Anatomy
The adrenal glands are retroperitoneal structures weighing approximately 5–7 g that have a characteristic golden colour due to cholesterol in their cortex (see Fig. 3.1 ). Their position and shape are slightly different on each side. The right gland is pyramidal and lies at the upper pole of the right kidney, between the right crus of the diaphragm and the inferior vena cava. The left adrenal gland is crescentic and is situated on the upper medial aspect of the left kidney. The tail of the pancreas lies anterior and the diaphragm crus is posterior to the left adrenal gland. Each gland consists of two distinct parts, which have different structures and functions – the outer cortex and the inner medulla.
Blood supply and lymphatic drainage
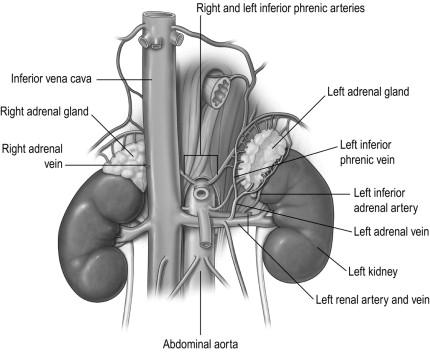
The blood supply is derived from branches of three vessels, the inferior phrenic artery, the ipsilateral renal artery and the aorta. These branches often subdivide into a leash of vessels, before entering the gland. Venous return is via a single adrenal vein that drains into the renal vein on the left and directly into the inferior vena cava on the right. The right vein is short and an important landmark for the surgeon. The veins drain from the medulla and therefore the venous return from the cortex flows through the medulla. This is relevant as glucocorticoid hormones secreted by the cortex activate phenylethanolamine N -methyltransferase (PNMT), an enzyme involved in the synthesis of catecholamines. Medullary and subcapsular lymphatic plexuses drain into lymphatics that follow the arterial supply to the para-aortic lymph nodes.
Nerve supply
The adrenal cortex receives a few vasomotor fibres, but the medulla is richly supplied with fibres derived from the splanchnic nerves (T5–9) and should be regarded as a modified sympathetic ganglion in which the axon has been replaced by secretory cells, called phaeochromocytes or chromaffin cells, filled with granules containing catecholamines.
Microscopic anatomy
The adrenal medulla accounts for 15% of the volume of the adrenal gland and consists of vascular spaces and eosinophilic phaeochromocytes of variable size containing polymorphic nuclei. Phaeochromocytes are characterised histologically by their uptake of dichromate salts and hence are referred to as chromaffin cells.
The adrenal cortex is divided into three layers: the outer zona glomerulosa, the zona fasciculata and the innermost zona reticularis. The zona glomerulosa produces mineralocorticoids. It consists of columnar cells, with relatively little cytoplasm compared with their nuclei, organised into clusters. The zona fasciculata is the largest of the cortical zones, occupying approximately 75% of the cortex, that contains poorly staining, polyhedral cells organised in radial columns and is primarily responsible for the production of glucocorticoids. The innermost zona reticularis is characterised by rounded branching cords of cells and produces sex hormones, particularly dehydroepiandrosterone sulphate (DHEAS).
Embryology
The cortex and medulla have different embryological origins. The adrenal cortex is mesodermal in origin and starts to appear in the fifth week of gestation as two clefts on either side of the embryonic dorsal mesentery that enlarge to form the primitive or foetal cortex. This is surrounded at the seventh week by a second wave of mesothelial cells to form the secondary cortex that eventually becomes the adult adrenal cortex. Concordantly, cells migrating from the neural crest invade the developing adrenal gland to form the adrenal medulla, which is therefore neuroectodermal in origin. The adrenal glands are large at birth, but reduce in size thereafter due to regression of the primary cortex, and do not regain their original size until puberty. The zona glomerulosa starts to appear before delivery, followed by the zona fasciculata and finally the zona reticularis a few months after birth. Neural crest cells, outside the adrenal medulla, are widely present in the embryo, but regress after birth.
Physiology
Adrenal medulla
Catecholamine synthesis and metabolism
The adrenal medulla synthesises the catecholamines dopamine, noradrenaline (norepinephrine) and adrenaline (epinephrine) from tyrosine by a series of steps via 3,4-dihydroxyphenylalanine (DOPA). The enzyme tyrosine hydroxylase, which controls the rate-limiting step, is largely confined to the central and sympathetic nervous systems, and the adrenal medulla. The final step in the pathway of catecholamine synthesis (to adrenaline) is catalysed by PNMT, which is induced by cortisol as previously discussed (see Fig. 3.2 ).
