The Basis for Platelet Adhesion
Brian Savage
Zaverio M. Ruggeri
PERSPECTIVE
Mammalian platelets are circulating fragments of bone marrow megakaryocytes1,2 that function primarily to maintain the integrity of the vasculature by stopping hemorrhage after vascular injury and tissue trauma.3 Although platelets lack genomic DNA, they contain megakaryocyte-derived messenger RNA and are capable of protein synthesis.4,5 During their 10-day lifetime in the human circulation, most platelets never participate in irreversible adhesion, and are cleared from the circulation by macrophages in the spleen and liver. However, in response to alterations of endothelial cells lining the vessel wall or exposure of subendothelial matrix components following vascular injury, platelet adhesion, activation, and thrombus formation occur,3 events that are crucial for maintaining primary hemostasis and preventing posttraumatic blood loss. On the other hand, thrombus formation in diseased vessels can impede blood flow to vital organs, for example, in heart coronary arteries and cerebral arteries, leading to myocardial infarction and stroke, respectively. In addition to their role in hemostasis and thrombosis, platelets are thought to contribute to other processes, such as atherogenesis6,7,8 and inflammation,9,10,11,12 by maintaining normal vascular permeability and directing other effector cells to sites where microbial and viral pathogens induce inflammatory responses.13,14 Evidence also suggests a role for platelets in tumor angiogenesis15 since tumor cells can activate platelets and induce release of angiogenic factors that directly affect the tumor endothelium.16
To ensure normal hemostasis without the undesired thrombus formation, the adhesive properties of platelets must be tightly regulated to respond to a precise set of stimuli. A comprehensive review of the signaling mechanisms that regulate platelet adhesion is not presented in detail here, but can be found elsewhere.17,18,19,20 In this chapter, we focus on the principal substrates and receptors that mediate platelet adhesion, activation, and aggregation, and the role of blood flow in modulating platelet adhesive functions.
INDUCTION OF PLATELET ADHESION RESPONSES
Platelets circulating in close proximity to intact, unperturbed endothelial cells do not attach to them, at least in part because endothelial cells actively maintain an antiadhesive phenotype.21,22,23 However, disruption of the endothelium by mechanical trauma or pathogenic stimuli resulting from endovascular or extravascular disease processes leads to platelet adhesion at the injured site. Initiation of platelet adhesion in posttraumatic hemostasis involves platelet interaction with exposed extracellular matrix components, consisting of a large number of adhesive macromolecules including various types of collagen,24,25 von Willebrand factor (vWF),26,27 fibronectin,28 laminin,29 fibulin,30 and thrombospondin.31 In addition, platelets can interact indirectly with extracellular matrix proteins that upon exposure to blood bind plasma-derived proteins such as fibrinogen/fibrin32 vWF,26 and vitronectin33 that become immobilized onto subendothelial surfaces at sites of vascular damage. Depending on the prevailing rheologic conditions and the types of matrix proteins exposed to the flowing blood, platelet adhesion requires the synergistic function of distinct platelet receptors, ultimately leading to platelet activation and aggregation.34 Following activation, the platelet membrane presents a negatively charged phospholipid surface required for the assembly of activated coagulation factor complexes, which in turn leads to thrombin generation and the formation of a fibrin network stabilizing the growing thrombus.35
PLATELET ADHESION TO vWF
The Essential Role of Immobilized vWF
