patients. At present, ten countries and two European consortia have already initiated cancer genome projects coordinated by the ICGC. These projects will deal with at least 500 samples per cancer type from cancers affecting a variety of human organs and tissues, including blood, brain, breast, kidney, liver, pancreas, stomach, oral cavity, and ovary.135 All participating countries and scientists have adhered to a series of predetermined procedures for ethical approval, sample quality, clinical annotation, study design, statistical issues, data storage, and intellectual property. In this regard, the ICGC has made the commitment to make the data available to the entire research community as rapidly as possible to accelerate the understanding of cancer biology and translate these discoveries into clinical practice.
All these coordinated projects have already provided new insights into the catalog of genes mutated in cancer and have unveiled specific signatures of the mutagenic mechanisms, including carcinogen exposures or DNA-repair defects, implicated in the development of different malignant tumors.92,93,136 Furthermore, these cancer genome studies have also contributed to define clinically relevant subtypes of tumors for prognosis and therapeutic management, and in some cases have identified new targets and strategies for cancer treatment.66,73,95,137,138 Nevertheless, and similar to the doubts raised at the first stages of the Human Genome Project, the proposal to sequence large numbers of cancer genomes has also generated some controversy because of the high cost of these projects, the lack of novel functional hypotheses driving these projects, or their failure to characterize the mutational heterogeneity within individual tumors.139,140 However, the rapid technological advances in DNA sequencing will likely drop the costs of sequencing cancer genomes to a small fraction of the current price and will allow researchers to overcome some of the current limitations of these global sequencing efforts. Hopefully, worldwide coordination of cancer genome projects with those involving large-scale functional analysis of genes in both cellular and animal models will likely provide us with the most comprehensive collection of information generated to date into the causes and molecular mechanisms of cancer.
THE GENOMIC LANDSCAPE OF CANCERS
Examination of the overall distribution of the identified mutations redefined the cancer genome landscapes whereby the mountains are the handful of commonly mutated genes and the hills represent the vast majority of genes that are infrequently mutated. One of the most striking features of tumor genomic landscape is that it involves different sets of cancer genes that are mutated in a tissue-specific fashion.141,142 To continue with the analogy, the scenery is very different if we observe a colorectal, a lung, or a breast tumor. This indicates that mutations in specific genes cause tumors at specific sites, or are associated with specific stages of development, cell differentiation, or tumorigenesis, despite many of those genes being expressed in various fetal and adult tissues. Moreover, different types of tumors follow specific genetic pathways in terms of the combination of genetic alterations that it must acquire. For example, no cancer outside the bowel has been shown to follow the classic genetic pathway of colorectal tumorigenesis. Additionally, KRAS mutations are almost always present in pancreatic cancers but are very rare or absent in breast cancers. Similarly, BRAF mutations are present in 60% of melanomas but are very infrequent in lung cancers.1 Another intriguing feature is that alterations in ubiquitous housekeeping genes, such as those involved in DNA repair or energy production, occur only in particular types of tumors.
In addition to tissue specificity, the genomic landscape of tumors can also be associated with the gender and the hormonal status. For example HER-2 amplification and PIK3C2A mutations, two genetic alterations associated with breast cancer development, are correlated with the estrogen-receptor hormonal status.143 The molecular basis for the occurrence of cancer mutations in tissue- and gender-specific profiles is still largely unknown. Organ-specific expression profiles and cell-specific neoplastic transformation requirements are often mentioned as possible causes for this phenomenon. Identifying tissue and gender cancer mutations patterns is relevant as it may allow the definition of individualized therapeutic avenues.
THE CANCER GENOME AND THE NEW TAXONOMY OF TUMORS
The deciphering of the cancer genome has already impacted clinical practice at multiple levels. On the one hand, it allowed the identification of new cancer genes such as IDH1, a gene involved in glioma, which was discovered recently (see above), and on the other hand, it is redesigning the taxonomy of tumors.
