FIGURE 4.1 Human telomere structure. A: Human telomeres form telomere loop (T loop) and displacement loop (D loop) secondary structures. Long stretches of telomeric repeats create a loop-back structure (T loop), completed by the invasion of the single GT-rich 3’ overhang into the double-stranded DNA molecule (D loop), thus protecting the chromosome terminus. B: In human cells, double-stranded telomeric repeats are bound directly by two proteins, TRF1 (TTAGG repeat binding factor 1) and TRF2. Cell culture studies have suggested that the main function of TRF1 is to regulate telomere length, whereas TRF2 functions to protect telomeres from activating nonhomologous end-joining (NHEJ) and other DNA repair or DNA damage response pathways. TRF2 also interacts with the human Rap1 protein (hRap). Biochemical studies also suggest that the formation of the T loop depends on TRF2. Another protein, POT1 (protection of telomere 1), has been shown to bind to the single-stranded human telomeric 3’ overhang. Two shelterin proteins, TIN2 and TPP1, connect POT1 to TRF1 and TRF2. POT1 has been proposed to interact with TRF1 complexes to regulate telomere length. Thus, there is significant interplay between telomeric binding proteins and the formation of the secondary/tertiary structures that protect the ends of chromosomes.
Many proteins involved in DNA DSB repair, including nonhomologous end-joining and homologous recombination processes, have been found to be physically associated with the telomeres.7–10 These findings have fueled speculation that DSB repair proteins provide a protective role at the telomere; for example, by sequestering the telomere end from the DNA damage surveillance/repair machinery. Experimental support for this hypothesis has emerged from the mouse, in which germ line inactivation of various repair proteins (e.g., Ku and DNA-PK) results in reduced telomere length or loss of capping function, or both, leading to increased end-to-end fusions.11 Correspondingly, in cultured human cells, experimental disruption of telomere-binding proteins results in the unraveling of higher-order nucleoprotein structure and telomere localization of DNA DSB surveillance/repair proteins (e.g., 53BP1, gamma-H2AX, Rad17, ATM, and Mre11), establishing that dysfunctional telomeres can indeed serve as substrates for the classic DNA repair machinery.12 Recently, elegant in vitro and mouse genetic experiments have shown that subunits of the shelterin complex actively repress the ATM and ATR DNA damage signaling pathways.7,13
A further understanding of the molecular mechanisms governing the repression versus activation of the DNA DSB surveillance/repair apparatus at the telomere could lead to the development of novel cancer therapeutic options. For example, the design of agents that can uncap telomeres while preserving the DNA damage checkpoint response yet neutralize the actual DNA damage repair process would be ideal because they would produce unrepaired DSBs and elicit cell-cycle arrest or apoptosis responses. Lastly, in the near future, agents designed to uncap the telomeres could be used in combination with conventional chemotherapeutic agents that create DSB for cancer treatment, thereby simultaneously targeting these intertwined pathways.
Telomerase
Conventional DNA polymerases operating in the S phase of the cell cycle require an RNA primer for reverse-strand synthesis, resulting in incomplete DNA replication of telomeres during each cell division. The solution to this “end-replication problem” is the telomere-synthesizing telomerase enzyme, a specialized ribonucleoprotein complex with reverse transcriptase activity. The functional telomerase holoenzyme is a large multisubunit complex that includes an essential telomerase RNA (hTERC) component serving as a template for the addition of telomere repeats and a telomerase reverse transcriptase (hTERT) catalytic subunit.14 In some normal human somatic cells, telomerase levels are insufficient to maintain telomere length, resulting in progressive attrition with each cell division. This forms the basis for the theory that the metered loss of telomeres can serve as a cellular mitotic clock that ultimately limits the number of cell divisions and cellular lifespan. In support of this view, shortening of telomere length with aging can be demonstrated in human peripheral blood cells,15–17 and the rate of shortening can be associated with conditions of increased hematopoietic stem cell turnover (e.g., in paroxysmal nocturnal hemoglobinuria).18
Many normal somatic human cells and differentiated tissues express readily detectable levels of the hTERC component. In contrast, hTERT expression and activity are more restricted because of stringent regulation on the levels of transcriptional initiation, alternative RNA processing, posttranslational modification, and subcellular localization. With the identification of an increasing number of TERT-associated proteins, it is likely that additional regulatory mechanisms will surface, such as those governing the accessibility of the telomerase holoenzyme onto the telomere end.19 Here again, a more complete elucidation of these regulatory mechanisms may provide additional therapeutic strategies that can preferentially target telomerase-mediated telomere maintenance in cancer cells. Indeed, the development of such selective strategies may become paramount and more challenging as recent studies have revealed low telomerase levels in cycling somatic human cells that were previously thought to have no telomerase activity.20 Eradication of residual telomerase function in these primary cells alters the maintenance of the 3’ single-strand telomeric overhang without changing the rate of overall telomere shortening, resulting in diminished proliferation rates and overall reduction in proliferative capacity. These studies support an additional protective function of telomerase at the telomeres21 and raise concerns that generalized antitelomerase therapy could lead to the immediate uncapping of telomeres in normal cells, thus limiting the use of antitelomerase therapy in cancer patients.
Lastly, in addition to forming the telomerase holoenzyme complex with TERC, TERT was recently shown to be able to interact with the RNA component of mitochondrial RNA processing endoribonuclease (RMRP). This distinct TERT/RMRP ribonucleoprotein complex has RNA-dependent RNA polymerase activity and produces double-stranded RNAs that can be processed into small interfering RNAs.22 Also, there is compelling experimental evidence that TERT can interact and engage the Wnt signaling pathway.23,24 These results suggest that TERT contributes to cell physiology independently of its ability to elongate telomeres, a fact that further complicates efforts to specifically target telomerase enzymatic activity as an anticancer therapy.