vWF is a glycoprotein (GP) that circulates in plasma as large multimers ranging in size from 500 to 20,000 kDa36,37 that contain binding sites for collagen, as well as the platelet receptors GPIbα and the integrin αIIbβ3(GPIIb/IIIa).38 The monomeric subunit of mature vWF consists of 2,050 amino acids and is composed of four distinct repeating domains that are crucial for the function of the molecule.39,40 Megakaryocytes and endothelial cells are the only cells that synthesize vWF. Megakaryocyte-derived vWF is stored in α-granules and is only released during agonist-induced platelet secretion. By contrast, endothelial cell vWF is either constituently secreted or directed to Weibel-Palade body storage granules for agonist-induced secretion.36 Plasma vWF is almost exclusively derived from endothelial cells.41 The multimers secreted from endothelial cell Weibel-Palade bodies are extremely large (ultra large vWF [ULvWF]) and their ability to bind to extracellular matrix proteins42 and to the platelet receptor, GPIbα in the GPIb-IX-V complex is enhanced compared to the smaller vWF multimers present in plasma.43 The metalloprotease, ADAMTS13, cleaves the secreted ULvWF to functionally less active, smaller multimers.44,45
Under conditions of elevated shear stress, typically present in arteries and small arterioles, vWF plays an essential role in initiating platelet adhesion and sustaining platelet aggregation.26,32 It is present as a constitutive component of the extracellular matrix of endothelial cells46 where it is associated with collagen type VI filaments,47,48,49 and can directly support platelet adhesion.50,51,52,53,54 In addition, circulating plasma vWF can interact with exposed extracellular matrix components, particularly collagen types I
and III in deeper layers of the vessel wall55 and microfibrillar collagen type VI in the subendothelial matrix.49,56,57,58 Other potential binding sites for plasma vWF include heparin, sulfatides, and fibrin.3 The main site for vWF binding to collagen types I and III is located in the vWF A3 domain,59 with a minor role, but possibly an essential one under high blood flow conditions, attributed to A1 domain binding to collagen type VI.55,60 Additionally, plasma vWF can become immobilized by self-association; a process involving the reversible interaction of circulating vWF multimers with matrix bound and endogenous subendothelial vWF.61 vWF self-association is modulated by multiple domains,62 although none have been identified as essential, including A1 and A361 (FIGURE 28.1).
and III in deeper layers of the vessel wall55 and microfibrillar collagen type VI in the subendothelial matrix.49,56,57,58 Other potential binding sites for plasma vWF include heparin, sulfatides, and fibrin.3 The main site for vWF binding to collagen types I and III is located in the vWF A3 domain,59 with a minor role, but possibly an essential one under high blood flow conditions, attributed to A1 domain binding to collagen type VI.55,60 Additionally, plasma vWF can become immobilized by self-association; a process involving the reversible interaction of circulating vWF multimers with matrix bound and endogenous subendothelial vWF.61 vWF self-association is modulated by multiple domains,62 although none have been identified as essential, including A1 and A361 (FIGURE 28.1).
Platelet Capture by Immobilized vWF
When circulating platelets encounter immobilized vWF under arterial blood flow conditions, that is, wall shear rates exceeding 800/second (value for the human circulation; approximately double in the mouse circulation), they are captured and tethered by a mechanism involving the transient interaction of platelet GPIbα with the A1 domain of immobilized vWF.32 Although this interaction is short-lived and insufficient by itself to mediate stable platelet adhesion, it maintains platelets in close contact with the blood vessel wall as they translocate in the direction of blood flow. This reduces their velocity and facilitates their subsequent adhesion and activation32 (FIGURE 28.2). It is during the translocation phase that platelets establish contact with collagen in the extracellular matrix via platelet glycoprotein VI (GPVI) and integrin α2β1, proteins that are unable to mediate platelet adhesion by themselves under high flow velocity conditions. Engagement of GPVI and α2β1 with collagen triggers intracellular signals that activate platelets and shift platelet integrins to a high-affinity state, as well as the release of the agonists ADP and thromboxane A2 (TXA2) that contribute synergistically to induce full platelet activation. Following platelet activation, stable adhesion is reinforced by the β1 integrins, α2β1, α5β1, and α6β1, that bind to collagen, fibronectin, and laminin respectively, as well as by integrin αIIbβ3 that binds to fibronectin and collagenbound vWF.
