Chapter 22 Targeted Radionuclide Therapy—Expanded Content
Selective delivery of radionuclides to cancer cells using an antibody or other conjugate has been under investigation for more than 20 years. In 2002, the Food and Drug Administration (FDA) granted its first approval for a radiolabeled antibody. Initially, this form of radionuclide therapy mainly involved the use of antibodies or antibody-derived constructs as carriers of radionuclides and is therefore called radioimmunotherapy (RIT). However, because the concept also includes binding to nonantigen receptors, targeted radionuclide therapy (TaRT) is a more comprehensive term. Paul Wallner coined the acronym STaRT for systemic targeted radionuclide therapy. The development of TaRT has required the cooperation of basic scientists in the areas of radiation biology, chemistry, physics, and immunology with multiple clinical specialists. With the exception of some gene therapy approaches, TaRT differs from external beam radiation therapy in that selective targeting can be at the cellular level rather than the target volume level. Among its potential applications, TaRT provides a means of irradiating multiple tumor sites throughout the body with relative sparing of normal tissues. A number of challenges hampering the use of TaRT have been overcome, and others are areas of active investigation. Many of these are covered in more detail in other reviews.1–15
Tumor Targeting
Antibody Targeting
The efficacy of RIT is dependent on a number of factors, including properties of the targeted antigen or receptor, tumor, and targeting agent. Antigen/receptor variables include affinity, avidity, density, availability, shedding, and heterogeneity of expression.16 Tumor factors include vascularity, blood flow, and permeability. Antibody features to consider are specificity of the binding site, which affects selective tumor uptake; immunoreactivity, which can affect localization; stability in vivo; and both avidity and affinity.16–18 Affinity can be described by an intrinsic association constant K that characterizes binding of a univalent ligand (formation of a stable antibody-antigen complex) and can be calculated from the ratio of the rate constants for association and dissociation. Because intact antibodies and most antigens are multivalent, the tendency to bind depends upon the affinity, number of binding sites, and other nonspecific factors involved in aggregation. Therefore the term avidity is used to describe the overall tendency of antibody to bind to antigen.
The optimal level of binding affinity for maximal in vivo effectiveness is controversial, and binding affinity does not always correlate with activity in vivo.17,19 One might expect that high-affinity antibodies compared with low-affinity antibodies would result in increased tumor uptake, increased retention, and improved efficacy, as has been shown in some studies.20–22 However, high-affinity antibodies may preferentially bind to perivascular regions in the periphery of tumors, whereas antibodies of lower affinity are able to penetrate deeper into tumors.23 The optimal amount of unlabeled antibody remains undetermined for most agents, but relatively high doses have been as good as or better than lower doses in most imaging studies.24,25 Efforts by the Seattle transplant team to optimize the amount of unlabeled antibody for individual patients by tracer studies with varying amounts often showed the highest concentration studied to be as good as or better than direct RIT with iodine-131 (131I) antibody conjugates.26 Recent modeling with alpha-emitting conjugates attempts to maximize tumor cell kill while remaining within renal tolerance by individualization based on antigen ratio, adjuvant chemotherapy cytoreduction, and dose fractionation.13,27 A wide variety of antibodies have been made against a number of tumor-specific and tumor-associated antigens, which, although present on some normal cells, are usually expressed at lower levels on normal cells than on the targeted tumor cells. Most RIT trials have used monoclonal antibodies, and many have used intact murine immunoglobulin G (IgG) antibodies. Early on, the immunogenicity of the nonhuman antibodies was recognized as a serious limitation of RIT.28 With the exception of patients with lymphoma, who are less prone to develop an immune response to murine antibodies (human antimouse antibody [HAMA] response), more than 80% of patients usually develop an immune response against a therapeutically administered murine or other species antibody after a single injection of antibody.28 Such an immune response can occur even after doses as small as 1 mg used for imaging studies or following administration of antibody fragments or smaller constructs (but with less frequency than is found following