SENESCENCE
Primary human cells, even when cultured under optimal conditions, will eventually encounter a cell division barrier, termed cellular senescence, triggered by critically shortened telomeres. Senescence is a specific cell biologic phenotype composed of a permanent and durable growth arrest, alterations in cellular morphology, expression of characteristic markers of senescence such as senescence-associated (SA) β-galactosidase activity, and alterations of chromatin structure to a growth-repressive state.25 Induction of senescence is intimately associated with p16INK4a and p53 activation, and when induced in vitro as a result of telomere dysfunction, this barrier is termed the Hayflick limit (M1) in honor of the discoverer of senescence.26 Because loss of p16INK4a-RB and/or p53 pathway function in primary human cells permits additional cell divisions beyond the Hayflick limit, these pathways appear to be involved in the activation of this senescence program brought about by the “shortened telomere” signal.
Beyond the connection with telomeres, cellular senescence appears to be a general anticancer mechanism, induced by a variety of oncogenic stresses. In addition to telomere erosion or structural uncapping (see later discussion), senescence is also induced by forms of DNA damage, oxidative stress, suboptimal growth conditions, and activation of certain oncogenes (reviewed in refs. 8 and 13). Senescence requires activation of the Rb and/or p53 protein; and expression of their regulators such as p16INK4a, p21CIP, and ARF (Fig. 4.2).27–30 An important form of senescence is induced by p53, which has several antiproliferative activities including stimulation of the expression of p21CIP, a cyclin-dependent kinase inhibitor. These inhibit progression through the cell cycle by inhibiting cyclin-dependent kinases that phosphorylate and thereby inactivate Rb and related proteins p107 and p130.31 The activation of p53 is predominantly effected by specific posttranslational modifications and its stabilization, which are prompted by the same stimuli that induce its expression, including telomere dysfunction, DNA damage, and oncogene activation (reviewed in refs. 18 through 20), as well as inappropriate cell cycle entry.32,33 A major sensor of oncogene activation and inappropriate cell cycle entry is ARF (also designated p14ARF in the human or p19ARF in the mouse), which binds to and blocks MDM2-mediated degradation of p53.33–36
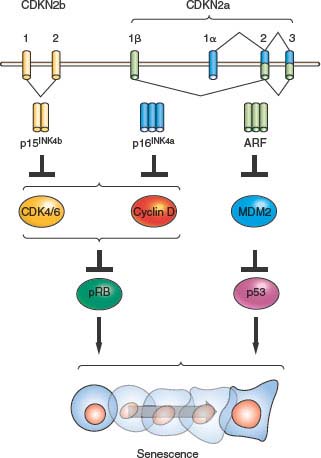
FIGURE 4.2 The INK4a/ARF/INK4b locus (also called CDKN2a and CDKN2b) and downstream pathways. The locus contains three open reading frames encoding the ARF, p15INK4b, and p16INK4a tumor suppressor proteins. p16INK4a and p15INK4b inhibit the activity of the proliferative kinases CDK4/6, which phosphorylate RB and related proteins p107 and p130. Therefore, INK4 expression induces RB-family hypophosphorylation, which in turn represses E2F-regulated transcription and cell-cycle arrest. ARF inhibits the MDM2-mediated degradation of p53; and p53 stabilization in turn induces a number of targets including many proteins involved in cell-cycle arrest or apoptosis. The entire locus spans a mere 35 kb in the human genome, and inactivation of all three genes by a single genetic deletion is common in many human and murine cancers.
Another prominent molecular correlate of senescence is up-regulation of the cyclin-dependent kinase inhibitor, p16INK4a, which increases markedly in senescent cells on passage in culture or advancing age in tissues.37 Correspondingly, ectopic expression of p16INK4a is sufficient to induce senescence in some cell types,38 and senescence can be delayed or prevented in some cell types by p16INK4a silencing or neutralization by antisense or siRNA.39–43 The regulation of p16INK4a is not as well understood as that of p53, although it appears to be induced by several stimuli, including oncogene activation and growth in culture.37 Activation of p53 (and hence p21CIP1) and/or accumulating levels of p16INK4a is able to produce Rb-family member protein hypophosphorylation and activation, which leads to repression of cell-cycle progression,27,30 enabling initiation of the senescence process. Recent data have suggested that Rb may be of particular importance in the promotion of senescence compared with its related family members p107 and p130, likely explaining the frequent inactivation of Rb in human cancers relative to the other Rb-family members.44
Senescence as a Cancer Prevention Mechanism
Several lines of evidence have suggested an important role for senescence in the prevention of cancer in vivo. It is important to note that the field has been limited by the lack of robust in vivo biomarkers of senescence. Although (SA)-β-galactosidase and p16INK4a expression have been used as markers of in vivo senescence, both have certain limitations and neither can be considered unequivocal proof of senescent state in vivo. These technical shortcomings notwithstanding, a growth arrest important for the prevention of tumorigenesis with characteristic features of senescence (p16INK4a expression and (SA)-β-galactosidase expression) has been noted in several murine and human in vivo tumor systems, and we believe the data suggest bona fide senescence occurs in the intact organism.