Role of Receptor GPIbα
There are two physiologically relevant platelet receptors for vWF63,64: GPIbα, a component of the GPIb-IX-V complex65 that binds to the vWF A1 domain,66,67 and the integrin αIIbβ368 that binds to the Arg-Gly-Asp motif located in vWF C1 domain.69 A third receptor, the integrin αvβ3,70 may participate in vWF-mediated platelet adhesion to endothelial cells.71 GPIbα is a highly abundant, extensively glycosylated protein expressed exclusively on megakaryocytes and platelets. It is a component of the GPIb-IX-V complex composed of four gene products: GPIbα, GPIbβ, GPIX, and GPV.72,73,74,75 Resting platelets express 25,000 copies of GPIb-IX and 12,000 copies of GPV. Human GPIbα has a 290-residue N-terminal domain containing 8 tandem leucine-rich repeats73,76 that binds vWF34 and α- thrombin.77,78 GPIbα has also been reported to contain binding sites for coagulation factors XI and XII,79 high molecular weight kininogen,80 P-selectin,81 and the leukocyte receptor Mac-1,82,83 but the precise identity of these sites remains to be determined. The interaction between GPIbα and vWF under dynamic conditions of blood flow supports the activationin-dependent capture and tethering of platelets to immobilized vWF32 and this occurs despite forces opposing platelet adhesion
at elevated flow rates.32,84 GPIbα-mediated platelet adhesion to vWF is functionally similar to selectin-mediated leukocyte rolling on inflamed vascular surfaces,85 and presumably shares similar biophysical characteristics, such as rapid on-rates86,87,88 and rapid dissociation-rates.84,86,88,89,90 In addition, both interactions support flow-enhanced adhesion91 and are thought to involve “catch” bonds, rather than “slip” bonds.92 Catch bonds are characterized by a lifetime that increases directly with applied shear force up to a maximum threshold value. As applied shear force increases above the threshold, the lifetime of the bond decreases, resulting in the transition to a more typical slip bond for which the lifetime decreases with increasing shear stress.92 Recent studies have provided a better understanding of the biomechanical properties of vWF A1 domain binding to GPIbα.93 A method that enables the repeated measurement of the binding and unbinding of interacting receptor and ligand domains engineered within a single molecule has revealed the existence of two states of the vWF A1 domain-GPIbα bond and has thus been named a flex-bond. One state is seen at low force, while the second state begins to engage at higher force (10 pN) with approximately 20-fold longer lifetime and greater force resistance.
at elevated flow rates.32,84 GPIbα-mediated platelet adhesion to vWF is functionally similar to selectin-mediated leukocyte rolling on inflamed vascular surfaces,85 and presumably shares similar biophysical characteristics, such as rapid on-rates86,87,88 and rapid dissociation-rates.84,86,88,89,90 In addition, both interactions support flow-enhanced adhesion91 and are thought to involve “catch” bonds, rather than “slip” bonds.92 Catch bonds are characterized by a lifetime that increases directly with applied shear force up to a maximum threshold value. As applied shear force increases above the threshold, the lifetime of the bond decreases, resulting in the transition to a more typical slip bond for which the lifetime decreases with increasing shear stress.92 Recent studies have provided a better understanding of the biomechanical properties of vWF A1 domain binding to GPIbα.93 A method that enables the repeated measurement of the binding and unbinding of interacting receptor and ligand domains engineered within a single molecule has revealed the existence of two states of the vWF A1 domain-GPIbα bond and has thus been named a flex-bond. One state is seen at low force, while the second state begins to engage at higher force (10 pN) with approximately 20-fold longer lifetime and greater force resistance.