administration of intact IgG). Administration of antibody in a patient with a HAMA response can result in a severe immune reaction and rapid blood clearance, limiting tumor uptake of radiolabeled antibody.29 Several approaches have been employed in an effort to ameliorate the problem of antibody immunogenicity, such as (1) the development of less immunogenic (chimeric and humanized) antibodies, (2) the use of immunosuppression by cyclosporine or deoxyspergualin to prevent an immune response, (3) the removal of HAMA directed to the therapeutic antibody by administration of an excess of unlabeled antibody before the administration of the radioimmunoconjugate, or (4) post-treatment removal of immune complexes by passing the patient’s blood through an extracorporeal immunoadsorption column.30–33
Genetic engineering has been successful in providing numerous targeting agents with reduced immunogenicity and has allowed for optimization of other aspects of the therapy.34–36 Genetic constructs have been produced that vary in specificity and can be altered by conjugating them to other agents (e.g., cytokines, toxins, or radiosensitizers) to form fusion proteins.37,38
Technologic advances have provided a wide variety of antibodies and constructs varying in specificity, size, and number of antigen combining sites, rapidity of distribution, immunogenicity, and immunologic function.39 Use of small antibody constructs such as single-chain antigen-binding proteins may be most useful for diagnostic studies or for multistep targeting strategies.40 The decreased immunogenicity of fragments/constructs and human and humanized antibodies now allows for administration of repeat courses or fractionated RIT dosing.41 As immunogenicity of antibodies has been ameliorated, some investigators have had concern about the potential immunogenicity of other components of therapy. For example, as more macrocyclic chelators have been developed to improve stability of conjugates, some data suggest that these molecules may be immunogenic as has been the streptavidin used in some pretargeting schemes.42–44 Therefore work continues to diminish immunogenicity problems from these other components.
Nonantibody Targeting
Several of the factors affecting antibody development/efficacy for TaRT are also pertinent to nonantigen targeting, such as receptor density, availability, affinity, shedding, and heterogeneity of receptor expression, as well as features of the tumor, including vascularity, blood flow, and permeability. The discovery made more than a decade ago that many tumors overexpress receptors for peptide hormones opened the field for TaRT and other forms of targeted therapy. Radiolabeled peptide analogs such as those for somatostatin, bombesin, calcitonin, oxytocin, and epidermal growth factor have been studied. Analogs of somatostatin are the most studied to date, with radiolabeled peptides being used for detection and treatment of a variety of neuroendocrine tumors.10,45,46 Initial treatment of tumors expressing somatostatin receptor subtype 2 was with higher doses of the diagnostic agent (111In-DTPA)-octreotide.47 Subsequently, the higher-affinity (DOTA, Tyr3) octreotide and (DOTA, Tyr3) octreotate were introduced and used with yttrium-90 (90Y) and lutetium-177 (177Lu) labeling as the therapeutic beta emitter for peptide receptor radionuclide therapy (PRRT). (Tyr3) octreotate differs from octreotide by substitution of tyrosine as the third residue and the replacement of threoninol by threonine. Early studies show impressive response rates and also show that fractionation of the administered dose as well as infusion of amino acids has ameliorated the dose-limiting renal toxicity.3,48,49 Various multicenter studies have been conducted for optimization of 90Y-(DOTA,Tyr3) octreotide, 177Lu-(DOTA,Tyr3) octreotate, and 90Y-(DOTA) lanreotide, another somatostatin analog.50 In addition to objective clinical response rates that are often higher than 25% and improved survival rates, clinical benefit in terms of decreased symptoms has usually been observed in the majority of patients with progressive neuroendocrine tumors.14,46,49,51 Radiolabeled peptides targeting tumor neovasculature are also under investigation and may provide therapy for nonendocrine cancers.52 Another targeting entity under investigation is the use of liposomes or nanoparticles as carriers of radionuclides.53–55 There continues to be progress on strategies that are not aimed at one tumor type, such as the targeting of tumor blood vessels and the evolving applications of nanoparticles.56–58
Selection of Radionuclides
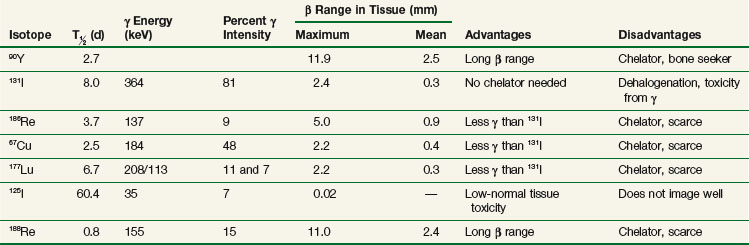