The lines of evidence for senescence as a tumor suppressor mechanism are quite strong. First, the aforementioned minimal residual disease data showing frequent oncogenic translocations and other mutagenic events demonstrate a constant need for tumor suppression, even in young animals. Additionally, several of the initially described “tumor suppressor” proteins that are mutated in familial cancer syndromes (e.g., p16INK4a, p53, Rb) are intimately involved in the induction of senescence. Mice lacking p16INK4a or p53 are prone to spontaneous cancers,45–47 and mice with severe compromise of the senescence pathway due to combined p16INK4a and p53 inactivation die of cancer, often harboring multiple synchronous primary tumors, with a median age of 8 weeks (compared with a normal murine lifespan of more than 100 weeks).19 Importantly, mice and humans with impaired p16INK4a and/or p53 function develop with only modest phenotypic alterations other than an age-dependent increase in cancer and an increased susceptibility to cancer following carcinogen exposure. Several groups have demonstrated a senescencelike growth arrest in murine and human tissues in association with somatic oncogenic events.48–54 For example, some forms of benign cutaneous nevi appear to be collections of senescent melanocytes, suggesting that these common benign neoplasms would transform into melanomas were it not for the successful interdiction of this process by the senescence tumor suppressor mechanism. In aggregate, these data establish that senescence-promoting molecules are critical to the prevention of mammalian cancer with advancing age.
Lastly, although the concept of “tumor maintenance” is becoming well established with regard to oncogene-activation,55 a similarly important role for the persistent inactivation of the senescence checkpoint has been established in cancer. Several groups have established in genetically engineered murine models, for example, that persistent p53 inactivation is required for tumor maintenance.51,52,56 Therefore, just as the finding that tumors in murine models require persistent RAS activation presaged the successful development of therapeutic compounds such as epidermal growth factor receptor inhibitors that target pathways required for tumor maintenance in vivo, similarly, these data support the notion that reactivation of senescence-promoting mechanisms such as p53 could be of therapeutic benefit in some cancers. In fact, it is likely that certain chemotherapeutics exert their therapeutic effects through the promotion of senescence by activating p53 and related senescence-inducing pathways.57
Dysfunction of self-renewing somatic stem cell compartments has also been suggested to play a role in organismal aging.25,58 In this model, the activation of p16INK4a and p53 in response to cellular stresses including telomere dysfunction causes a decline in tissue-regenerative capacity (Fig. 4.3). This model is supported by murine studies59–63 and makes several predictions relevant to human disease. For example, this hypothesis suggests that heritable differences in regulation of the senescence-promoting machinery should alter individual susceptibility to human age-associated diseases, a concept that has been supported by a plethora of recent genomewide association studies.64 With regard to oncology, this model predicts that some agents and ionizing radiation used to treat cancer, for example, by inducing DNA damage, also can potentially induce senescence of important self-renewing cells of nonmalignant tissues such as the bone marrow. Studies in irradiated or chemotherapy-treated mice support such a role of senescence in the long-term hematopoietic toxicities of these therapeutic approaches.65,66 Likewise, somatic attrition of regenerative self-renewing cells as a result of senescence activation may place an increased replicative demand on the remaining functional cells of a given tissue, which may increase the rate of telomere dysfunction and speed transformation in other stem cells of a tissue in a cell nonautonomous manner (see later discussion).
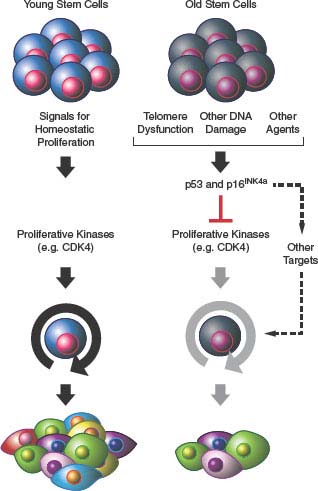
FIGURE 4.3 Senescence and aging. Activation of p53- and/or p16INK4a-mediated senescence pathways in stem cell compartments in response to DNA damage, telomere dysfunction, or other unknown stimuli leads to attrition of tissue-specific stem cells (e.g., hematopoietic stem cell and pancreatic β-cells) with attendant compromise of organ function and aging.
In aggregate, these genetic and in vivo data support the view that senescence prevents cancer in the intact organism on a near-daily basis and that reactivation of this mechanism in fully established cancers can effect dramatically beneficial responses, but also has the potential to produce long-term toxicity in nonmalignant tissues. It stands to reason that an improved understanding of the molecular basis of senescence could lead to therapeutic approaches designed to beneficially reawaken this potent tumor suppressor mechanism.
The INK4a/ARF/INK4b Senescence-Promoting Locus
Senescence is intimately associated with activation of the INK4a/ARF locus (also known as CDKN2a). This locus possesses an unusual gene structure that dually encodes p16INK4a and ARF (or p14ARF in humans and p19ARF in mice) in nonoverlapping open reading frames (Fig. 4.2). The locus also harbors the neighboring CDKN2b gene, which encodes p15INK4b, a protein highly related to p16INK4a that also activates Rb, which is located a short physical distance (10 kb) from the first exon of ARF. In addition to the links of p15INK4b/p16INK4a and ARF to Rb and p53 pathways, respectively, data showing that these proteins play prominent roles in the prevention of human cancer are strong. Activation of the locus in response to stimuli, which may be both independent and dependent on telomere dysfunction, is thought to promote tumor suppression through induction of senescence.