Until the genomic revolution, tumors had been classified based on two criteria: their localization (site of occurrence) and their appearance (histology). These criteria are also currently used as primary determinants of prognosis and to establish the best treatments. For many decades it has been known that patients with histologically similar tumors have different clinical outcomes. Furthermore, tumors that cannot be distinguished based on histological analysis can respond very differently to identical therapies.144
It is becoming increasingly manifest that the frequency and distribution of mutations affecting cancer genes can be used to redefine the histology-based taxonomy of a given tumor type. Lung and colorectal tumors represent paradigmatic examples. Genomic analysis led to the identification of activating mutations in the receptor tyrosine kinase EGFR in lung adenocarcinomas.145 The occurrence of EGFR mutations molecularly defines a subtype of non–small cell lung cancer (NSCLC) that occur mainly in nonsmoker women, tend to have a distinctly enhanced prognosis, and typically respond to EGFR-targeted therapies.146–148 Similarly, the recent discovery of the EML4-ALK fusion identifies yet another subset of NSCLC that is clearly distinct from those that harbor EGFR mutations, have distinct epidemiologic and biological features, and respond to ALK inhibitors.119,149
The second example is colorectal cancers (CRC), the tumor type for which the genomic landscape has been refined with the highest accuracy. CRCs can be clearly categorized according to the mutational profile of the genes involved in the KRAS pathway (Fig. 1.9). It is now known that KRAS mutations occur in approximately 40% of CRCs. Another subtype of CRC (approximately 10%) harbors mutations in BRAF, the immediate downstream effectors of KRAS.15
In CRC and other tumor types, KRAS and BRAF mutations are known to be mutually exclusive. The mutual exclusivity pattern indicates that these genes operate in the same signaling pathway. Large epidemiologic studies have shown that the prognosis of tumors harboring wild type KRAS/BRAF genes is distinct, typically more favorable, than that of the mutated ones.150,151 Of note, KRAS and BRAF mutations have been recently shown to impair responsiveness to the anti-EGFR monoclonal antibodies therapies in CRC patients.152–154 Clearly distinct subgroups can be genetically identified in both NSCLC and CRC with respect to prognosis and response to therapy. It is likely that, as soon as the genomic landscapes of other tumor types are defined, molecular subgroups like those described above will also become defined.
In conclusion, the taxonomy of tumors is being rewritten using the presence of genetic lesions as major criteria. Genome-based information will improve diagnosis and will be used to determine personalized therapeutic regimens based on the genetic landscape of individual tumors.
CANCER GENOMICS AND DRUG RESISTANCE
Cancer genomics has dramatically impacted disease management, as its application is helping researchers determine which patients are likely to benefit from which drug. As discussed in great detail in Chapters 12 and 13, good examples for such treatment include targeted therapy using imatinib for chronic myeloid leukemia (CML) patients and use of gefitinib and erlotinib for NSCLC patients.
Key to the successful development and application of anticancer agents is a better understanding of the effect of the therapeutic regimens and of resistance mechanisms that may develop. In most tumor types, a fraction of patients’ tumors are refractory to therapies (intrinsic resistance). Even if an initial response to therapies is obtained, the vast majority of tumors subsequently become refractory (i.e., acquired resistance) and patients eventually succumb to disease progression. Secondary resistance should therefore be regarded as a key obstacle to treatment progress. The analysis of the cancer genome represents a powerful tool both for the identification of chemotherapeutic signatures as well as to understand resistance mechanisms to therapeutic agents. Examples for each of these are described below.
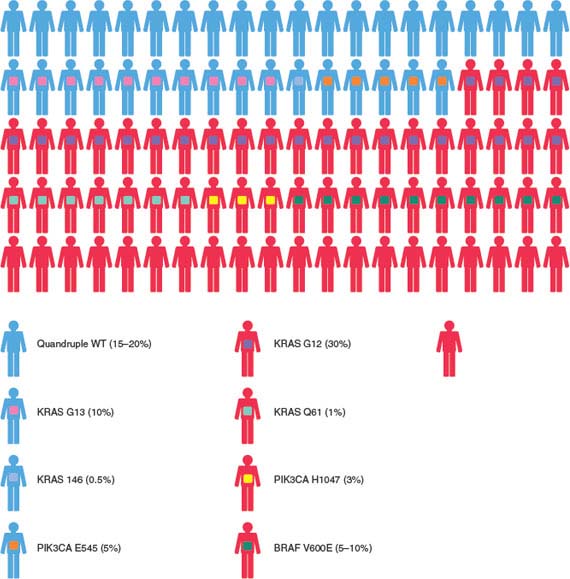
FIGURE 1.9 Graphic representation of a cohort of 100 patients with colorectal cancer treated with cetuximab or panitumumab. The genetic milieu of individual tumors and their impacts on the clinical response are listed. KRAS, BRAF, and PIK3CA somatic mutations as well as loss of PTEN protein expression are indicated according to different color codes. Molecular alterations mutually exclusive or coexisting in individual tumors are indicated using different color variants. The relative frequencies at which the molecular alterations occur in colorectal cancers are described. (Redrawn from Bardelli A, Siena S. Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. J Clin Oncol 2009;22:6043.)