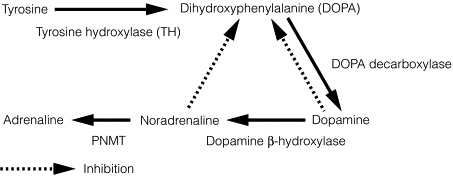
After stimulation of the adrenal medulla, catecholamine release occurs by a calcium-dependent process in which secretory granules fuse with the cell membrane (exocytosis). The majority of the released catecholamines are taken back up into the presynaptic terminals of the chromaffin cells and deaminated by monoamine oxidase. The remaining catecholamines enter the systemic circulation, where they have diverse effects and are methylated by carboxy- O -methyltransferase to methoxytyramine, metanephrine and normetanephrine. Vanillylmandelic acid (VMA) is produced in the liver following deamination and methylation of catecholamines, and is excreted along with sulphate-conjugated metanephrines in the urine.
Catecholamine physiological effects
Basal secretion of catecholamines is low but rises substantially in response to certain stimuli. Catecholamines exert their characteristic effects by binding to cell-membrane-bound α- and β-adrenoreceptors present in most tissues and organs. The physiological effects are typified as the ‘flight or fight’ response, i.e. preparing the organism for optimal performance in times of threat, and include an increase in heart rate, blood pressure and cardiac output, excitation of the central nervous system, increase in blood flow to muscles and decrease in splanchnic perfusion – as well as metabolic effects such as lipolysis, gluconeogenesis and glycogenolysis – to provide substrates for this action to occur.
Adrenal cortex
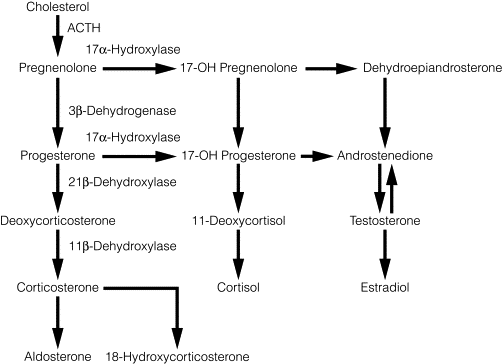
The zones of the adrenal cortex synthesise steroid hormones from cholesterol via a common pathway illustrated in Fig. 3.3 .
Mineralocorticoids
Aldosterone, the main mineralocorticoid hormone, acts on the distal renal tubule to increase resorption of sodium (and water) by an active transport mechanism, in which Na + is exchanged for K + or H + ions, thus leading to an increase in the circulatory volume. The secretion of aldosterone is controlled primarily by angiotensin II, which in turn is generated through the activity of renin and angiotensin-converting enzyme on angiotensinogen. Falls in the circulatory volume or blood pressure, or increases in sympathetic output, stimulate renin secretion from the juxtaglomerular apparatus of the kidney. Other factors that stimulate aldosterone secretion include adrenocorticotropic hormone (ACTH) and elevated plasma potassium levels. A feedback loop controlling renin and aldosterone secretion occurs to regulate intravascular volume and electrolyte balance.
Glucocorticoids
Cortisol and corticosterone are the main glucocorticoids. Their effects on glucose and protein metabolism cause hyperglycaemia by promoting hepatic gluconeogenesis and glycogenolysis, protein catabolism in muscle and lipolysis in fat. They also have weak mineralocorticoid effects. Osteoporosis occurs with supraphysiological levels of glucocorticoids due to decreased intestinal absorption and increasing urinary excretion of calcium. Cortisol, which is secreted under the action of ACTH, regulates its own secretion via a negative-feedback mechanism on the hypothalamus and pituitary. The hypothalamus controls ACTH secretion from the pituitary via corticotropin-releasing hormone (CRH) and is also responsible for the diurnal rhythm of serum cortisol, which is highest in the early morning and lowest at night.
Sex steroids
The adrenal cortex produces a number of weakly androgenic sex steroids under the action of ACTH, of which DHEAS is quantitatively the most important. They are converted in peripheral tissues to dihydrotestosterone and oestradiol via the aromatase enzyme system.