As the INK4a/ARF/INK4b locus at chromosome 9p21 is the most frequent site of single copy or homozygous deletion in human cancers,67,68 extensive analysis of this cytogenetic region has been performed. As somatic deletions in cancer frequently abrogate expression of all three INK4a/ARF/INK4b proteins (p15INK4b, p16INK4a, and ARF), debate has focused on which member or members of the locus represents the principal tumor suppressor activity located at human chromosome 9p21. A substantial body of human and murine data has now unequivocally shown that all three proteins are human tumor suppressors.37 For example, p15INK4b appears mainly important in the suppression of hematopoietic malignancies, whereas p16INK4a and ARF appear to play more general anticancer roles in several tumor types. Elegant murine studies69 have further shown that these tumor suppressor genes can play “back-up” roles to each other, suggesting that combined inactivation of the locus is more oncogenic than deletion of any single member. Therefore, although the human and murine genetic data considered as a whole establish that the INK4a/ARF/INK4b locus encodes three major human tumor suppressor proteins, their relative and combinatorial importance in a particular tumor type is a subject of ongoing study.
Crisis, Telomerase Reactivation, and Alternative Lengthening of Telomeres
Under circumstances of extended cell divisions beyond the Hayflick limit with inactivation of the p16INK4a and p53 pathway, progressive telomere erosion ultimately leads to loss of telomere capping function, resulting in increasing chromosomal instability. This leads to progressive loss of cell viability and proliferative capacity across the cell population, ultimately resulting in “cellular crisis.” The cellular phenotypes of massive cell death and growth arrest are likely by-products of DNA damage checkpoint responses and rampant chromosomal instability with associated loss of essential genetic material. Emergence from crisis is a rare event in human cell culture and requires restoration of telomere function either by up-regulation of telomerase activity or activation of the alternative lengthening of telomeres (ALT) mechanism.70 The restoration of functional telomeres serves to quell DNA damage signaling and high levels of chromosomal instability, thereby enhancing the viability of cells with procancer genotypes. Finally, the extent to which normal tissues experience telomere-associated Hayflick and crisis transitions continues to be an area of ongoing investigation. Nevertheless, although clear evidence of the presence of these events is still lacking, strong support is mounting for telomere-based crisis, particularly during early stages of neoplastic development.
Transcriptional up-regulation of the TERT gene seems to be a key rate-limiting step in telomerase reactivation, whereas the telomerase-independent ALT pathway appears to be executed via a poorly understood process involving activation of the homologous recombination pathway.71,72 The analysis of pathways regulating TERT gene transcription has forged links to well-known oncoproteins and tumor suppressors including Myc, Mad, and Menin, among others, demonstrating the capacity of these proteins to engage the TERT gene promoter directly.73–75 In contrast, the enigmatic ALT process has been variously associated with p53 deficiency and with tumors of mesenchymal origin.76
Studies in yeast have also shown that ALT is enhanced in mismatch repair-deficient cells, owing to increased homologous recombination between chromosomes. The rare use of ALT by epithelial-derived tumors, coupled with functional comparisons of telomerase versus ALT-mediated telomere maintenance, has shown that ALT may not be as biologically robust in advancing malignancy, a finding that diminishes the theoretical concern that ALT may provide a robust resistance mechanism to antitelomerase therapy in advanced malignancy.77 The idea that ALT may be a less effective telomere maintenance mechanism derives additional support from studies in human cell culture and the mouse revealing that telomerase per se is needed for full malignant transformation, including metastatic potential.78 The fundamental mechanistic differences between ALT and telomerase reactivation in telomere maintenance may provide an explanation for the report of more favorable clinical outcomes for ALT-positive compared with telomerase-positive glioblastomas,79 although analysis of 71 human osteosarcoma cases failed to show a more favorable clinical outcome for the ALT-positive subset.80 However, it should be noted that, in the latter, the absence of any telomere maintenance mechanism was more associated with improved survival than stage or response to chemotherapy, further emphasizing the general importance of telomere maintenance in cancer.
TELOMERE MAINTENANCE AND CANCER
Robust telomerase activity is observed in more than 80% of all human cancers,81 a profile consistent with its role in promoting malignant progression. However, another side to the telomerase-cancer connection has emerged from mouse models and correlative data in staged human tumors. These data have indicated that a lack of telomerase and associated telomere attrition during the early stages of neoplastic growth provides a potent mutator mechanism that enables would-be cancer cells to achieve the high threshold of cancer-promoting changes required to traverse the benign to malignant transition.
Indeed, telomeres of human cancer cells are often significantly shorter than their normal tissue counterparts, suggesting that telomere attrition has occurred during the life history of these cancer cells, apparently at very early phases of the transformation process when telomerase activity is low. The subsequent reactivation of telomerase restores telomere function, albeit at a shorter set length. Thus, although reactivation of telomerase is critical to the emergence of immortal human cells, this preceding and transient period of telomere shortening and dysfunction promotes the carcinogenic process through the generation of chromosomal rearrangements. These chromosomal rearrangements are brought about through breakage-fusion-bridge (BFB) cycles (Fig. 4.4). A DSB created by these BFB cycles is now known to provide a nidus for amplification and/or deletion at the site of breakage for the resulting daughter cells. The broken chromosome may become fused to another chromosome, generating a second dicentric chromosome and perpetuating the BFB cycle. The accumulation of wholesale genetic changes via aneuploidy, nonreciprocal translocations, amplifications, and deletions by the BFB cycles coupled with the reactivation of telomerase enables rare cells incurring a threshold number of carcinogenic changes needed to initiate the transformation process.
