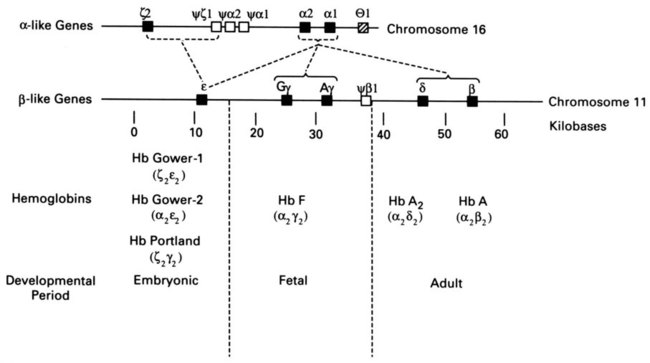
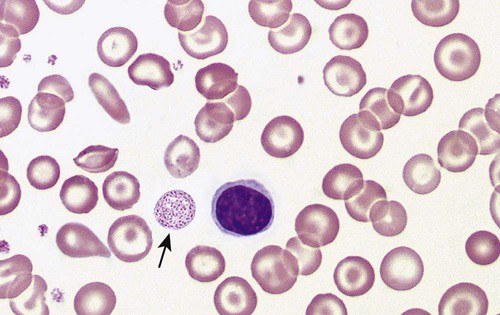
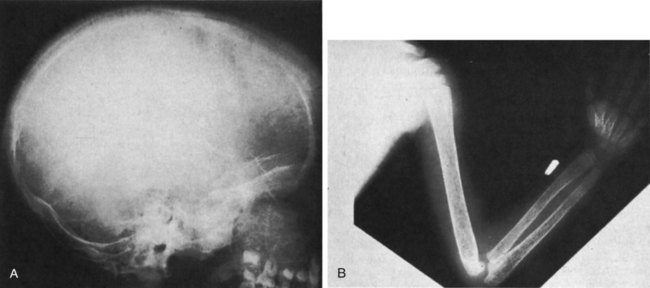
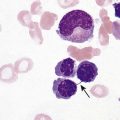
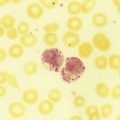
After completion of this chapter, the reader will be able to: 1. Describe the hemoglobin (Hb) defect found in thalassemias. 2. Name the chromosomes that contain the α-globin gene and the β-globin gene clusters and the globin chains produced by each. 3. Discuss the geographic distribution of thalassemia and explain its association with malaria. 4. Explain the pathophysiologic effects caused by the imbalance of globin chain synthesis in thalassemia. 5. List the clinically defined thalassemic syndromes associated with genetic defects of the β-globin gene cluster and describe the clinical expression of each heterozygous and homozygous form. 6. Describe the type of genetic mutations that result in β-thalassemias, including hereditary persistence of fetal hemoglobin (HPFH). 7. Recognize the pattern of laboratory findings in heterozygous and homozygous β-thalassemias, including HPFH. 8. Describe the treatment of homozygous β-thalassemias, the risks involved, and the reason it is necessary to monitor iron levels. 9. Correlate the clinical syndromes of α-thalassemia with the number of α genes present. 10. Recognize the laboratory findings associated with various α-thalassemia syndromes. 11. Justify genetic counseling for those of Southeast Asian descent who have the α0-thalassemia haplotype. 12. Describe the clinical syndromes of thalassemia associated with structural hemoglobin variants. 13. Specify the tests that may be helpful in screening for β-thalassemia minor. 14. Correlate the results of hemoglobin electrophoresis or high-performance liquid chromatography and quantitation of Hb F and Hb A2 with the various thalassemia syndromes. 15. Differentiate β-thalassemia minor from iron deficiency anemia. 1. Why was the family history so important in this case, and what diagnosis did it suggest? 2. What laboratory values helped confirm the diagnosis? 3. From what other disorders should this anemia be differentiated? What laboratory tests would be helpful? Why is differentiation important? 4. If this individual was planning to have children, what genetic counseling should be done? The thalassemias are a diverse group of inherited disorders caused by genetic alterations that reduce or preclude the synthesis of one or more of the globin chains of the hemoglobin (Hb) tetramer. Since thalassemia was first described in 1925 by Cooley and Lee,1 investigators have worked to define and better understand this fascinating but often confusing condition. Cooley and Lee1 described four children with anemia, splenomegaly, mild hepatomegaly, and mongoloid facies. These characteristics would later become typical findings in young children with untreated β-thalassemia major, often referred to as Cooley’s anemia. Seven years later, Whipple and Bradford published a paper outlining the detailed autopsy studies of children who died of this disorder.2 Because of the high incidence of patients of Mediterranean descent with this disorder, Whipple called the disease Thalassic (Greek for “great sea”) anemia, which was subsequently changed to thalassemia.2 Several investigators in the 1940s demonstrated the genetic basis for this anemia and were able to show that in patients who were homozygous for this condition (thalassemia major) the disease had a severe course. The heterozygotes, however, not only were carriers, but also had a milder anemia (thalassemia minor). In the 1950s, thalassemias resulting from defects in the α chain were described.2 Together the thalassemias are the most common single-gene disorder in humans. The clinical outcomes associated with the many genetic mutations are extremely wide ranging, with certain defects causing no anemia and other