Adrenal incidentaloma
Case study 1
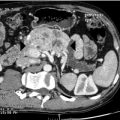
Consider the management of a 65-year-old woman who has an incidental 3-cm right adrenal adenoma on unenhanced computed tomography (CT) scan (see Fig. 3.4 ) performed following a fall in which she suffers a femoral shaft fracture. There is a recent history of hypertension. After surgery for her fracture a bone mineral density scan shows severe osteoporosis. Biochemical evaluation including 24-hour urinary free cortisol, plasma free metanephrines, plasma aldosterone and renin actitivity are unremarkable, but plasma cortisol fails to suppress after an overnight low-dose dexamethasone test and plasma ACTH is suppressed.
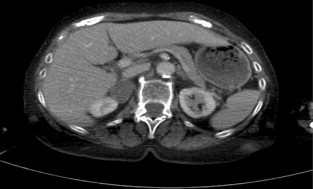
Definition and incidence
Incidentalomas are tumours identified inadvertently during investigation of an unrelated condition. Overall adrenal incidentalomas are present on 4% of abdominal CT scans, though their incidence increases with age from < 1% in the third decade to 7% in the eighth decade of life. The current widespread use of cross-sectional abdominal imaging has created the clinical dilemma of how to manage these incidentalomas. The objective is to identify those that are potentially malignant, or harmful due to excessive hormonal secretion.
Aetiology
The majority of adrenal incidentalomas are benign non-functioning adenomas. The incidence of clinically significant adrenal incidentaloma varies between studies, but about 15% are found to secrete excess hormones. Adrenocortical carcinoma and phaeochromocytoma each account for 5–10%. Excluding studies of oncology patients, metastases account for a small (2.5%) proportion of incidentalomas. Lung, breast, ovary, kidney and melanoma are the most common primary tumours that metastasise to the adrenal gland. The remaining incidentalomas are due to myleolipoma, haemorrhage, adrenal cortical cyst, ganglioneuroma, neuroblastoma, lymphoma, congenital adrenal hyperplasia, haemangioma, granulomatous disease and other rare diagnoses.
Investigation
Initial assessment of a patient with an incidentaloma should start with the patient and not by focusing on the imaging. Reflex biopsy and unconsidered investigation of coincidentally found adrenal masses can lead to disasters. A past medical history of malignant disease or family history of endocrine disease should be sought. History and clinical examination should be carefully undertaken, looking for evidence of hormone excess such as obesity, hypertension, diabetes, osteoporosis, virilisation or feminisation. Features of pituitary–adrenal axis imbalance may suggest Cushing’s syndrome, and hypokalaemia in hypertensive patients suggests primary hyperaldosteronism.
Biochemistry
Biochemical investigation needs to exclude phaeochromocytoma by measuring fractionated urinary metanephranes or plasma free metanephranes. The diagnosis of Cushing’s syndrome is more difficult to exclude as more subtle forms, so-called subclinical Cushing’s syndrome, occur and 24-hour urinary free cortisol, low-dose (1 mg) overnight dexamethasone suppression test and midnight salivary cortisol may all be required to confirm or exclude the diagnosis. Primary hyperaldosteronism is suggested in hypertensive patients with elevated plasma aldosterone:renin activity ratio >20. Hypokalaemia is present in only half the patients with primary hyperaldosteronism. Serum DHEAS and 17-hydroxyprogesterone are measured to exclude adrenal androgen hypersecretion that occurs in some adrenocortical carcinomas or, when bilateral adrenal masses are present, congenital adrenal hyperplasia.
Biopsy
Adrenal biopsy is generally unhelpful, as differentiating between benign and malignant primary adrenocortical lesions is rarely possible, and biopsy may precipitate a ‘phaeochromocytoma crisis’ in unblocked phaeochromocytoma.
Adrenal biopsy is not recommended, except in patients with a history of extra-adrenal primary malignant disease, following exclusion of a phaeochromocytoma, when cells resembling those of the primary tumour retrieved by guided biopsy may establish the presence of metastatic disease.
Imaging
Adrenal cysts, myelolipoma and haemorrhage have characteristic features on CT enabling diagnosis; differentiating adrenal adenoma from malignant tumours, phaeochromocytoma and other causes of incidentaloma can be more problematic, as some overlap occurs in the appearance of these lesions on cross-sectional imaging.