Although at first glance the cancer-promoting effects of telomere-based crisis seem to counter the established role of telomerase activation in cancer progression, this mechanism is less paradoxical if one considers that many early-stage cancers deactivate pathways essential for telomere checkpoint responses, thus increasing the survival and proliferation of cells experiencing increasing chromosomal instability.75,82 This hypothesis of “episodic instability” derives additional support from genetic studies in the mouse showing that telomere-based crisis coupled with loss of the p53-dependent DNA damage response can act cooperatively to effect malignant transformation. In humans, the accumulation of oncogenic lesions during normal aging or accelerated accumulation of DNA damage (e.g., environmental carcinogen exposure or oxidative damage) may deactivate the telomere checkpoint response, accelerate telomere attrition, and drive the affected premalignant cells into crisis. It is the rare transformed cell that emerges from this process, often with reactivated telomerase. Thus, telomeric shortening can be viewed as a barrier to cancer development in the presence of intact checkpoint response and as a facilitator for numerous genetic changes necessary for the emergence of nascent cancer cells in the absence of the checkpoint response pathways.
It has also been suggested that telomere dysfunction can be oncogenic in a cell nonautonomous process.83 Murine data in the hematopoietic system suggest that telomere dysfunction leads to stem cell dysfunction.84,85 Therefore, telomere dysfunction could induce premature loss of stem cells (as described in Fig. 4.3), which might induce a compensatory hyperproliferation of remaining functional stem cells. This increased proliferative drive might facilitate mutagenesis in the remaining functional self-renewing cells, and in turn select for clones with damaged genomes, in particular those harboring defects in the senesce-promoting machinery.
Several recent lines of evidence have suggested that the oncogenic effects of telomere dysfunction are an important determinant of susceptibility to human cancer. For example, several human kindreds have been identified with congenital telomerase deficiency syndromes due to inactivating mutations of TERT or other members of the shelterin complex.4 Such patients exhibit age-associated pathologies such as bone marrow failure and pulmonary fibrosis, but also appear to be at increased risk for several cancers including acute myelogenous leukemia and cutaneous carcinomas.4,83,86–88 Likewise, human genomewide association studies of large human cohorts have pointed to sequence variants in the chromosome 5p15.33 locus as a susceptibility locus for many types of cancer, including tumors of the skin, lung, bladder, prostate, and cervix.89 The single nucleotide polymorphisms associated with these cancers are near to both the CLPTM1L (cisplatin resistance-related protein CRRP9) gene and the TERT gene. It is unclear whether one or both of these genes are responsible for the association as there is limited functional biological validation. These human data suggest that telomere dysfunction could be oncogenic, and that hypomorphic alleles of TERT could contribute to human cancer susceptibility on a population basis.
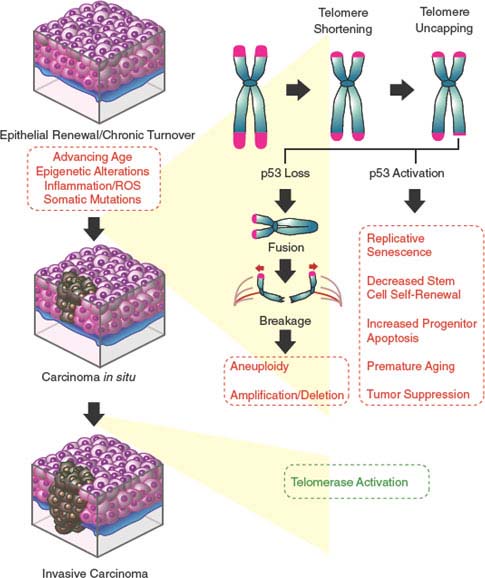
FIGURE 4.4 Dysfunctional telomere-induced genomic instability model of epithelial carcinogenesis. Continuous epithelial turnover during aging coupled with somatic mutations inactivating checkpoint responses is thought to lead to critical telomere erosion, resulting in telomere uncapping and the initiation of breakage-fusion-bridge (BFB) cycles. The double-strand breaks created by the BFB cycles are nidi for amplifications and deletions for the resulting daughter cells. The broken chromosome may become fused to another chromosome, generating a second dicentric chromosome and perpetuating the BFB cycle. This facilitation of the accumulation of genetic changes (via aneuploidy, nonreciprocal translocations, amplifications, and deletions) by the BFB cycles coupled with the reactivation of telomerase enables cells to emerge from crisis and proceed to malignancy.
Telomere-Induced Chromosomal Instability
The study of senescence and telomeres has provided some insights into the link between advancing age and increased cancer risk. In humans, there is a dramatic escalation in cancer risk between the ages of 40 and 80, resulting primarily from a marked increase in epithelial malignancies such as carcinomas of the breast, lung, colon, and prostate. A conventional view is that the cancer-prone phenotype of older humans reflects the combined effects of cumulative mutational load, decreased DNA repair capabilities, increased epigenetic gene silencing, and altered hormonal and stromal milieus. Although these factors are almost certain to contribute to increasing cancer incidence in aged humans, it is less evident why such processes would spur the preferential development of epithelial cancers. Moreover, these mechanisms do not readily explain one of the cardinal features of adult epithelial carcinomas—namely, a radically altered genome typified by marked aneuploidy and complex nonreciprocal chromosomal translocations.
The study of telomere dynamics in normal and neoplastic cells of the mouse has provided a potential explanation for the observed tumor spectrum and associated cytogenetic profiles in aged humans. In Terc p53 compound mutant mice, the presence of telomere dysfunction results in a dramatic shift in the tumor spectrum toward epithelial cancers, including those of the lung, colon, and skin.90 Moreover, in contrast to the largely normal cytogenetic profiles of cancers arising in mice with intact telomeres, the cancers generated in the Terc p53 compound mutant mice had highly complex cytogenetic profiles with a striking resemblance to human epithelial cancer genomes.