Early reviews provide detail on radionuclide selection, primarily with regard to the radiolabeling of whole immunoglobulins with beta emitters.59–61 Some later publications have focused on the selection of radionuclides for use with smaller engineered, antibody-derived constructs,62–64 and small nonantibody targeting delivery entities.65 Alpha emitters are also of increasing use.53,66–70 The emission profile of radionuclides determines their suitability for therapy and imaging. Particle energy and mean path length in tissue are important determinants of therapeutic efficacy, whereas the presence of gamma emissions at an appropriate energy is best for imaging. Some radionuclides such as (131I) have beta and gamma emissions that allow for both therapy and imaging. The therapeutic effect from such radionuclides is primarily because of the beta radiation, whereas the gamma component allows for imaging but contributes substantially more to normal tissue toxicity than efficacy. Important considerations for selection of radionuclides for TaRT include the size and biologic features of the tumor and antibody (or other targeting agent). In a one-step RIT administration using directly labeled antibodies, matching of the physical half-life of the radionuclide with the initial peak of tumor to nontumor antibody concentration was reported to be the most important factor for optimal radionuclide selection.59 For example, a good therapeutic ratio would not be obtained if one used a radionuclide that decays within several hours linked to an antibody that has a circulation time of several days, because most of the radiation exposure would have occurred before the radiolabeled antibody localized in the tumor. In this case, the resulting radiation dose to normal tissues would be nearly as great as that to the targeted tumor. Pretargeting strategies, discussed in more detail in the optimization section, allow for use of radionuclides with shorter half-lives because unlabeled methods of tumor targeting precede administration of the radionuclide.
The development of labeling and chelation chemistry for the production of stable radioconjugates has greatly facilitated TaRT. 131I can be directly attached to antibody without a chelator. However, if a targeted antigen/receptor undergoes modulation (internalization), the tumor cells can dehalogenate the conjugate and release free iodine. 131I can also be attached by a secondary molecule conjugated through an iodinatable linker rather than directly linked to antibodies. These radioimmunoconjugates appear to be more resistant to dehalogenation.71 A variety of increasingly stable chelators have been developed for 90Y and other metal radionuclides, but some have been found to be immunogenic (probably due in part to aggregation).42,44 The chemistry, stability, and in vivo processing of radioimmunoconjugates also have implications for toxicity. The filtration of small radioconjugates or their metabolites through the kidneys may result in considerable amounts of radiation being delivered to the kidneys. Renal toxicity has been reported to be the dose-limiting toxicity for some small-molecular-weight peptide conjugates and alpha emitter conjugates.68,72 Hepatic uptake, metabolism of radioimmunoconjugates, and retention of 90Y-complexes in the liver are potentially dose limiting in myeloablative TaRT.73 With some antibodies, comparison of radionuclides for optimization purposes has been studied using human tumors in xenograft models or theoretical modeling based on prior patient pharmacokinetics data.74–76 In summary, there are a variety of radionuclides that are suitable for TaRT. These include alpha emitters, low-, medium-, and high-energy beta emitters, and those that work through electron capture and/or internal conversion (Auger electrons). Beta emitters (e.g., 131I and 90Y) have been the most popular radionuclides for clinical TaRT trials to date. These radionuclides have the advantage of not needing to target every individual tumor cell, because the mean radiation range is approximately 0.4 mm and 2.5 mm, respectively. Features of some of these radionuclides are compared in Table 22-1. Alpha emitters offer promise for the future, especially when they are used to treat microscopic disease or are administered intratumorally. Their suitability includes the relatively short half-lives for most alpha emitters (e.g., 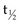 = 45.6 minutes for bismuth-213 (213Bi) and 7 hours for astatine-211 (211At) and a short range for the high-LET alpha particles (e.g., limited to 40 to 85 microns). These radionuclides may prove to be advantageous for the treatment of small-volume disease.77 Use of some alpha emitters with a longer half-life will facilitate more widespread application as it will be feasible to ship products.68,78,79