Most adrenal adenomas are lipid rich and appear as low-density (or attenuation) masses on unenhanced CT. They are characteristically homogeneous with a regular outline, and adrenal incidentaloma <4 cm in size with these features, and an attenuation value of <10 Hounsfield units on unenhanced CT require no further diagnostic imaging.
Malignant lesions, phaeochromocytoma and up to 30% of adenomas are lipid poor and have high attenuation on unenhanced CT. Other features that increase the risk of malignancy include heterogeneity, irregular outline and size >4 cm. CT with contrast enhancement and washout characterise these lesions. Both adrenocortical carcinomas and medullary tumours show rapid contrast enhancement but adenomas have a rapid washout of contrast, resulting in an attenuation value of <30 Hounsfield units at 10 minutes, in contrast to malignant adrenocortical tumours and phaeochromocytoma where contrast is retained.
Magnetic resonance imaging (MRI) can distinguish between benign and malignant tumours in 90% of cases. Malignant tumours usually have a higher fluid content than lipid-rich adenomas, which results in high signal intensity on T2-weighted MRI. Loss of signal intensity on out-of-phase MRI with chemical shift imaging, also due to the high lipid content of adenomas, can be used to distinguish benign from malignant tumours. Homogeneous enhancement following intravenous gadolinium-enhanced MRI is characteristic of adrenal adenoma.
[ 18 F]Fluorodeoxyglucose (FDG) positron emission tomography (PET) combined with CT is highly accurate at differentiating benign from malignant incidentalomas and is helpful in lesions with an indeterminate appearance on CT or MRI. Malignant tumours characteristically have high [ 18 F]FDG uptake compared to adenomas.
Management
The evidence base for the management of incidentalomas is based on expert opinion due to the lack of prospective studies. Potentially malignant and functional adrenal incidentaloma should generally be excised.
Size is the major determinant of malignant potential, as less than 2% of tumours <4 cm will be adrenocortical carcinoma (ACC), whereas 25% of tumours >6 cm are ACC.
Guidelines from North America state that non-functional tumours <4 cm in size with a benign appearance on imaging can be managed conservatively with regular follow-up: adrenalectomy is advised for tumours >6 cm in size or in smaller lesions when malignancy is suspected; adrenalectomy is reasonable for tumours 4–6 cm in size, though conservative management with close radiological and biochemical follow-up may equally be undertaken in this group, with surgery if rapid growth rate or malignant features develop during surveillance.
The natural history of adrenal incidentalomas is unknown. Although approximately a quarter of incidentalomas increase in size with time, the risk of malignant change is thought to be low. Annual surveillance imaging is often performed though there is limited evidence to recommend the optimum length or frequency of radiological follow-up. To avoid excessive radiation exposure that in itself may be tumour inducing, MRI is the preferred method for serial surveillance. Secretory hyperfunction may also develop during follow-up in up to 9% of incidentalomas, though further data are needed to establish the benefit, length and frequency of follow-up hormonal evaluation.
Although clinically evident Cushing’s syndrome is rare in patients presenting with incidentalomas, more subtle elevations of cortisol secretion, termed ‘subclinical’ Cushing’s syndrome, are detected in 5–20%. These patients may exhibit some signs of glucocorticoid excess such as obesity, hypertension, diabetes mellitus or osteoporosis, but lack the characteristic features of the full-blown syndrome. Often one or more of the biochemical tests for Cushing’s syndrome are normal in patients with subclinical Cushing’s syndrome, commonly the urinary free cortisol. The low-dose overnight dexamathasone test or late-night salivary cortisol are more sensitive tests, and progression to full-blown Cushing’s syndrome is unpredictable.
Efforts to establish evidence-based management of subclinical Cushing’s syndrome are hampered by any agreed diagnostic criteria and the lack of randomised studies comparing medical to surgical management. Uncontrolled surgical series have reported improvements in hypertension, obesity and diabetes mellitus following adrenalectomy in patients with subclinical Cushing’s syndrome, though these studies did not compare surgery with best medical management.
A single prospective randomised trial of 45 patients, comparing surgery with medical management of subclinical Cushing’s syndrome, showed greater improvement in diabetes mellitus, hypertension, hyperlipidaemia and obesity, but not bone mineral density in the surgical arm. Given the lack of evidence at present a pragmatic approach is to offer surgery to young patients with subclinical Cushing’s syndrome who have diseases attributable to cortisol excess.