In attempting to assign relevance of these murine studies to humans, it is worth considering that the typical adult cancer, an epithelial carcinoma, derives from a compartment that has undergone continued renewal throughout the human lifespan. Against this backdrop of physiologic cell turnover, combined with the occasional pro-proliferative oncogenic mutation, telomere lengths would shorten in self-renewing progenitor cells of these epithelial tissues. If somatic mutations also neutralize Rb/p16INK4a/p53-dependent senescence checkpoints, continued growth beyond the Hayflick limit further drives telomere erosion and loss of the capping function, culminating in cellular crisis with attendant genomic instability. In this manner, telomere-based crisis provides the means to generate many additional mutations required to reach the early stages of malignant transformation. The subsequent reactivation of telomerase in transformed clones would serve to stabilize the genome to a level compatible with cell viability, allowing these initiated neoplasms to mature further.91 It is unclear whether additional somatic mutations, beyond telomerase activation, would be needed to produce a fully malignant phenotype that includes invasive and metastatic potential. Thus, a transient period of explosive chromosomal instability before telomerase reactivation appears to be required for the stochastic acquisition of the relatively high number of mutations thought to be required for adult epithelial carcinogenesis. Another line of support is the fact that a proportion of early-stage epithelial cancers are hardwired for lethal metastatic progression, suggesting that many cancers acquire a full profile of genome change early in their life history.
The episodic instability model of epithelial carcinogenesis fits well with current knowledge regarding the timing of telomerase activation and evolving genomic changes during various stages of human carcinoma development, particularly those of the breast, esophagus, and colon. Comparative genome hybridization has demonstrated that dysplastic human breast, esophageal, and colon lesions sustain widespread gains and losses of regions of chromosomes early in their development, often well before these tissues exhibit carcinoma in situ or invasive growth.92–94 The ploidy changes detected by comparative genome hybridization appear to correlate tightly with the presence of complex chromosomal rearrangements, and these markers of genomic instability are evident in the stages of advanced dysplasia of these tissues (e.g., ductal carcinoma in situ, Barrett’s esophagus). As these cancers progress through invasive and metastatic stages, genomic instability continues, apparently at a moderate rate, but further mutations would be predicted to derive from non–telomere-based mechanisms. Correspondingly, the measurement of telomerase activity in adenomatous polyps and colorectal cancers has established that telomerase activity is low or undetectable in small and intermediate-sized polyps, reflecting less intact telomere function. In contrast, telomerase increases markedly in large adenomas and colorectal carcinomas, reflecting stabilization of telomere function.95 Therefore, it appears that widespread and severe chromosomal instability is present early on during human tumorigenesis at a time when telomerase activity is low.
Additional support for this episodic instability model derives from the documentation of anaphase bridging (a correlate of telomere-based crisis) in evolving human colorectal cancers and in genomically unstable pancreatic cancers.96,97 This suggests that the DSB-induced conditions (including but not limited to telomere dysfunction), coupled with mutations that allow survival in the face of a DSB, could provide amplification/deletion mechanisms across the genome. Biologic selection forces would in turn lead to the emergence of clones with the amplifications and deletions that target cancer-relevant loci. Studies in the telomerase mutant mouse have begun to provide mechanistic insight into how BFB leads to cancer-relevant changes. In particular, telomerase-p53 compound mutant mice with telomere dysfunction have increased end-to-end fusions, and the ensuing BFB process is associated with chromosomal regional gains and losses that appear linked to nonreciprocal translocations.55,75
In future human studies, it will be important to document telomere attrition in renewing epithelial stem cells and to perform a simultaneous comparison of telomere status, telomerase activity, and chromosomal instability in the same tumor samples, particularly during the earliest stages of human epithelial cell transformation. Defining the temporal point at which telomerase is reactivated in the genesis and progression of the different cancers may also lead to the development of biomarkers for diagnosis, prognosis, and outcomes prediction. Such studies are needed to more firmly establish a causal link between telomere dysfunction and early chromosomal instability in human neoplasms.
Telomere Dynamics, Inflammatory Diseases, and Cancer
The telomere dysfunction-induced genomic instability model also suggests some unanticipated opportunities for the therapies of other human diseases. For example, this model provides a potential explanation for the high cancer incidence associated with diseases characterized by chronic cell destruction and renewal as well as inflammation. One of the most notable examples of this tight link is the high incidence of hepatocellular carcinoma in late-stage cirrhotic livers. Cirrhosis is the phenotypic end point of prolonged cycles of hepatocyte destruction and regeneration, and cirrhotic livers show a documented reduction in telomere length over time. Humans with congenital telomerase deficiency may be predisposed to fibrotic liver disease, including cirrhosis.87 Mouse models involving the telomerase-deficient mouse have shown that critical reductions in telomere length and function can accelerate the development of cirrhosis and hepatocellular carcinoma in chronic liver injury experiments.98–100 Another example of a telomere-based pathogenic relationship between chronic tissue turnover, telomere-based crisis, and increased cancer risk is ulcerative colitis, a condition characterized by rapid cell turnover and oxidative injury to the intestines, and a high incidence of intestinal dysplasia or cancer.97 In addition to the progressive telomere attrition resulting from the cell turnover, accelerated telomere attrition might occur via increased oxidative stress and from the altered inflammatory microenvironment milieu. Together, such observations suggest the intriguing possibility that early somatic reconstitution of telomerase could attenuate telomere attrition and paradoxically reduce the occurrence of cancers in these high-turnover disease states, a theory that requires additional preclinical studies. In addition, serial analyses of telomere length from these tissues may provide prognostic information regarding the rising risk of cancer development. Thus, progress in our understanding of telomere biology has mechanistically connected diverse fields in medicine involving chronic inflammatory diseases, degenerative diseases, geriatrics, and oncology.