= 45.6 minutes for bismuth-213 (213Bi) and 7 hours for astatine-211 (211At) and a short range for the high-LET alpha particles (e.g., limited to 40 to 85 microns). These radionuclides may prove to be advantageous for the treatment of small-volume disease.77 Use of some alpha emitters with a longer half-life will facilitate more widespread application as it will be feasible to ship products.68,78,79
Designing chelators with special characteristics can also affect radionuclide “matching” with the targeting agent. For example, selective degradation of a cathepsin-sensitive chelate reduced liver uptake of radiometal.80 The use of such chelators may allow for dose escalation of radiometal conjugates such as curium-67 (67Cu) that otherwise would be limited by hepatic toxicity. Other conditionally cleavable chelators are also under study.81 The synthesis of the 2-(4-isothiocyanotobenzyl)-1,4,7,10-tetraaza-1,4,7,10-tetra-(2-carbamonyl methyl)-cyclododecane (TCMC) chelator has allowed progress in the use of lead-212 (212Pb).82 Macrocyclic chelators such as DOTA, which have been developed for stable chelation to 90Y, and other beta-emitting radiometals have shown acid lability with 212Pb and likely with other alpha emitters.83 To overcome this, the bifunctional chelating agent TCMC was synthesized, characterized, and subsequently used in several animal studies.82
Radiobiology
Biologic effects of TaRT may be associated with both the radionuclides and their carriers, either alone or in combination.84,85 This is particularly true for certain antibody carriers that may have antitumor efficacy alone (e.g., anti-CD20 antibodies). Antibody-induced responses can include apoptosis, complement-dependent cytotoxicity, and antibody-dependent cell-mediated cytotoxicity.86 Radiation effects can result in cell death during mitosis or by apoptosis, necrosis, and/or a metabolic cell death. The relative importance of these types of cell death depends on factors such as the inherent genetic makeup of the cells. For lymphomas, apoptosis is an important mechanism of cell death, whereas apoptosis usually plays less of a role in killing solid tumor cells. Lymphomas are especially sensitive to apoptosis induced not only by radiation but by a variety of cytotoxic agents, including antibody therapy alone.87–90 Several signal transduction pathways have been found to be associated with radiation-induced apoptosis. These have been reviewed by Hernandez and Knox.84 Antibody-induced intracellular signaling may also function to enhance the effects of RIT, and dose rate effects may be very important to the efficacy of RIT as well.91,92
Several studies have shown that cells exposed to either continuous or exponentially decreasing low-dose-rate (LDR) radiation are arrested in the radiosensitive G2/M phase of the cell cycle and undergo apoptosis.93–96 This G2/M block from LDR radiation as delivered via TaRT may also sensitize tumor cells to killing by other therapies such as external beam irradiation.97 Data from some experiments showing this effect suggest that therapy may be optimized by using TaRT before external beam irradiation.97,98 External beam irradiation has also been given before TaRT to increase tumor uptake of radionuclide conjugates, as will be discussed later. In addition, regulating cell cycle distribution or altering the underlying apoptotic potential by drugs or biologic response modifiers in tumor cells can affect the susceptibility of such cells to radiation-induced apoptosis.99 Recent studies include exploration of the use of gene transfer technology to induce expression of high-affinity membrane receptors to enhance the specificity of radioligand localization or the use of genetically engineered antibody molecules (e.g., diabodies: noncovalent single-chain Fv dimers) as carriers for TaRT that may be able to enhance the efficacy of this therapy.100–103
Although there is usually a positive correlation between the level of radiation-induced cell killing and the dose rate, an inverse dose rate effect has been reported with low-dose-rate irradiation in the range of about 0.3 Gy/hour, with some cell types being more sensitive to this inverse dose rate effect than others.104 Knox and colleagues105 showed lymphoma cells to be very sensitive to 131I-labeled antibody low-dose-rate radiation in a direct comparison to high-dose-rate external beam irradiation. At least one mechanism for this observation appears to be greater arrest in the G2/M phase of the cell cycle. Another mechanism of lymphoma radiation sensitivity may be less efficient DNA repair among lymphomas compared with some other cell types.84,85 Ongoing studies will help elucidate the radiobiology of TaRT, which will facilitate optimization of TaRT therapy in the future and provide the prerequisite understanding of underlying biologic mechanisms to determine how best to use TaRT in combination with other treatment modalities.