An important application of systematic sequencing experiments is identification of the effects of chemotherapy on the cancer genome. For example, gliomas that recur after temozolomide treatment have been shown to harbor large numbers of mutations with a signature typical of a DNA alkylating agent.155,156 Since these alterations were detected using Sanger sequencing, which as described above has limited sensitivity, the data suggested that the detected alterations were clonal. The model that unfolds from this study indicates that although temozolomide has limited efficacy, almost all of the cells in a glioma respond to the drug. However, a single cell that was resistant to the chemotherapy proliferated and formed a cell clone. Later genomic analyses of the cell clone allowed the identification of the underlying mutated resistance genes.155,156
Single-molecule targeted therapy is almost always followed by acquired drug resistance.157–159 Genomic analyses can be successfully exploited to decipher resistance mechanisms to such inhibitors. Below a few paradigmatic examples are presented that will be discussed extensively in other chapters. Despite the effectiveness of gefitinib and erlotinib in EGFR mutant cases of NSCLC,160 drug resistance develops within 6 to 12 months after initiation of therapy. The underlying reason for this resistance was identified as a secondary mutation in EGFR exon 20, T790M, detectable in 50% of patients who relapse.161–163 Importantly, some studies have shown the mutation to be present before the patient was treated with the drug,164,165 suggesting that exposure to the drug selected for these cells.166 As the drug resistant EGFR mutation is structurally analogous to the mutated gatekeeper residue T315I in BCR-ABL, T670I in c-KIT, and L1196M in EML4-ALK, which have been shown previously to confer resistance to imatinib and other kinase inhibitors,158,167,168 this mechanism of resistance represent a general problem that needs to be overcome.
A recent elegant study, which also represents the use of genomics in understanding drug resistance mechanisms, focused on the inhibition of activating BRAF (V600E) mutations, which occur in 7% of human malignancies and 60% of melanomas.14 Clinical trials using PLX4032, a novel class I RAF-selective inhibitor, showed an 80% antitumor response rate in melanoma patients with BRAF (V600E) mutations, however, cases of drug resistance were observed.169 Use of microarray and sequencing technologies showed that in this case the resistance was not due to secondary mutations in BRAF, but due rather to either up-regulation of PDGFRB or NRAS mutations.170
It was, however, the introduction of two anti-EGFR monoclonal antibodies, cetuximab and panitumumab, for the treatment of metastatic colorectal cancer that provided the largest body of knowledge on the relationship between tumors’ genotypes and response to targeted therapies. The initial clinical analysis pointed out that only a fraction of metastatic colorectal cancer patients benefited from this novel treatment. Different from the NSCLC paradigm, it was found that EGFR mutations do not play a major role in the response. On the contrary, from the initial retrospective analysis it became clear that somatic KRAS mutations, thought to be present in 35% to 45% of metastatic colorectal cancers, are important negative predictors of efficacy in patients who are given panitumumab or cetuximab.152–154 Among tumors carrying wild type KRAS, mutations of BRAF or PIK3CA, or loss of PTEN expression may also predict resistance to EGFR-targeted monoclonal antibodies, although the latter biomarkers require further validation before they can be incorporated into clinical practice. From these few examples, it is clear that future deeper genomic understanding of targeted drug resistance is crucial to the effective development of additional as well as alternative therapies to overcome this resistance.