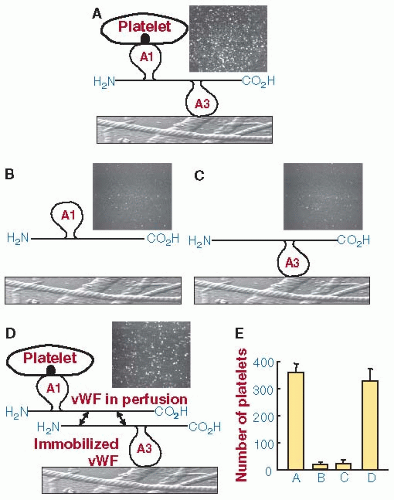 FIGURE 28.1 Functional self-association of vWF multimers. Suspensions of washed blood cells containing ethylenediaminetetraacetic acid (EDTA) to block integrin function and prostaglandin (PG) E1 to block platelet activation were perfused over immobilized collagen type I fibrils at the wall shear rate of 1,500/s. A: Normal multimeric vWF was added to a cell suspension. Its A3 domain mediates vWF binding to collagen and the A1 domain interacts with platelet GPIbα. In the electron micrograph, tethered platelets are seen rolling on the collagen fibrils. B: vWF devoid of the A3 domain (ΔA3-vWF) which cannot bind to collagen or (C) vWF devoid of the A1 domain (ΔA1-vWF), which cannot interact with platelet GPIbα was added to the cell suspension. In neither case, were platelets tethered to the immobilized collagen. D: Collagen fibrils were precoated with ΔA1-vWF multimers and then exposed to the blood cell suspension containing ΔA3-vWF. Although the latter cannot bind directly to collagen, they can associate with the ΔA1-vWF multimers to support platelet tethering. The two-sided arrows between multimers indicate that the association is reversible. The images are single frames from a real time recording representing an area of 65,536 mm2. The bar graph shows the number of platelets tethered to the surface under the different experimental conditions described above (mean ± SEM of two separate experiments). E: quantifies the results shown in A–D. (Modified from Savage B, Sixma JJ, Ruggeri ZM. Functional self-association of von Willebrand factor during platelet adhesion under flow. Proc Natl Acad Sci U S A 2002;99:425-430, and reprinted with permission. Copyright (2002) National Academy of Sciences, U.S.A.) |
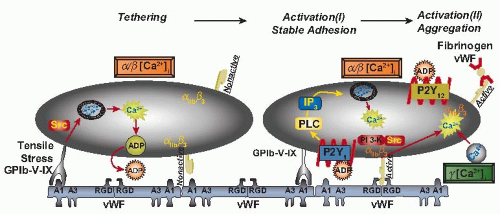 FIGURE 28.2 Schematic representation of the sequential signaling events induced by the interaction of platelets with immobilized vWF under high shear stress. On the left, a platelet is shown during the initial tethering to the A1 domain of immobilized vWF mediated by the GPIb-IX-V complex. An α/β [Ca2+]i elevation is elicited as a consequence of this interaction and leads to ADP release from intracellular storage granules. Src family kinases may be involved at this stage and cAMP/cGMP levels modulate this and other downstream responses. Subsequent events are shown in the platelet on the right. Released ADP binds to the Gq-coupled P2Y1 receptor, leading to PLC activation and enhanced Ca2+ release from internal stores during α/β oscillations. At this stage, there is a first level of localized αIIbβ3 activation that supports more prolonged platelet adhesion mediated by the RGD sequence in the vWF C1 domain. PI 3-K activation may enhance this response. Further PI 3-K activation, and possibly the involvement of Src family kinases, contribute to more generalized αIIbβ3 activation that permits soluble ligand binding and supports formation of platelet-platelet aggregates. This second level of αIIbβ3 activation is concurrent with or subsequent to a type γ [Ca2+]i elevation dependent on a transmembrane ion flux. The second ADP receptor, P2Y12, supports the formation of larger platelet aggregates through mechanisms that occur after the measured Ca2+ oscillations. The TXA2 pathway inhibited by aspirin appears to have a very limited role in the successive stages of platelet adhesion, activation, and aggregation induced by the interaction with immobilized vWF. IP3, inositol-1,4,5-trisphosphate; Src, Src family tyrosine kinases; PLC, phospholipase C; PKC, protein kinase C; PI 3-K, phosphatidylinositol 3-kinase. (Adapted from Mazzucato M, Cozzi MR, Pradella P, et al. Distinct roles of ADP receptors in von Willebrand factor-mediated platelet signaling and activation under high flow. Blood 2004;104:3221-3227.) |
Mutations in both vWF A1 domain and GPIbα can change the mechanical properties of the flex bond. A gain-of-function point mutation in GPIbα at Met239Val results in an approximately sixfold increase in affinity for the interaction with vWF A1 domain due to an increased on-rate.94 By contrast, a gainof-function point mutation in vWF A1 at Arg543Gln does not appear to form catch bonds with GPIbα, and has an increased affinity for GPIbα due mainly to a decreased off-rate, resulting from the formation of slip bonds even at low shear stresses.95 Regulation of the GPIbα/vWF interaction appears to involve conformational changes in both the receptor and the ligand,96 and phosphorylation of Ser166 of GPIbβ may negatively regulate vWF binding.97
GPIb-IX-V-Dependent Signaling