Case study 1 (discussion)
This patient has subclinical Cushing’s disease with hypertension and osteoporosis that may be due to glucocorticoid excess. The size and imaging characteristics of the incidentaloma do not merit surgery alone, as these suggest a benign adrenal adenoma. If conservative management is pursued then medical management of hypertension and osteoporosis should be undertaken, with surveillance to (a) exclude enlargement of the adrenal tumour or development of malignant features, (b) monitor continued hormone excess and (c) detect any deterioration in bone mineral density. It is very reasonable to offer adrenalectomy; the laparoscopic transabdominal or retroperitoneal approaches are both suitable. Age and medical comorbidities influence the decision to operate in these circumstances. Perioperative steroids should be given to prevent an Addisonian crisis postoperatively.
Adrenocortical carcinoma
Case study 2
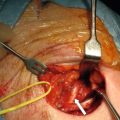
Consider the management of a 50-year-old man who on investigation for abdominal discomfort is found to have a large non-functioning adrenal mass (see Fig. 3.5 ).What is the differential diagnosis? Staging investigations are negative. What is the surgical approach and which adjuvant therapy is indicated?
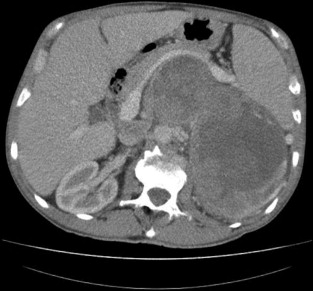
Incidence and aetiology
Adrenocortical carcinoma (ACC) is one of the most lethal endocrine tumours but fortunately is rare, with an incidence of 1–2 per million per year. Most ACC is sporadic in origin but it may be associated with a number of rare syndromes, including: multiple endocrine neoplasia type 1 due to mutation in a gene on chromosome 11 encoding the protein menin; Beckwith–Wiedemann syndrome (exophthalmos, macroglossia and nephromegaly) in childhood due to mutation of two genes on chromosome 11 leading to overproduction of insulin-like growth factor (IGF) 2; and Li–Fraumeni syndrome (breast carcinomas, osteosarcomas and brain tumours) due to inactivating mutations of tumour suppressor gene p53 on chromosome 17.
Clinical features
The majority of ACCs are hormonally functional and present with Cushing’s syndrome, primary hyperaldosteronism, virilisation or feminisation. Secretion of multiple hormones is characteristic of ACC. In our own series, in which the frequency of clinical presentation was quite typical, 57% of ACCs were functioning, 48% presented with Cushing’s syndrome (18% secreted multiple hormones – Cushing’s syndrome and virilisation), 6% presented with pure virilisation and 3% feminisation; 43% were non-functioning, of which 34% had symptoms (abdominal discomfort) and 9% were found incidentally.
Biochemistry
Biochemical evaluation to establish the steroid secretory profile of ACC should be undertaken as described elsewhere in this chapter. Certain biochemical findings are characteristic of ACC: excess secretion of multiple steroid hormones, particularly glucocortocoids and androgens, elevated DHEAS, raised oestradiol in men and postmenopausal women, and as enzyme function is often defective in ACC, accumulation of steroid precursors such as 17α-hydroxyprogesterone and androstenedione. Phaeochromocytoma should also be excluded biochemically, as this is not possible on imaging alone.
Imaging
ACCs typically appear as large (usually >5 cm), heterogeneous lesions with an irregular margin on unenhanced CT. They are denser than lipid-rich adenomas so they have attenuation >10 Hounsfield units on unenhanced CT and display delayed washout of contrast. Local invasion or distant metastases may be apparent on CT. The incidence of malignancy, which increases with size, is 2% in adrenal masses < 4 cm, 6% for those 4–6 cm and 25% in lesions >6 cm.
MRI can be helpful as ACCs have high signal intensity on T2-weighted images, heterogeneous enhancement and delayed washout with gadolinium contrast, and do not exhibit loss of signal intensity on out-of-phase chemical shift imaging seen with lipid-rich adenomas. MRI, in addition to an inferior vena cava (IVC) contrast study, is useful if vascular invasion or tumour thrombosis is suspected.