defects leading to fetal death. Thalassemia results from a reduction in the rate of synthesis of one or more of the globin chains. This decreased globin production leads to imbalanced globin chain synthesis, defective hemoglobin production, and damage to the red blood cells (RBCs) or their precursors by the excess accumulation of the globin chain that is not affected.3 Usually, the synthesis of either the α or the β chain of Hb A (α2β2) is impaired. Thalassemias are named according to the chain with reduced or absent synthesis. Although approximately 7% of the world’s population has a thalassemia or hemoglobinopathy, the distribution of thalassemia is concentrated in the “thalassemia belt” that extends from the Mediterranean east through the Middle East and India to Southeast Asia and south to Northern Africa (see Figure 26-3).4,5 The carrier frequency of β-thalassemia depends on the region, with Sardinia, Cyprus, and Greece having the highest frequency in Europe (6% to 19%) and India, Thailand, and Indonesia having the highest frequency in Asia and Southeast Asia (0.3% to 15%).5 The carrier frequency of α-thalassemia varies considerably. In Europe, Cyprus has the highest carrier frequency at 14%. The carrier frequency reaches 50% to 60% in Eastern Saudi Arabia and parts of Asia and Africa, and may be as high as 75% to 80% in certain groups in Nepal, India, Thailand, and Papua New Guinea.5 The geographic location of the thalassemia belt coincides with areas in which malaria is prevalent (see Figure 26-3). Thalassemia minor (heterozygous thalassemia) appears to impart resistance to malaria. This allowed the selective advantage that established thalassemia in high frequency in areas in which malaria is endemic.6,7 Several case-control studies evaluated the incidence of thalassemia in subjects with severe malaria compared with a control population and consistently found a lower incidence of thalassemia in the population with malaria than in the population without malaria. One study demonstrated that the risk of death from malaria was 40% lower in patients with αα/– α thalassemia and more than 60% lower in those with – α/– α thalassemia.7 The mechanism of this resistance is still not fully elucidated; however, two major theories have been put forward: defective growth of the parasite in the affected cell and increased phagocytosis of the infected cell.8,9 Although the exact mechanism is not known, the geographic distribution and the case-control studies corroborate the protective nature of the thalassemias in promoting resistance to malaria. The normal hemoglobin molecule is a tetramer (double dimer) of two α-like chains (α or ζ) with two β-like chains (β, γ, δ, or ε). Combinations of these chains produce six normal hemoglobins. Three are embryonic hemoglobins: Hb Gower-1 (ζ2ε2), Hb Gower-2 (α2ε2), and Hb Portland (ζ2γ2). The others are fetal hemoglobin (Hb F, α2γ2) and two adult hemoglobins (Hb A, α2β2, and Hb A2, α2δ2). The α-like globin chain genes are located on chromosome 16, whereas the β-like globin gene cluster is on chromosome 11 (Figure 27-1). The α-like globin gene cluster contains the functional embryonic globin gene (ζ) and two functional adult genes (α1 and α2). The β-like globin gene cluster contains five functional genes, ε, Gγ, Aγ, δ, and β.3,10 These genes are positioned in the order that corresponds with their developmental stage of expression. During weeks 6 to 11 of gestation, the ζ and ε genes are expressed, generating Hb Gower-1 (ζ2ε2). Expression of the α genes begins close to the eleventh week, and the ζ gene is gradually switched off so that Hb Gower-2 (α2ε2) is produced. Around the eleventh week, the ε gene is switched off, and γ chain synthesis begins. The γ chains combine either with the remaining ζ chains to make Hb Portland (ζ2γ2) or with the α chains to make Hb F (α2γ2), the predominant hemoglobin