Clinical Results
Several routes of administration have been employed, including intravenous, intra-arterial, intrathecal, intraperitoneal, intratumoral, and, rarely, intrapleural administration. The most prevalent route of administration has been the intravenous route for systemic therapy. However, for localized disease, regional or intracavitary administration may be advantageous. Intraperitoneal RIT has generally been more efficacious than intravenous RIT for the treatment of small-volume disease confined to the peritoneal cavity, but there are conflicting reports concerning the relative merits of arterial and venous routes of administration of radiolabeled antibodies for other sites.106–110
Radioimmunotherapy of Nonhematologic Malignant Disease
Radioimmunotherapy (RIT) for solid tumors continues to be actively investigated as a form of systemic targeted radiation therapy in a variety of tumor types and clinical settings. Initial clinical efforts were with polyclonal antibodies produced in immunized rabbits and other species.111 Beginning in the 1970s with the development of hybridoma technology, large-scale production of clinical grade monoclonal antibodies (Mabs) selected against specific tumor-associated antigens became possible and greatly expanded the number of agents evaluated in clinical trials. Subsequent molecular engineering efforts allowed for the customized design of antibody constructs with properties better optimized for therapy.
RIT demonstrated initial promise in animal models, with dozens of studies in tumor-bearing animal models documenting significant tumor growth delay and cures in a wide variety of solid tumor types.112–115 Tumor doses often exceeded 10 to 15 Gy after a single infusion.116,117 In some preclinical studies, RIT achieved comparable tumor growth delays as equivalent doses of single-fraction or conventionally fractionated external beam radiation therapy116–122 or equitoxic doses of chemotherapy.123 Initial clinical trials evaluating biodistribution and pharmacokinetics confirmed tumor targeting and demonstrated that therapeutic doses of radiation to tumor were potentially achievable.124–129 However, RIT in solid tumors has not realized the same results in the clinic as had been predicted from preclinical studies. Table 22-2 provides an overview of Mabs that have been evaluated in clinical trials. RIT has been evaluated in most major disease sites, including gastrointestinal, breast, brain, ovarian, head and neck, medullary thyroid, melanoma, renal, lung, and prostate cancers. Results from selected solid tumor trials evaluating systemically administered nonmyeloablative doses of RIT as a single modality are summarized in Table 22-3. Early efforts used Mabs labeled with 131I because of availability and ease of labeling, with subsequent trials using radiometals such as 90Y, rhenium-186 (186Re), and 177Lu. Almost all of the antibodies studied have been administered at low protein doses (~5 to 50 mg), have had no significant clinical immunomodulatory or antitumor properties, and have therefore served primarily as a delivery vehicle for radiation. Dose-limiting toxicities have primarily been thrombocytopenia and leukopenia. Results have been comparable regardless of disease site or the particular radionuclide-antibody combination.