When adrenal tumours cannot be characterised on conventional cross-sectional imaging, [ 18 F]fluorodeoxyglucose positron emission tomography (FDG-PET) with CT has a sensitivity of 100% and specificity of 88% in differentiating malignant from benign lesions. FDG-PET may also be useful to detect recurrent or metastatic disease not seen on CT.
Diagnosis and staging
Percutaneous biopsy is not recommended for the work-up of ACC due to the difficulty in differentiating primary malignant from benign adrenal disease, and the risk of seeding tumour cells along the biopsy track. The only absolute criterion for the diagnosis of ACC is presence of extensive invasion of local structures or metastases. The most widely used algorithm for the diagnosis of ACC was described by Weiss and is based on nine histological features, the presence of three or more indicating malignant potential. Ki67 immunohistochemistry may also help differentiate benign from malignant adrenocortical tumours.
The 2004 International Union Against Cancer TNM stages ACC according to size, presence of lymph node metastases, local invasion and distant metastases, and is based on the original staging system described by MacFarlane in 1958 and Sullivan et al. in 1978. The European Network for the Study of Adrenal Tumours (ENSAT) has recently proposed further refinements of this staging system, which is the basis for prognostic and treatment stratification in ACC, as well as enabling comparison of outcomes between treatment centres.
Treatment
Surgery
Open surgery is recommended when malignancy is strongly suspected on the basis of preoperative investigations. Laparoscopic surgery in expert hands has been advocated in some centres for large, potentially malignant adrenal tumours, provided there is no evidence of local invasion, but is generally not advised for ACC, due to the risk of recurrence and peritoneal carcinomatosis.
Surgery offers the only potential cure, and prognosis in patients with incompletely resected ACC remains poor, due to lack of response of ACC to systemic therapy. For tumour resection a transabdominal or, if necessary, thoraco-abdominal approach should enable good access to allow vascular control of the aorta, IVC and renal vessels. En bloc resection of the perinephric fat, regional lymph nodes and adjacent organs including kidney, pancreas, liver or spleen maybe required to achieve complete resection. Following surgery, close radiological and biochemical follow-up is undertaken as early identification and re-excision of recurrent disease may improve survival. Palliative surgery may also have a role, particularly in functional tumours.
Medical
Medical therapy with mitotane (1,1-dichloro-2-( o -chlorophenyl)-2-( p – chlorophenyl)ethane) is used in advanced ACC. Mitotane, originally used as an insecticide, is a lipophilic agent concentrated in the adrenal cortex,where it induces necrosis by mitochondrial degeneration. Tumour response occurs at a therapeutic range of 14–20 mg/L in up to one-third of patients, though gastrointestinal and neurological side-effects are common and adrenaline sufficiency may occur.
Adjuvant therapy with mitotane has been shown to improve recurrence-free survival in ACC and has recently been recommended for use in patients with ACC at high risk of recurrence, based on resection status, presence of vascular or capsular invasion, or Ki67 proliferative index. Adjuvant radiotherapy to the tumour bed may also reduce the risk of local recurrence in high-risk patients with involved surgical margins.
Mitotane potentiates the cytotoxic activity of some chemotherapeutic drugs so combinations can be used in advanced and metastatic disease. Overall, the results of cytotoxic chemotherapy for ACC are disappointing, with the best-reported (partial) response rate being 49%. The results of the FIRM-ACT trial to establish the optimum chemotherapy regime should shortly be available. Novel systemic therapies such as IGF receptor and tyrosine kinase inhibitors are currently under investigation.
Prognosis
The reported overall 5-year survival for ACC varies between 16% and 38% depending on the stage at diagnosis. Median survival following non-curative surgery or diagnosis of metastatic disease is less than 12 months.
Case study 2 (discussion)
The CT scan shows an ACC. Phaeochromocytoma or neuroblastoma may give a similar appearance but these diagnoses can be excluded on the basis of negative urinary or plasma metanephrines. As staging was negative open surgery is indicated. The patient should be warned of the possible need to resect adjacent organs; in this case the left kidney was excised en bloc with the adrenal tumour. Histopathology showed a 2.3-kg ACC. Tumour thrombus was present in the renal vein. Postoperative mitotane and chemotherapy were given in this case.