Until recently it was thought that the interaction of GPIbα with immobilized vWF only provides a mechanism for recruiting platelets to sites of vascular damage, thereby reducing their velocity to enable other platelet receptors to elicit intracellular activation signals. However, it is now clear that GPIb-IX-V, in addition to its role as an adhesion receptor, can mediate intracellular signals leading to activation of the integrins αIIbβ3 and α2β1, allowing platelets to firmly attach, spread, and aggregate.26,98,99,100,101 Several intracellular signaling pathways are involved in GPIb-IX-V-mediated platelet activation, including the Syk/phospholipase C (PLC) pathway,101,102 the phosphatidylinositol 3-kinase (PI 3-K)-Akt pathway,103,104,105 the cGMP106 and mitogenactivated protein kinase pathways,107 and the TXA2 and ADP pathways.108,109 Members of the Src family kinases appear to
play an important role because Src inhibitors prevent GPIb-IX-V-induced αIIbβ3 activation,110,111 Ca2+ mobilization,112 PI 3-K activation,113,114 and TXA2 synthesis.108 The Src family tyrosine kinase Lyn has been shown to be crucial in GPIb-IX-V-mediated activation of the cGMP pathway.111
play an important role because Src inhibitors prevent GPIb-IX-V-induced αIIbβ3 activation,110,111 Ca2+ mobilization,112 PI 3-K activation,113,114 and TXA2 synthesis.108 The Src family tyrosine kinase Lyn has been shown to be crucial in GPIb-IX-V-mediated activation of the cGMP pathway.111
Engagement of GPIb-IX-V with the vWF A1 domain also leads to the tyrosine phosphorylation of adhesion and degranulation promoting adaptor protein (ADAP),110 a potential linker between cell activation events at the plasma membrane and intracellular events. ADAP is required for the modulation of αIIbβ3 affinity induced by GPIb-IX-V interaction with the vWF A1 domain.115 Despite the many signaling pathways attributed to the interaction of GPIb-IX-V with vWF, the relative importance of this interaction in vivo compared to other receptor/ligand signaling pathways remains to be established. Indeed, platelet translocation on immobilized vWF ex vivo may last for several minutes before stable αIIbβ3-mediated attachment occurs,32 indicating that the activating signal is relatively weak compared with those induced by more thrombogenic surfaces like collagen.
As recently demonstrated, platelet adhesion and aggregation can be initiated independently of platelet or integrin activation through a mechanism that involves the interaction with immobilized as well as soluble vWF under high shear stress conditions116 (FIGURE 28.3). A key feature of this mechanism is that sustained platelet attachment and aggregation only occur above a threshold shear rate of 10,000/second, a pathologically relevant shear rate in arterial thrombosis.116 These studies have been extended using experimental mouse models of vascular injury, demonstrating that thrombi initially develop through the progressive stabilization of discoid platelet aggregates.117 These discoid platelets adhere preferentially in low-shear zones downstream of forming thrombi, indicating that disturbed blood flow, creating gradients of increasing and decreasing shear stress, may play an important role in initiating the process of thrombogenesis in arteries.
The Role of vWF in Venous Thrombosis
Although the participation of vWF in arterial thrombosis has been repeatedly demonstrated, its role in platelet recruitment during the initiation of venous thrombosis has only recently been documented.118 Unlike arterial thrombosis, thrombosis in deep veins is not initiated by injury to the endothelial layer,119 but rather by blood flow disturbances resulting in complete or partial flow restriction.120 Although increased procoagulant states are known risk factors for deep vein thrombosis,121,122,123 disturbed blood flow due to long-term immobilization can also provoke deep vein thrombosis in the absence of hypercoagulability.124,125 Flow disturbance-induced venous thrombosis in mice due to prolonged flow restriction in the inferior vena cava was significantly reduced in mice genetically deficient in vWF.118 Furthermore, infusion of a mutant GPIbα-IgG chimera that inhibits the interaction of vWF A1 domain with platelets prevented thrombosis in wild-type mice with normal vWF levels. Although circulating vWF is coupled to factor VIII, infusions of recombinant factor VIII did not rescue thrombosis in vWF-deficient mice. These observations suggest that vWF-platelet adhesive interactions may play a key pathogenic role in initiating venous thrombosis following flow restriction.