TABLE 22-2 Examples of Monoclonal Antibodies Evaluated for RIT
| Malignancy | Antigen | Antibody |
|---|---|---|
| Colorectal cancer | CEA | cT84.66, hMN-14, A5B7, F6 |
| TAG-72 | B72.3, CC49 | |
| A33 | anti-A33 | |
| EpCAM | NR-LU-10, NR-LU-13, 17-1A | |
| DNA histone H1 | chTNT-1/B | |
| Breast cancer | MUC1 | huBrE-3, m170 |
| L6 | chL6 | |
| TAG-72 | CC49 | |
| CEA | cT84.66 | |
| Ovarian cancer | MUC1 | HMFG1 |
| Folate receptor | cMov18 | |
| TAG-72 | B72.3, CC49 | |
| Prostate cancer | PSMA | huJ591, CYT-356 |
| TAG-72 | CC49 | |
| Lung cancer | DNA histone H1 | chTNT-1/B |
| TAG72 | CC49 | |
| Head and neck cancer | CD44v6 | U36, BIWA4 |
| Gliomas | EGFR | 425 |
| Tenascin | 816C, BC4 | |
| Melanoma | p97 | 96.5 |
| Renal cancer | G250 glycoprotein | cG250 |
| Medullary thyroid | CEA | cT84.66, hMN-14, NP-4 |
| Neuroblastoma | Ganglioside GD2 | 3F8 |
| NCAM | UJ13A, ERIC-1 |
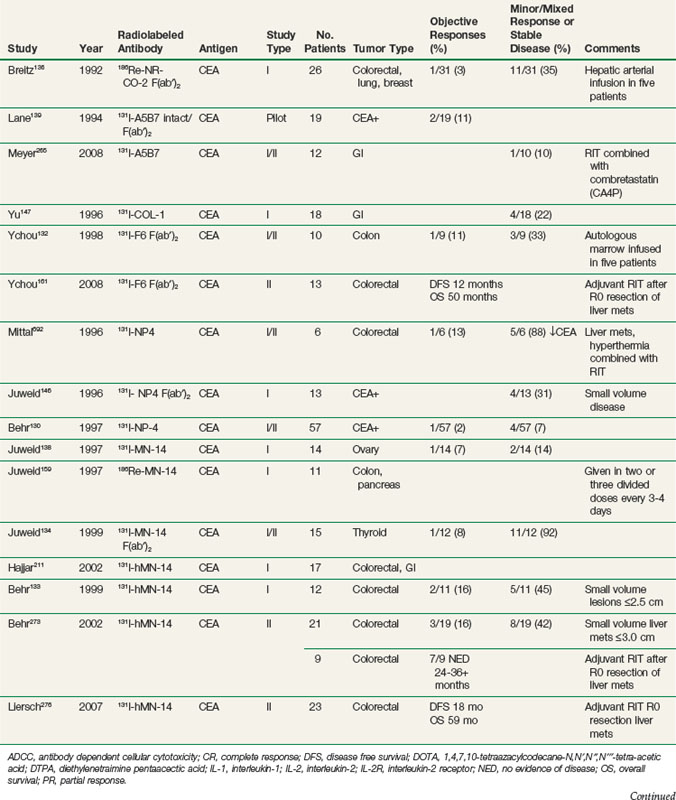
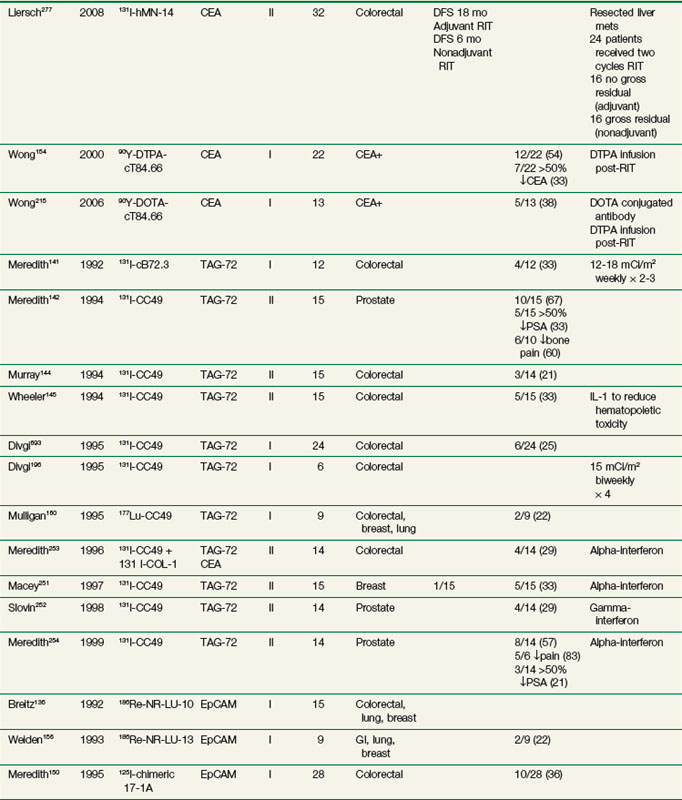
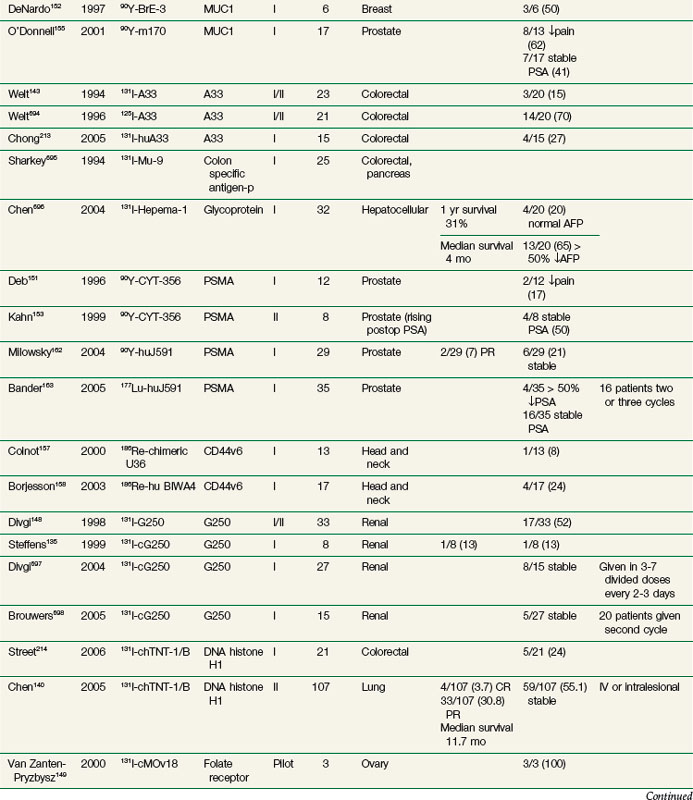
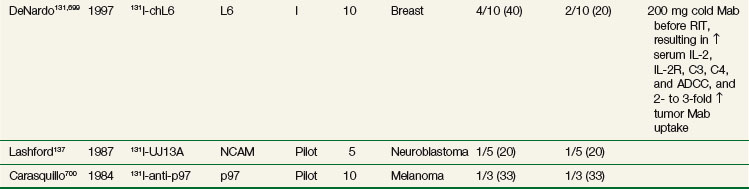
Until recently, most of the clinical experience involved phase I trials, evaluating RIT as monotherapy in patients with advanced, bulky, chemotherapy-refractory disease progressing into therapy. As expected in this patient population, partial and complete responses have been infrequent regardless of disease type,130–140 with most antitumor effects reported as stable disease and serologic, mixed, or minor responses.137,141–163 This is in contrast to clinical results observed with RIT in lymphomas with objective responses ranging from 30% to 85%.164–172
A number of factors have been put forth to explain the limited responses seen. With hematologic malignant disease, at least some of the observed response rates can be attributed to the immunologic effects of the antibody itself,173 which is not the case for most radiolabeled antibodies directed against solid tumors. Other factors involve those that restrict antibody targeting and uptake to tumor more so for solid tumors than for lymphomas. These include differences in tumor physiology, tumor vascularity, antigen accessibility, and heterogeneity of antigen expression.174,175 However, these factors probably play a minor role because the clinical literature demonstrates no significant differences in estimated RIT dose to tumor between solid tumors and hematologic cancers. This suggests that the difference in radiosensitivity between solid tumors and hematologic cancers is the major factor in explaining the difference in RIT response rates.