Phaeochromocytoma and paraganglioma
Case study 3
Consider the management of a 60-year-old hypertensive woman investigated for symptoms of palpitations. Twenty-four-hour electrocardiogram and echocardiography are unremarkable and she is commenced on beta-blockers. Subsequently overnight urinary fractionated metanephrines and normetanephrines are found to be markedly elevated. Anatomical and functional localisation studies show a right adrenal phaeochromocytoma (see Fig. 3.9 ).
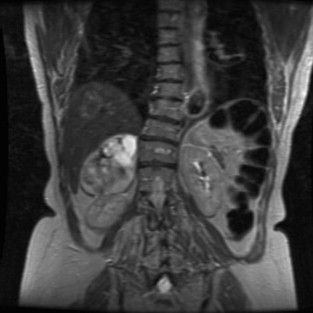
Incidence and aetiology
Phaeochromocytoma and extra-adrenal paraganglioma are tumours derived from catecholamine-producing chromaffin cells of neural crest origin arising in either the adrenal medulla (phaeochromocytoma) or extra-adrenal autonomic ganglia (paraganglioma). Phaeochromocytoma has an incidence of 3–8 per million per year and usually presents in middle age, though hereditary forms occur at a younger age. Phaeochromocytoma is very rare in children. Studies suggest 0.05% of all autopsies harbour an undiagnosed phaeochromocytoma and it is probable that this rare tumour is under-recognised in life. Traditionally termed the ‘10% tumour’ (10% bilateral, extra-adrenal, familial or malignant), this description has now been challenged by recent advances in genetics and diagnosis.
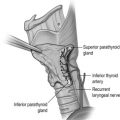
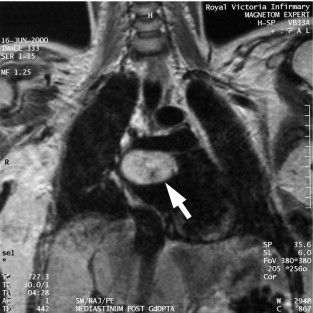
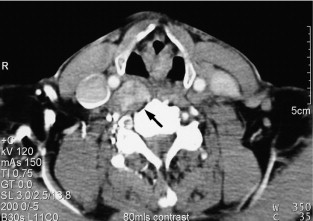
The majority of extra-adrenal sympathetic paragangliomas occur in the abdomen, either arising from sympathetic ganglia in the organ of Zuckerkandl, which is situated along the lower abdominal aorta and its bifurcation, or around the renal hilum ( Fig. 3.6 ). Less common sites for paraganglioma include the urinary bladder or the mediastinum, where they may even arise in the nerves supplying the myocardium (see Fig. 3.7 ). One-quarter of phaoechromocytomas managed in our unit are extra-adrenal, and a similar high proportion of extra-adrenal tumours are reported from other endocrine surgical centres, though this may reflect referral bias. In contrast to sympathetic paraganglioma, those arising from parasympathetic ganglia occur mostly in the head and neck (see Fig 3.8 ), and rarely produce catecholamines.
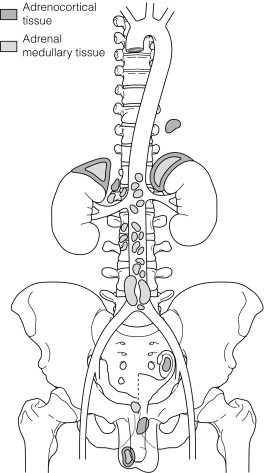
Although the majority of phaeocytochromas are sporadic, it is now thought that up to one-third of patients with phaeocytochromas carry germ-line mutations in predisposing genes, three of which are well known: RET (multiple endocrine neoplasia types 2A and 2B), VHL (von Hippel–Lindau syndrome) and NF1 (neurofibromatosis type 1). The remaining mutations occur in genes ( SDHB and C ) encoding subunits of the succinate dehydrogenase (SDH) enzyme responsible for the ‘paraganglioma–phaeocytochroma syndrome’.
The diagnosis of phaeochromocytoma may be the presenting episode of a hereditary syndrome; therefore, a careful medical and family history should be taken and examination performed to identify related features of a predisposing syndrome.