of fetal life. Shortly before birth, γ chain synthesis begins to decrease, whereas β and δ chain synthesis increases, so that by 6 months of age and through adult life, Hb A (α2β2) is the predominant hemoglobin, with a low percentage of Hb A2 (α2δ2) (Table 27-1; see Figure 10-6).3,11 TABLE 27-1 Reference Ranges for Normal Hemoglobins (Hbs) in Adults The γ genes Gγ and Aγ code for two globin chains that differ by a single amino acid (glutamic acid or alanine). Both of these globin chains are expressed in fetal hemoglobin, and it appears that there is no functional difference between them. Similarly, the α gene loci are duplicated on each chromosome 16 and are denoted α1 and α2. Either of these genes can contribute to the two α-globin chains in the hemoglobin tetramer, and no functional difference seems to exist between the two. Interspersed between the functional genes on these chromosomes are four functionless genelike loci or pseudogenes that are designated by the prefixed symbol ψ. The purpose of these pseudogenes is unknown.10 The organization of these genes on chromosomes 16 and 11 is shown in Figure 27-1. The thalassemias are divided into β-thalassemias, which include all the disorders of reduced globin chains arising from the β-globin gene cluster on chromosome 11, and α-thalassemias, which involve the α1 and α2 loci on chromosome 16. Various defects of deletion (absence of a portion of a gene) and nondeletion can cause each of these disorders. Even defects that seem the same clinically are often heterogeneous at the genetic level.10,12 α-Thalassemia, usually caused by gene deletion, involves the α gene complex (α1 or α2 genes) on chromosome 16. The designation α+ is used to indicate a deletion of either the α1 or the α2 gene in the α gene complex (also called the – α haplotype), which results in a decreased production of α chains from that chromosome. The designation α0 is used for a deletion of both the α1 and α2 genes in the α gene complex (also called the – –haplotype), which results in no production of α chains from that chromosome.10,12 In cases in which there is a mutation in the α gene rather than a deletion, α-thalassemia can still result. The designation αT is used for the conditions that result from these mutations.10 The major gene designations in thalassemia are summarized in Table 27-2. TABLE 27-2 Genetic Designations in Thalassemia The clinical sequelae of thalassemia stem not only from a decreased production of a particular globin chain but also, more importantly, from an imbalance of all of globin synthesis.10,13 For α-thalassemia and β-thalassemia, the resultant decrease in hemoglobin produces microcytic, hypochromic RBCs. The unequal production of either the α- or β-globin chains leads to decreased RBC survival that is a significant contributor to the anemia. Importantly, the mechanism and the degree of shortened RBC survival are different for the α-thalassemias and β-thalassemias. In the β-thalassemias, the unpaired, excess α chains precipitate in the developing RBCs and damage their surfaces. These damaged RBCs are subsequently destroyed by the macrophages in the bone marrow or in the circulation. The premature death of RBCs in the bone marrow leads to ineffective erythropoiesis. In this situation, the bone marrow attempts to produce RBCs, but is not able to release viable cells into the circulation. Some of these cells leave the bone marrow only to be destroyed in the spleen through extravascular hemolysis. Therefore in β-thalassemia the anemia is multifactorial and results from ineffective production and increased destruction. Typically, individuals with β-thalassemia are asymptomatic during fetal life and through 4 to 6 months of age because Hb F (α2γ2) is the predominant circulating hemoglobin. They become symptomatic only after the γ to β switch.11 In α-thalassemia, accumulation of non-α chains