Telomerase and Telomere Maintenance as Therapeutic Targets
Some evidence supports the view that telomerase-mediated telomere maintenance represents a near-universal therapeutic target for cancer. Indeed, cell culture–based studies of human cancer cells have established that inhibition of telomerase culminates in cell death after extended cell divisions. The past few years have witnessed intense efforts to design therapeutic strategies capable of targeting telomere structure and the telomerase holoenzyme function.14,101,102 Unfortunately, most of these compounds and agents are still in preclinical and early clinical development and thus their safety and efficacy profiles in human patients are not fully known.
Presently, the only clinically advanced telomerase-related cancer treatment strategy is immunotherapy, targeting immune recognition and the destruction of cells that express telomerase. Immune responses, specifically cytotoxic T-cell responses, have been generated against peptide sequences of the hTERT protein, and it has been demonstrated that these cytotoxic T cells are capable of selectively lysing target cells that express TERT peptides presented on the cell surface in the context of major histocompatibility complex class I molecules. There have been several promising completed phase 1/2 trials using peptides from telomerase as vaccines.103,104 A large randomized phase 3 trial comparing gemcitabine alone versus gemcitabine with a telomerase peptide vaccine (GV1001) showed no difference in survival benefit in the first 360 enrolled patients, and the trial was stopped. A second large 1,110 pancreatic cancer patient trial comparing gemcitabine/capecitabine combination therapy with concurrent and sequential gemcitabine/capecitabine therapy with GV1001 is still ongoing. Lastly, other TERT-based immune approaches, such as infusion of patient’s primed antigen-presenting dendritic cells ex vivo with TERT mRNA, are also currently in early clinical trials.105
As for the ongoing design of rational clinical trials of telomere-based therapeutics, such efforts will be informed by the considerable body of knowledge accumulated in telomere biology. Experience with the telomerase mutant mouse model and human cell culture systems should serve to guide the design of human clinical trials. These studies suggest that inhibitors of telomerase activity might be expected to exhibit a long lag time and might promote malignancy in some circumstances, but also may be particularly useful in the setting of minimal residual disease after the administration of standard chemotherapeutic agents and surgery. In addition, pharmacodynamic assays capable of assessing inhibition of telomerase activity in individual patients are needed. Moreover, given that the activation of senescence-promoting mechanisms such as p53 and p16INK4a has been associated with aging-like pathologies in several tissues,59–63 some caution is warranted regarding the toxicity resulting from the induction of premature senescence in nondiseased tissues. This potential is underlined by evidence of germ cell defects, defects in proliferative homeostasis of certain tissues, and an increased rate of spontaneous malignancy in mice with telomere dysfunction, suggesting that clinical trials of such agents will need to be actively monitoring patients for these sequelae.
Furthermore, it seems prudent that the genetic profile of tumors enlisted into clinical trials should be determined to assess the integrity of p53. This caution relates to mouse models showing that the combination of p53 deficiency and telomere dysfunction drives greater genomic instability and thus potential for emergence of therapeutic resistance. In contrast, when p53 responses are intact, critical telomere shortening should induce p53-dependent senescence and apoptosis. The final answers to these safety questions reside in the analyses of current and future clinical trials with humans.
Conversely, the telomerase-deficient mouse model has also informed that cells and animals with telomere dysfunction are more sensitive to ionizing radiation and DNA DSB chemotherapeutic agents; thus, telomerase activity inhibitors may be more effective when paired with radiation or certain classes of chemotherapy that produce DSBs, as they might produce synergistic cytogenetic catastrophe. Again, however, particular care is warranted here as the combination of increased DNA damage with reduced capacity for normal repair may also produce marked increases in the toxicity of chemoradiotherapy.
Recent years have witnessed significant progress in the telomere biology field that is now maturing into new opportunities for improved diagnostics and novel therapeutic applications in human diseases, including cancer. Discoveries in telomere biology, rewarded with the 2009 Nobel Prize, have provided new mechanistic insights into the pathogenesis of human cancer and of inherited and acquired degenerative disorders. The role of telomere dysfunction driving episodic genomic instability in epithelial cancers—first seen in the telomerase-deficient mouse—has now been substantiated in the study of several human cancer types, with further support from genomewide association studies and kindreds with congenital telomerase deficiency. The pivotal role of telomere attrition in the pathogenesis of cancer and tissue aging provides potential avenues for the development of cancer risk biomarkers, diagnostics, and rationally designed therapeutics.
Selected References
The full list of references for this chapter appears in the online version.
3. Sahin E, Depinho RA. Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature 2010;464:520.
4. Calado RT, Young NS. Telomere diseases. N Engl J Med 2009;361:2353.
6. O’Sullivan RJ, Karlseder J. Telomeres: protecting chromosomes against genome instability. Nature Rev 2010;11: 171.
7. de Lange T. How telomeres solve the end-protection problem. Science 2009;326:948.
8. de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev 2005;19: 2100.
10. Zhu XD, Kuster B, Mann M, Petrini JH, de Lange T. Cell-cycle-regulated association of RAD50/MRE11/NBS1 with TRF2 and human telomeres. Nat Genet 2000;25:347.