PERSPECTIVES OF CANCER GENOME ANALYSIS
The completion of the human genome project has marked a new beginning in biomedical sciences. As human cancer is a genetic disease, the field of oncology has been one of the first to be impacted by this historic revolution. Knowledge of the sequence and organization of the human genome allows the systematic analysis of the genetic alterations underlying the origin and evolution of tumors. High throughput mutational profiling of common tumors, including lung, skin, breast, and colorectal cancers, and the application of next-generation sequencing to whole genome, whole exome, and whole transcriptome of cancer samples has allowed substantial advances in the understanding of this disease by facilitating the detection of all main types of somatic cancer genome alterations. These have also led to historical results such as the identification of genetic alterations that are likely to be the major drivers of these diseases.
However, the genetic landscape of cancers is by no means complete, and what has been learned so far has raised new and exciting questions that must be addressed. There are still important technical challenges for the detection of somatic mutations.78 Clinical tumor samples often contain large amounts of nonmalignant cells, which makes the identification of mutations in cancer genomes more challenging when compared with similar analyses of peripheral blood samples for germline genome studies. Moreover, the genomic instability inherent to cancer development and progression largely increases the complexity and diversity of genomic alterations of malignant tumors, making it necessary to distinguish between driver and passenger mutations. Likewise, the fact that malignant tumors are genetically heterogeneous and contain several clones simultaneously growing within the same tumor mass raises additional questions regarding the quality of the information currently derived from cancer genomes. Hopefully, in the near future, advances in third-generation sequencing technologies will make it feasible to obtain high-quality sequence data of a genome isolated from a single cell, an aspect of crucial relevance for cancer research.
One of the next imperatives is the definition of the oncogenomic profile of all tumor types. Particularly the less common—though not less lethal—ones are still largely mysterious to scientists and untreatable to clinicians. For some of these diseases few new therapeutically amenable molecular targets have been discovered in the past years. For example, identification of druggable genetic lesions associated with pancreatic and ovarian cancers could help define new therapeutic strategies for these aggressive diseases. To achieve this, detailed oncogenomic maps of the corresponding tumors must be drafted. The latter will hopefully be completed in the coming years, thanks to the systematic cancer genome projects that are presently being performed.
Even in the case of common cancers, a lot of genomic profiling efforts still lay ahead. For example, in a significant fraction of breast and lung tumors the mutations that are likely to be drivers have not yet been found. This is not surprising considering that even in these tumor types only a limited number of samples have been systematically analyzed so far. Therefore, low incidence mutations that could represent potentially key therapeutic targets in a subset of tumors might have escaped detection. Consequently, the scaling up of the mutational profiling to large number of specimens for each tumor type is warranted.
Finally, understanding the cellular properties imparted by the hundreds of recently discovered cancer alleles is another area that must be developed. As a matter of fact, compared to the genomic discovery stage, the functional validation of putative novel cancer alleles, despite their potential clinical relevance, is substantially lagging behind. To achieve this, high-throughput functional studies in model systems that accurately recapitulate the genetic alterations found in human cancer must be developed.
To conclude, the eventual goal of profiling the cancer genome is not only to further understand the molecular basis of the disease, but also to discover novel diagnostic and drug targets. One might anticipate that the most immediate application of these new technologies will be noninvasive strategies for early cancer detection. Considering that oncogenic mutations are present only in cancer cells, screening for tumor-derived mutant DNA in patients’ blood holds great potential and will progressively substitute current biomarkers, which have poor sensitivity and lack specificity.171 Further improvements in next-generation sequencing technologies are likely to reduce their cost as well as make these analyses more facile in the future. Once this happens, most cancer patients will undergo in-depth genomic analyses as part of their initial evaluation and throughout their treatment. This will offer more precise diagnostic and prognostic information, which will affect treatment decisions. Although many challenges remain, the information gained from next-generation sequencing platforms is laying a foundation for personalized medicine, in which patients are managed with therapies that are tailored to the specific gene mutations found in their tumors. Ultimately, these should lead to therapeutic successes similar to the ones attained for chronic myelogenous leukemia patients with imatinib,172,173 melanoma patients with PLX4032,169 and NSCLC patients with gefitinib and erlotinib.160 Clearly, this is the absolute goal for all of this work.
Selected References
The full list of references for this chapter appears in the online version.
1. Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med 2004;10(8):789.