has different consequences. Because α chains are shared by fetal and adult hemoglobins, decreased α chain production results in excess γ chains in the fetus and excess β chains from 6 months of age through adult life. In the fetus, the excess γ chains accumulate even if only one α gene is deleted or nonfunctional. The amount of γ chain accumulation is proportional to the number of deleted or nonfunctional α genes. These excess γ chains form γ4 tetramers of hemoglobin, called Hb Bart. Because these tetramers are soluble, they do not precipitate to any significant degree in the developing RBCs in the bone marrow and do not cause severe ineffective erythropoiesis. As the mature RBCs age in the circulation, however, these tetramers precipitate and form inclusion bodies. The spleen removes the cells with these inclusions, which results in extravascular hemolysis. The RBCs are microcytic and hypochromic in α-thalassemia owing to the decrease in hemoglobin synthesis.10,14 The anemia is a result not only of ineffective production, but also of shortened RBC survival. Hb Bart (γ4) has a high affinity for oxygen and cannot adequately deliver oxygen to tissues. The fetus cannot survive with only Hb Bart (found when all four α genes are absent); hydrops fetalis develops, and the fetus ultimately dies in utero or shortly after birth.14 In the adult, β chain tetramers form Hb H. This is discussed in the α-thalassemia section later in the chapter. Molecular cloning, sequencing of deoxyribonucleic acid (DNA), and functional analysis of cloned genes have revealed many different types of defects at the molecular level.10,15 Types of genetic defects that cause a decrease in or lack of production of a particular globin chain and thalassemia include single nucleotide (or point) mutations, small insertions or deletions, or large deletions. The mechanisms10 by which these mutations interfere with globin chain production include: • Reduced or absent transcription of messenger ribonucleic acid (mRNA) due to mutations in the promoter region or initiation codon of a globin gene • mRNA processing errors due to mutations that add or remove splice sites resulting in no globin chain or altered globin chain production, as well as mutations in polyadenylation sites that decrease mRNA stability • Translation errors due to mutations that change the codon reading frame (frameshift mutations), substitute an incorrect amino acid codon (missense mutations), add a stop codon causing premature chain termination (nonsense mutations), or remove a stop codon, which results in an elongated and unstable mRNA that produces a dysfunctional globin chain • Deletion of one or more globin genes resulting in the lack of production of the corresponding globin chains10,15 Modern advances in genetic technology have revealed the heterogeneity of the β-globin gene cluster DNA defects that lead to the clinical syndrome of β-thalassemia.15 More than 200 genetic abnormalities have been discovered in the β-globin gene cluster, including mutations in each of the β, δ, and γ genes or in combined sections of the β-globin gene cluster.10,16 Although numerous different mutations exist, a small subset of mutations accounts for the majority of the mutant alleles within a single ethnic group or geographic area in which β-thalassemia is found.10 Because multiple mutations are present in each population, most individuals with severe β-thalassemia are genetic compound heterozygotes for two different thalassemia mutations. The frequency of specific β-thalassemia mutations has been compiled for numerous ethnic groups, and this information is particularly relevant for prenatal diagnosis of β-thalassemia.5 Historically, β-thalassemia has been divided into three clinical syndromes: β-thalassemia minor (heterozygous state), a mild microcytic, hypochromic hemolytic anemia; β-thalassemia major (homozygous