 FIGURE 28.3 Activation-independent platelet adhesion and aggregation at the interface of immobilized and soluble vWF. A: Blood containing 93 µM D-phenylalanyl-L-prolyl-L-arginine chloromethyl ketone dihydrochloride to inhibit α-thrombin, the fluorescent dye mepacrine at 10 µM, 10 µM PG E1, and 5 mM EDTA was perfused over immobilized vWF. The white line delimits vWF-coated (to the left) from uncoated glass. Single platelets adhere when the shear rate is 3,000/s (top); rolling aggregates (some identified by arrowheads) form at 20,000/s (bottom). B: Perfusion of washed blood cells suspended in buffer (20 mM Hepes, 150 mM NaCl, pH 7.4) over immobilized vWF. In the absence of soluble vWF, single platelets adhere when the shear rate is 3,000/s (upper left), and fewer single platelets adhere at 24,000/s (upper right). After adding 20 µg/mL soluble vWF, single platelets adhere at 3,000/s (lower left; an arrow points to a single platelet shown for reference), but aggregates form at 24,000/s (lower right; arrows point to a rolling aggregate and an inset highlights a stretched aggregate during stationary adhesion). C: Perfusion of washed blood cells with added soluble vWF and anti-vWF A1 domain monoclonal antibody (NMC-4, 20 µg/mL) over immobilized vWF. No platelet adhesion is detected. (From Ruggeri ZM, Orje JN, Habermann R, et al. Activation-independent platelet adhesion and aggregation under elevated shear stress. Blood 2006;108:1903-1910.) |
PLATELET ADHESION TO COLLAGEN AND PLATELET COLLAGEN RECEPTORS
The vessel wall contains several different types of collagen, particularly the fibrillar collagens type I, III, and VI. The collagens can associate with each other and with other matrix proteins,
and play an active role in the initiation of platelet adhesion and aggregation in response to injury.126 Experimental studies with collagens can be complicated by changes in collagen structure introduced during extraction of the protein from extracellular matrix. For example, acid-insoluble collagen type I fibers display a characteristic banded pattern due to the regular staggering of collagen monomers126,127 and react with platelets bearing GPVI even when α2β1 is inhibited. By contrast, pepsin-solubilized acid soluble collagen type I microfibrils lack the banded pattern and form spiral structures127 and optimally interact with platelets bearing functional α2β1, in addition to GPVI. As collagen is an insoluble matrix protein, preparations used in ex vivo experimental studies are often treated with proteases, such as pepsin, for solubilization resulting in functional behavior distinct from that of the native protein. Thus, the presence of spiral fibrils in pepsin-treated acid soluble collagen can lead to an overestimate of the role of α2β1 in the interaction of collagen with platelets.
and play an active role in the initiation of platelet adhesion and aggregation in response to injury.126 Experimental studies with collagens can be complicated by changes in collagen structure introduced during extraction of the protein from extracellular matrix. For example, acid-insoluble collagen type I fibers display a characteristic banded pattern due to the regular staggering of collagen monomers126,127 and react with platelets bearing GPVI even when α2β1 is inhibited. By contrast, pepsin-solubilized acid soluble collagen type I microfibrils lack the banded pattern and form spiral structures127 and optimally interact with platelets bearing functional α2β1, in addition to GPVI. As collagen is an insoluble matrix protein, preparations used in ex vivo experimental studies are often treated with proteases, such as pepsin, for solubilization resulting in functional behavior distinct from that of the native protein. Thus, the presence of spiral fibrils in pepsin-treated acid soluble collagen can lead to an overestimate of the role of α2β1 in the interaction of collagen with platelets.