2. Kinzler KW, Vogelstein B. Lessons from hereditary colon cancer. Cell 1996;87(2):159.
3. International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 2004;431(7011): 931.
5. Rous P. Transmission of a malignant new growth by means of a cell-free filtrate. JAMA 1911;56:198.
7. International HapMap Consortium. A haplotype map of the human genome. Nature 2005;437(7063):1299.
14. Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417(6892):949.
18. Bardelli A, Parsons DW, Silliman N, et al. Mutational analysis of the tyrosine kinome in colorectal cancers. Science 2003;300(5621):949.
19. Greenman C, Stephens P, Smith R, et al. Patterns of somatic mutation in human cancer genomes. Nature 2007; 446(7132):153.
20. Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004;304(5670):554.
21. Prickett TD, Agrawal NS, Wei X, et al. Analysis of the tyrosine kinome in melanoma reveals recurrent mutations in ERBB4. Nat Genet 2009;41(10):1127.
22. Sjoblom T, Jones S, Wood LD, et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006;314(5797):268.
23. Wang Z, Shen D, Parsons DW, et al. Mutational analysis of the tyrosine phosphatome in colorectal cancers. Science 2004;304(5674):1164.
33. López-Otín C, Hunter T. The regulatory crosstalk between kinases and proteases in cancer. Nat Rev Cancer 2010; 10(4):278.
35. López-Otín C, Matrisian LM. Emerging roles of proteases in tumour suppression. Nat Rev Cancer 2007;7(10):800.
36. Wood LD, Parsons DW, Jones S, et al. The genomic landscapes of human breast and colorectal cancers. Science 2007;318(5853):1108.
37. Palavalli LH, Prickett TD, Wunderlich JR, et al. Analysis of the matrix metalloproteinase family reveals that MMP8 is often mutated in melanoma. Nat Genet 2009;41(5):518.
43. Hanahan D, Weinberg RA The hallmarks of cancer. Cell 2000;100(1):57.
44. Teitz T, Wei T, Valentine MB, et al. Caspase 8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN. Nat Med 2000;6(5):529.
54. Ghavami S, Hashemi M, Ande SR, et al. Apoptosis and cancer: mutations within caspase genes. J Med Genet 2009;46(8):497.
56. Bignell GR, Warren W, Seal S, et al. Identification of the familial cylindromatosis tumour-suppressor gene. Nat Genet 2000;25(2):160.
59. Kato M, Sanada M, Kato I, et al. Frequent inactivation of A20 in B-cell lymphomas. Nature 2009;459(7247):712.
61. Harbour JW, Onken MD, Roberson ED, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 2010;330(6009):1410.
62. Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008;321(5897):1801.
63. Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008;321(5897):1807.
64. Jones S, Wang TL, Shih Ie M, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 2010;330(6001):228.
65. Parsons DW, Li M, Zhang X, et al. The genetic landscape of the childhood cancer medulloblastoma. Science 2010; (in press).
66. Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med 2009;360(8):765.
73. Mardis ER, Ding L, Dooling DJ, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 2009;361(11): 1058.
77. Mardis ER, Wilson RK. Cancer genome sequencing: a review. Hum Mol Genet 2009;18(R2):R163.
78. Meyerson M, Gabriel S, Getz G. Advances in understanding cancer genomes through second-generation sequencing. Nat Rev Genet 2010;11(10):685.
79. Metzker ML. Sequencing technologies—the next generation. Nat Rev Genet 2010;11(1):31.
80. Bell DW. Our changing view of the genomic landscape of cancer. J Pathol 2010;220(2):231.
81. Campbell PJ, Pleasance ED, Stephens PJ, et al. Subclonal phylogenetic structures in cancer revealed by ultra-deep sequencing. Proc Natl Acad Sci U S A 2008;105(35): 13081.
82. Kidd JM, Cooper GM, Donahue WF, et al. Mapping and sequencing of structural variation from eight human genomes. Nature 2008;453(7191):56.
85. Schadt EE, Turner S, Kasarskis A. A window into third-generation sequencing. Hum Mol Genet 2010;19(R2): R227.
89. Morozova O, Hirst M, Marra MA. Applications of new sequencing technologies for transcriptome analysis. Annu Rev Genomics Hum Genet 2009;10:135.