state), a severe anemia leading to dependence on transfusions; and β-thalassemia intermedia, with symptoms of severity between the other two.10,13,15 A fourth syndrome (designated as silent carrier state) is now recognized in patients with genetic changes in one of the two β genes that result in no hematologic abnormalities (Table 27-3).15 The clinical manifestations of the genetic changes depend on whether one or both of the β genes are affected and the extent to which the gene or genes are expressed. Some mutations result in the absence of production of the β chain, and these mutations are designated as β0 genes. Other mutations lead to production of the β chains, but at a significantly reduced rate, and these are designated as β+ genes.10 The range of β chain production in these β+ gene mutations varies from 10% to 50% of normal β chain synthesis. In addition, patients who are silent carriers have a normal hemoglobin level and mean cell volume (MCV), whereas other patients with defects that only minimally reduce β-globin production have minimal changes in their RBC profiles. TABLE 27-3 Some Clinical Syndromes of β-Thalassemias ↑, Increased; ↓, decreased; 0, absent; Hb, hemoglobin; N, normal; Sl, slight; V, variable. The designation βsilent includes the various heterogeneous β gene mutations that produce only a small decrease in production of the β chains. The silent carrier state (βsilent/β) results in nearly normal α-β chain ratios and no hematologic abnormalities.15,17 It was recognized through a study of families in which the affected children had a more severe β-thalassemia syndrome than a parent with typical β-thalassemia minor.13,15,17 The parents had normal levels of Hb A2 and a slight microcytosis. Several patients who are homozygous for a silent thalassemia gene (βsilent/βsilent) have been described. They have a moderate microcytic, hypochromic anemia with a hemoglobin level in the range of 7 to 9 g/dL. The Hb F values range from 2% to 19%, and the Hb A2 level is elevated to the range normally seen in individuals with thalassemia minor.18 β-Thalassemia minor (also called β-thalassemia trait) results when one of the two genes that produce the β-globin chains is defective (heterozygous state). It usually presents as a mild, asymptomatic anemia.10,12,15 One β gene is affected by a mutation that decreases or abolishes its function, whereas the other β gene is normal. The hemoglobin ranges from 10 to 13 g/dL, and the RBC count is normal or slightly elevated. The RBCs are microcytic and hypochromic, with some degree of poikilocytosis, including target cells and elliptocytes. Basophilic stippling may be seen in the RBCs on a Wright-stained peripheral blood film (Figure 27-2). The bone marrow shows mild to moderate erythroid hyperplasia, with slight ineffective erythropoiesis. Hepatomegaly and splenomegaly are seen in a few patients. In the most common β-thalassemia minor syndromes (β0/β and β+/β) the Hb A2 level is characteristically elevated and can vary from 3.5% to 8.0%. The Hb F level usually ranges from 1% to 5%. Less common variants of β-thalassemia minor exist, such as δβ0/β and δβLepore/β. Other rare variants have atypical features, such as one variant (Dutch β0-thalassemia minor) that shows the expected elevation in Hb A2 level but an Hb F level in the 5% to 20% range,19 and another variant found in a Sardinian family in which the Hb A2 level is normal.20 β-Thalassemia major is characterized by a severe anemia first detected in early childhood as the γ to β switch takes place. Normally at birth, the predominant hemoglobin is Hb F, but over the next 6 months Hb A levels increase and Hb F levels decrease (see Figure 10-6).10–12 Severe β-thalassemia is usually diagnosed when the patient is between 6 months and 2 years of age, because the Hb A level does not increase as expected. The natural history of this