Collagen activates platelets primarily through the membrane proteins GPVI and α2β155,128 (FIGURE 28.4); several other potential collagen “receptors” have been described, but none has been shown to convincingly play a role in platelet adhesion and aggregation. Even the relative contribution of GPVI and α2β1 to these processes remains a topic for debate. Ambiguities introduced during the isolation of collagen from extracellular matrices, as discussed above, as well as poorly understood differences between human ex vivo and mouse in vivo experimental systems may explain the lingering uncertainties.55,128 According to the prevalent current model, GPVI is essential for platelet activation and aggregation on collagen, whereas α2β1 is required for firm platelet adhesion with only a relatively minor role in platelet activation.128 More recent evidence, obtained studying platelet-collagen interactions under flow,129,130 supports the existence of a functional synergy between α2β1 and GPVI. Indeed, it was found that α2β1 can signal independently of GPVI or αIIbβ3 (GPIIb-IIIa), and that GPVI function is facilitated by preceding intracellular Ca2+ elevations resulting from α2β1 engagement. This does not exclude the possibility that the ligand-binding affinity of α2β1 is enhanced following GPVI interaction with collagen and/or αIIbβ3 activation. Accordingly, the functional activities of α2β1 and GPVI may be viewed as concurrent, or mutually reinforcing, rather than sequential, a concept supported by the finding that variations in the amount of α2β1 on the platelet surface influences the extent of GPVIdependent Ca2+ signaling.129
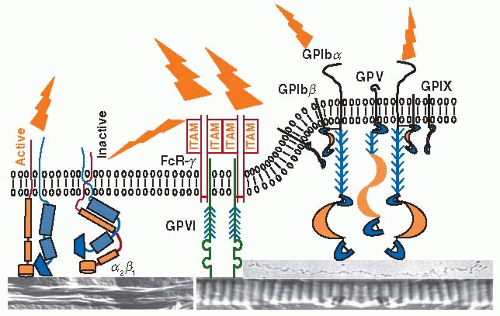 FIGURE 28.4 Schematic representation of the mechanisms of platelet adhesion to collagen. The collagen receptors, α2β1 and GPVI, are depicted along with the GPIb-IX-V complex. Collagen and vWF are represented by electron micrographs (not in scale). Two forms of collagen type I are shown, “native” acid-insoluble fibers (on the right) and pepsin-treated acidsoluble “spiraled” fibrils (on the left), with a vWF multimer shown as if adhering to the native collagen fiber. On a surface in which collagen and vWF form a functional unit, initial platelet tethering is mediated primarily by GPIbα binding to vWF-A1 under high flow conditions, allowing subsequent GPVI binding to collagen, specifically to the prototypic repeating sequence Gly-Pro-Hyp (where Hyp is hydroxyproline), typical of the fibril-forming collagens. Both receptors can signal, but the role of GPVI is predominant in this regard. The integrin α2β1 requires activation by an inside-out signal for high affinity binding to collagen; however, on partially degraded “spiraled” fibrils, it may be essential for the initial tethering through recognition of the sequence Gly-Phe-Hyp-Gly-Glu-Arg. The fact that GPVI is not sufficient for adhesion to this substrate attests to the conformational disruption of its recognition site in partially degraded collagen. |
Glycoprotein VI
Approximately 10% of collagens I and III consist of a repeating GPO (glycine-proline-hydroxyproline) motif, also known as collagen-related peptide131 that is the strongest platelet stimulus within the collagen molecule132 and that reacts specifically with platelet GPVI.133 The smallest collagen motif that recognizes and activates platelet GPVI consists of two GPO triplets.134 On the platelet surface, GPVI is present in a noncovalent complex with the Fc receptor-γ chain, an interaction that is essential for both GPVI expression and function.128,135 Functional GPVI is thought to be a dimer. Each GPVI monomer contains two extracellular Ig-C2-like domains, a mucinlike domain, a transmembrane region, and a short 51-residue cytoplasmic tail.136,137 The cytoplasmic tail has distinct sites
that interact with calmodulin to regulate Ca2+ signaling pathways and with the Src family tyrosine kinases Fyn and Lyn that signal independently of the Fc-receptor. GPVI can also signal in response to collagen or other ligands by an Fc- receptordependent pathway involving activation of the tyrosine kinase Syk.99 Signaling through GPVI upregulates platelet integrins, including α2β1 and αIIbβ3, events essential for shear-resistant platelet attachment to collagen.
that interact with calmodulin to regulate Ca2+ signaling pathways and with the Src family tyrosine kinases Fyn and Lyn that signal independently of the Fc-receptor. GPVI can also signal in response to collagen or other ligands by an Fc- receptordependent pathway involving activation of the tyrosine kinase Syk.99 Signaling through GPVI upregulates platelet integrins, including α2β1 and αIIbβ3, events essential for shear-resistant platelet attachment to collagen.
Related posts:
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Molecular Basis for Platelet Secretion
Molecular Basis for Platelet Secretion
 Acquired Nonimmune Thrombocytopenia
Acquired Nonimmune Thrombocytopenia
 Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