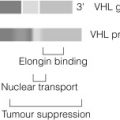
Phaeochromocytomas occur in approximately half of patients with multiple endocrine neoplasia type 2 (depending on the codon mutation), in association with medullary thyroid carcinoma (MTC) and hyperparathyroidism in multiple endocrine neoplasia type 2A (MEN2A) and multiple mucosal ganglioneuromas, megacolon and marfanoid habitus in MEN2B. Germ-line mutations in VHL , a tumour suppressor gene that regulates the accumulation of hypoxia-induced proteins and angiogenesis, lead to the rare VHL syndrome, characterised by central nervous system haemangioblastomas, renal cell cancer, cysts of kidney, testis and pancreas, and phaeochromocytoma (in up to a third). The clinical features of NF1 (also known as von Recklinghausen’s disease) include multiple neurofibromas, café-au-lait spots, skin-fold freckling and iris hamartomas. Phaeochromocytoma occurs in < 5% of NF1 patients.
Hereditary mutations in the SDH genes lead to failure of oxidative phosphorylation, the process by which adenosine triphosphate (ATP) is produced in mitochondria via the citric acid (Kreb’s) cycle. The SDH genes encode succinate dehydrogenase, a key component of aerobic glycolysis, ‘oxidative stress’ and accumulation of pro-oxidants leading to DNA damage. This is thought to underlie the pathogenesis of phaeochromocytoma and paragangliomas. Well-recognised genotype–phenotype correlations are seen with SDH mutations and, in contrast to RET , VHL and NF1 , patients with mutations in the SDH genes commonly present with paragangliomas. SDHB carriers characteristically present at a young age with solitary malignant abdominal paraganglioma, whereas SDHD carriers are more likely to present with multiple paragangliomas in the head and neck, abdomen and adrenal gland.
Patients carrying hereditary gene mutations predisposing to phaeochromocytoma are more likely to be young (< 50 years) or have multiple, bilateral and extra-adrenal tumours, so genetic screening should be undertaken in this group presenting with an apparently sporadic phaeochromocytoma. Genetic screening may not be cost-effective for all patients presenting with phaeochromocytoma, but should be undertaken in patients < 50 years where clinical features or family history suggest a hereditary syndrome.
Clinical presentation
The clinical symptoms and signs of phaeochromocytomas are numerous and are due to the paroxysmal excessive secretion of catecholamines. The classic symptoms are headaches, palpitations and sweating, but pallor, nausea, weight loss, tiredness and anxiety commonly occur. The characteristic feature of all presenting symptoms is that they are intermittent or paroxysmal in nature, which makes diagnosis difficult as the patient may be perfectly well between symptomatic episodes. Postural changes, exercise and anxiety often provoke symptoms. Hypertension is the commonest sign, along with diabetes mellitus. ‘Phaeochromocytoma crisis’ due to massive secretion of catecholamines into the circulation may present with sudden death, arrhythmia, heart failure, multi-organ failure or cerebrovascular accident. Anaesthesia, trauma, biopsy, haemorrhage or tumour manipulation during surgery may precipitate these crises. Phaeochromocytoma may also be found incidentally on cross-sectional imaging – approximately 5% of adrenal incidentalomas are phaeochromocytomas. Typically the diagnosis of phaeochromocytoma is delayed by several years as the symptoms overlap many common endocrine, cardiovascular or neurological disorders. The key to diagnosis is staying alert to the possibility of the disease.
Biochemical diagnosis
Phaeochromocytomas are rare tumours yet the consequences of missing the diagnosis may be catastrophic, so any screening test must be highly sensitive to minimise false-negative results. Plasma catecholamines are not a good diagnostic test as levels vary episodically in phaeochromocytoma, and may be elevated due to stress or medication in normal individuals. Measuring 24-hour or overnight urinary catecholamines improves diagnostic accuracy.
Plasma free or urinary fractionated metanephrines are now established as the best diagnostic test for phaeochromocytoma.
Metanephrines, produced continuously by the action of catechol- O -methyltransferase on catecholamines within tumour cells, leak into the circulation where their concentrations have been shown to accurately reflect tumour mass. When measuring plasma metanephrines, blood samples should be taken after 20 minutes of supine rest in order to avoid false-positive results. The sensitivity and specificity of plasma free metanephrines for the diagnosis of phaeochromocytoma are 96–100% and 80–100%, respectively. Twenty-four-hour or overnight urinary fractionated metanephrines have a similar diagnostic accuracy to plasma free metanephrines.