condition (in an untreated or inadequately treated patient) is characterized by increasing hepatosplenomegaly, jaundice, and marked bone changes due to an expanded marrow cavity caused by extreme erythroid hyperplasia. Skull radiographs may demonstrate a typical “hair on end” appearance. Radiographs of the long bones may exhibit a lacy appearance from marrow expansion, which causes a thin bony cortex that predisposes to pathologic fracture (Figure 27-3). A typical facies results, with prominence of the forehead (also known as frontal bossing), cheekbones, and upper jaw. Physical growth and development are delayed.10,12 In untreated β-thalassemia major, the hemoglobin level can fall to 2 or 3 g/dL.10 On evaluation of the peripheral blood film, marked microcytosis and hypochromia are seen, with a wide variety of RBC sizes (anisocytosis) and shapes (poikilocytosis), including target cells, teardrop cells, and elliptocytes. RBC fragments and microspherocytes are present as a direct consequence of unbalanced globin chain synthesis. Basophilic stippling and many nucleated RBCs are also found (Figure 27-4
Thalassemias
Case Study
Patient Results
Reference Range
RBCs (× 1012/L)
5.74
4.60-6.00
Hb (g/dL)
10.2
14.0-18.0
Hct (%)
35
40-54
MCV (fL)
61.0
80-100
MCH (pg)
17.8
26-32
MCHC (g/dL)
29.1
32-36
Definitions and History
Epidemiology
Genetic Control of Globin Synthesis
Hb A (α2β2)
95-100%
Hb A2 (α2δ2)
0-3.5%
Hb F (α2γ2)
0-2%
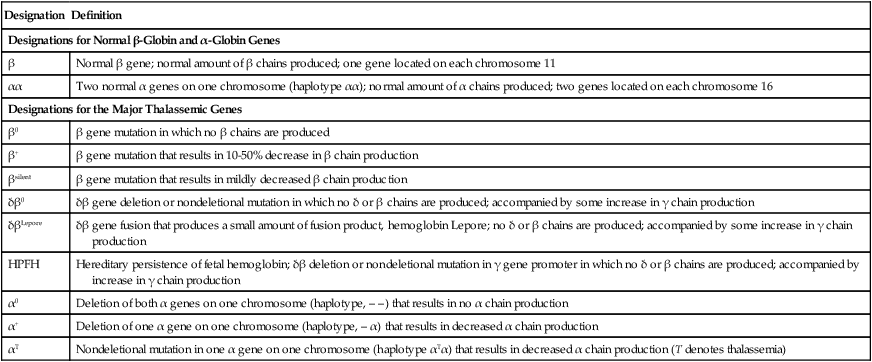
Categories of Thalassemia
Designation
Definition
Designations for Normal β-Globin and α-Globin Genes
β
Normal β gene; normal amount of β chains produced; one gene located on each chromosome 11
αα
Two normal α genes on one chromosome (haplotype αα); normal amount of α chains produced; two genes located on each chromosome 16
Designations for the Major Thalassemic Genes
β0
β gene mutation in which no β chains are produced
β+
β gene mutation that results in 10-50% decrease in β chain production
βsilent
β gene mutation that results in mildly decreased β chain production
δβ0
δβ gene deletion or nondeletional mutation in which no δ or β chains are produced; accompanied by some increase in γ chain production
δβLepore
δβ gene fusion that produces a small amount of fusion product, hemoglobin Lepore; no δ or β chains are produced; accompanied by some increase in γ chain production
HPFH
Hereditary persistence of fetal hemoglobin; δβ deletion or nondeletional mutation in γ gene promoter in which no δ or β chains are produced; accompanied by increase in γ chain production
α0
Deletion of both α genes on one chromosome (haplotype, – –) that results in no α chain production
α+
Deletion of one α gene on one chromosome (haplotype, – α) that results in decreased α chain production
αT
Nondeletional mutation in one α gene on one chromosome (haplotype αTα) that results in decreased α chain production (T denotes thalassemia)
Pathophysiology
Genetic Defects Causing Thalassemia
β-Globin Gene Cluster Thalassemias
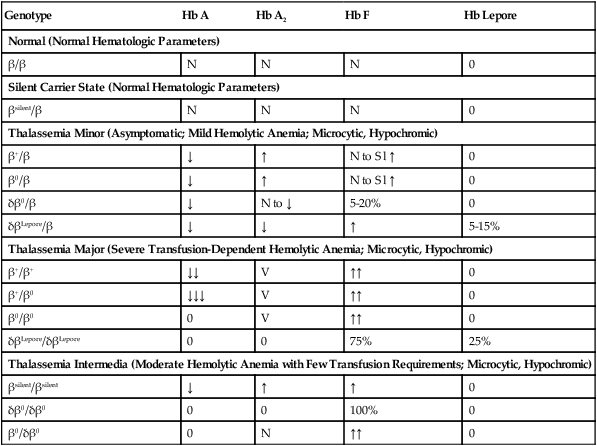
Clinical Syndromes of β-Thalassemia
Genotype
Hb A
Hb A2
Hb F
Hb Lepore
Normal (Normal Hematologic Parameters)
β/β
N
N
N
0
Silent Carrier State (Normal Hematologic Parameters)
βsilent/β
N
N
N
0
Thalassemia Minor (Asymptomatic; Mild Hemolytic Anemia; Microcytic, Hypochromic)
β+/β
↓
↑
N to Sl ↑
0
β0/β
↓
↑
N to Sl ↑
0
δβ0/β
↓
N to ↓
5-20%
0
δβLepore/β
↓
↓
↑
5-15%
Thalassemia Major (Severe Transfusion-Dependent Hemolytic Anemia; Microcytic, Hypochromic)
β+/β+
↓↓
V
↑↑
0
β+/β0
↓↓↓
V
↑↑
0
β0/β0
0
V
↑↑
0
δβLepore/δβLepore
0
0
75%
25%
Thalassemia Intermedia (Moderate Hemolytic Anemia with Few Transfusion Requirements; Microcytic, Hypochromic)
βsilent/βsilent
↓
↑
↑
0
δβ0/δβ0
0
0
100%
0
β0/δβ0
0
N
↑↑
0
Silent Carrier State of β-Thalassemia
β-Thalassemia Minor (β-Thalassemia Trait)
β-Thalassemia Major
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Thalassemias