91. Ley TJ, Mardis ER, Ding L, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature 2008;456(7218):66.
92. Pleasance ED, Cheetham RK, Stephens PJ, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 2010;463(7278):191.
93. Pleasance ED, Stephens PJ, O’Meara S, et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature 2010;463(7278):184.
94. Ding L, Ellis MJ, Li S, et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature 2010;464(7291):999.
95. Ley TJ, Ding L, Walter MJ, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med 2010;363(25): 2424.
99. Lee W, Jiang Z, Liu J, et al. The mutation spectrum revealed by paired genome sequences from a lung cancer patient. Nature 2010;465(7297):473.
100. Yachida S, Jones S, Bozic I, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010;467(7319):1114.
101. Shah SP, Morin RD, Khattra J, et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature 2009;461(7265):809.
102. Campbell PJ, Yachida S, Mudie LJ, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 2010;467(7319):1109.
110. Wiegand KC, Shah SP, Al-Agha OM, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med 2010;363(16):1532.
111. Farazi TA, Spitzer JI, Morozov P, et al. miRNAs in human cancer. J Pathol 2011;223(2):102.
112. Huarte M, Rinn JL. Large non-coding RNAs: missing links in cancer? Hum Mol Genet 2010;19(R2):R152.
113. Maher CA, Kumar-Sinha C, Cao X, et al. Transcriptome sequencing to detect gene fusions in cancer. Nature 2009;458(7234):97.
115. Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet 2009;10(10):669.
116. Laird PW. Principles and challenges of genome-wide DNA methylation analysis. Nat Rev Genet 2010;11(3):191.
118. Tomlins SA, Rhodes DS, Perner S, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005;310(5748):644.
119. Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007; 448(7153):561.
120. Palanisamy N, Ateeq B, Kalyana-Sundaram S, et al. Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer and melanoma. Nat Med 2010; 16(7):793.
121. Leary RJ, Kinde I, Diehl F, et al. Development of personalized tumor biomarkers using massively parallel sequencing. Sci Transl Med 2010;2(20):20ra14.
122. Stephens PJ, McBride DJ, Lin ML, et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature 2009;462(7276): 1005.
123. Feng H, Shuda M, Chang Y, et al. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 2008;319(5866):1096.
127. Kaminker JS, Zhang Y, Waugh A, et al. Distinguishing cancer-associated missense mutations from common polymorphisms. Cancer Res 2007;67(2):465.
128. Futreal PA. Backseat drivers take the wheel. Cancer Cell 2007;12(6):493.
129. Greenman C, Wooster R, Futreal PA, et al. Statistical analysis of pathogenicity of somatic mutations in cancer. Genetics 2006;173(4):2187.
134. The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008; 455(7216):1061.
135. Hudson TJ, Anderson W, Artez A, et al. International network of cancer genome projects. Nature 2010; 464 (7291):993.
136. Bignell GR, Greenman CD, Davies H, et al. Signatures of mutation and selection in the cancer genome. Nature 2010;463(7283):893.
137. Dalgliesh GL, Furge K, Greenman C, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature 2010;463(7279): 360.
142. Benvenuti S, Frattini M, Arena S, et al. PIK3CA cancer mutations display gender and tissue specificity patterns. Hum Mutat 2008;29(2):284.
144. Bleeker FE, Bardelli A. Genomic landscapes of cancers: prospects for targeted therapies. Pharmacogenomics 2007;8(12):1629.
145. Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004;304(5676):1497.
146. Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med 2008;358(11):1160.
149. Gerber DE, Minna JD. ALK inhibition for non-small cell lung cancer: from discovery to therapy in record time. Cancer Cell 2010;18(6):548.
152. Bardelli A, Siena S. Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. J Clin Oncol 2010;28(7):1254.
153. Siena S, Sartore-Bianchi A, Di Nicolantonio F, et al. Biomarkers predicting clinical outcome of epidermal growth factor receptor-targeted therapy in metastatic colorectal cancer. J Natl Cancer Inst 2009;101(19):1308.
158. Gorre ME, Mohammed M, Ellwood K, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 2001; 293(5531): 876.
169. Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 2010;363(9):809.
173. Druker BJ, Guilhot F, O’Brien SG, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med 2006;355(23):2408.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