12. Takai H, Smogorzewska A, de Lange T. DNA damage foci at dysfunctional telomeres. Curr Biol 2003;13:1549.
13. Deng Y, Chan SS, Chang S. Telomere dysfunction and tumour suppression: the senescence connection. Nat Rev Cancer 2008;8:450.
16. Valdes AM, Andrew T, Gardner JP, et al. Obesity, cigarette smoking, and telomere length in women. Lancet 2005; 366:662.
19. Artandi SE, DePinho RA. Telomeres and telomerase in cancer. Carcinogenesis 2010;31:9.
20. Masutomi K, Yu EY, Khurts S, et al. Telomerase maintains telomere structure in normal human cells. Cell 2003;114: 241.
22. Maida Y, Yasukawa M, Furuuchi M, et al. An RNA-dependent RNA polymerase formed by TERT and the RMRP RNA. Nature 2009;461:230.
24. Park JI, Venteicher AS, Hong JY, et al. Telomerase modulates Wnt signalling by association with target gene chromatin. Nature 2009;460:66.
25. Sharpless NE, DePinho RA. Telomeres, stem cells, senescence, and cancer. J Clin Invest 2004;113:160.
26. Hayflick L, Moorhead P. The serial cultivation of human diploid cell strains. Exp Cell Res 1961;25:585.
28. Kamijo T, Zindy F, Roussel MF, et al. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell 1997;91:649.
29. Sage J, Miller AL, Perez-Mancera PA, Wysocki JM, Jacks T. Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature 2003;424:223.
31. Classon M, Harlow E. The retinoblastoma tumour suppressor in development and cancer. Nat Rev Cancer 2002; 2:910.
36. Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell 1998;92:725.
37. Kim WY, Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell 2006;127:265.
43. Jacobs JJ, de Lange T. Significant role for p16INK4a in p53-independent telomere-directed senescence. Curr Biol 2004;14:2302.
44. Chicas A, Wang X, Zhang C, et al. Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. Cancer Cell 2010;17:376.
45. Donehower LA, Harvey M, Slagle BL, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992;356:215.
47. Sharpless NE, Bardeesy N, Lee KH, et al. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature 2001;413:86.
48. Braig M, Lee S, Loddenkemper C, et al. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 2005;436:660.
49. Collado M, Gil J, Efeyan A, et al. Tumour biology: senescence in premalignant tumours. Nature 2005;436: 642.
50. Chen Z, Trotman LC, Shaffer D, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005;436:725.
51. Ventura A, Kirsch DG, McLaughlin ME, et al. Restoration of p53 function leads to tumour regression in vivo. Nature 2007;445:661.
52. Xue W, Zender L, Miething C, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007;445:656.
53. Michaloglou C, Vredeveld LC, Soengas MS, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005;436:720.
55. Chin L, Artandi SE, Shen Q, et al. p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell 1999;97:527.
56. Martins CP, Brown-Swigart L, Evan GI. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell 2006; 127:1323.
57. Schmitt CA, Fridman JS, Yang M, Baranov E, Hoffman RM, Lowe SW. Dissecting p53 tumor suppressor functions in vivo. Cancer Cell 2002;1:289.
58. Campisi J. Suppressing cancer: the importance of being senescent. Science 2005;309:886.
61. Krishnamurthy J, Ramsey MR, Ligon KL, et al. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature 2006;443:453.
64. Sharpless NE, DePinho RA. How stem cells age and why this makes us grow old. Nature Rev 2007;8:703.
68. Beroukhim R, Mermel CH, Porter D, et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010;463:899.
69. Krimpenfort P, Ijpenberg A, Song JY, et al. p15Ink4b is a critical tumour suppressor in the absence of p16Ink4a. Nature 2007;448:943.
73. Blasco MA. Telomerase beyond telomeres. Nat Rev Cancer 2002;2:627.
74. Lin SY, Elledge SJ. Multiple tumor suppressor pathways negatively regulate telomerase. Cell 2003;113:881.
75. O’Hagan RC, Chang S, Maser RS, et al. Telomere dysfunction provokes regional amplification and deletion in cancer genomes. Cancer Cell 2002;2:149.
78. Chang S, Khoo CM, Naylor ML, Maser RS, DePinho RA. Telomere-based crisis: functional differences between telomerase activation and ALT in tumor progression. Genes Dev 2003;17:88.
81. Shay JW, Bacchetti S. A survey of telomerase activity in human cancer. Eur J Cancer 1997;33:787.
85. Rossi DJ, Bryder D, Seita J, Nussenzweig A, Hoeijmakers J, Weissman IL. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature 2007;447:725.
89. Rafnar T, Sulem P, Stacey SN, et al. Sequence variants at the TERT-CLPTM1L locus associate with many cancer types. Nat Genet 2009;41:221.
90. Artandi SE, Chang S, Lee SL, et al. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature 2000;406:641.
97. O’Sullivan JN, Bronner MP, Brentnall TA, et al. Chromosomal instability in ulcerative colitis is related to telomere shortening. Nat Genet 2002;32:280.
99. Farazi PA, Glickman J, Jiang S, Yu A, Rudolph KL, DePinho RA. Differential impact of telomere dysfunction on initiation and progression of hepatocellular carcinoma. Cancer Res 2003;63:5021.
100. Rudolph KL, Chang S, Millard M, Schreiber-Agus N, DePinho RA. Inhibition of experimental liver cirrhosis in mice by telomerase gene delivery. Science 2000;287:1253.
102. Shay JW, Wright WE. Telomerase therapeutics for cancer: challenges and new directions. Nat Rev Drug Discov 2006; 5:577.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