Table 22-4 compares tumor doses achieved after a single administration of nonmyeloablative activities of RIT for solid tumors and lymphomas and demonstrates that tumor doses are comparable for most antibodies evaluated in the clinic. Median and mean doses are approximately 10 to 20 Gy, with select tumors occasionally achieving doses of 20 to 70 Gy or greater. In our experience with 90Y-cT84.66 anti-CEA RIT, approximately 25% of tumors achieve tumor doses of more than 15 to 20 Gy after a single infusion. These data support the hypothesis that the differences in response rates between solid tumors and hematologic cancers are largely because of differences in radiosensitivity, not the result of differences in tumor antibody uptake. This is even after accounting for effects of unlabeled antilymphoma antibodies, because clinical trials have demonstrated no significant response rates with unlabeled antibodies such as Lym-1.176,177 In the case of 90Y-ibritumomab, a 24% increase in response rate was observed with the addition of radiolabeled antibody compared with unlabeled antibody alone in a recent randomized trial.173 Similarly for tositumomab, there was significant improvement in all therapeutic outcomes by additional 131I-labeled antibody.178
TABLE 22-4 Tumor Doses and Objective Response Rates from Selected Solid Tumor and Lymphoma RIT Trials
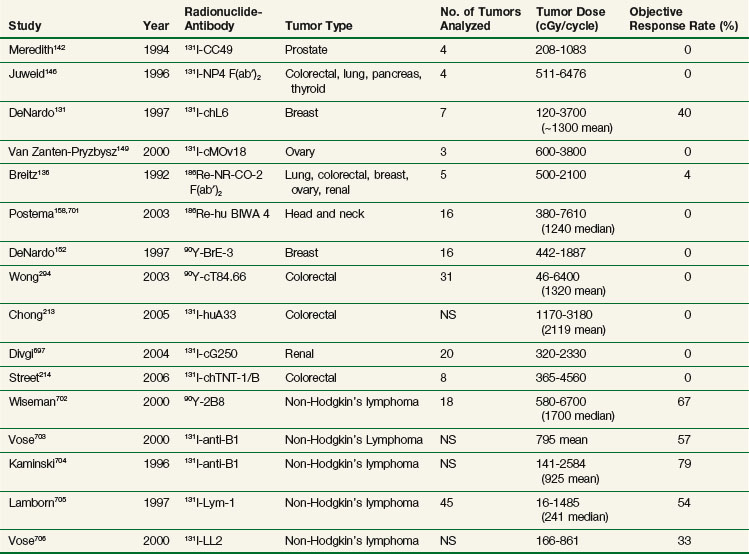
Although tumor doses with RIT are a fraction of what can be delivered by standard teletherapy or brachytherapy, doses currently achievable are at levels that can potentially result in clinically important antitumor effects in solid tumors if applied in the appropriate clinical setting, particularly in patients with subclinical or microscopic disease. With conventionally fractionated external beam radiotherapy, Withers and associates179 reviewed the clinical literature evaluating adjuvant radiation for solid tumors and noted an inverse linear relationship between radiation dose and tumor recurrence rate with doses ranging from 2000 to 5000 cGy. More importantly, there was no threshold effect and doses in the range of 2000 cGy had measurable effects in the adjuvant setting. Clinical trials have also demonstrated important antitumor effects in rectal cancer with doses in the range of 3000 cGy.180,181 Finally, doses as low as 3000 cGy conventionally fractionated have been combined with chemotherapy in esophageal and anal cancer, resulting in pathologic complete responses.182–184 Therefore centiGray doses to tumor achievable through RIT may have more clinical importance in low-tumor burden situations and/or as an additional agent added to a multiagent systemic therapy approach.
Autologous Stem Cell Support (see Table 22-3)
Because hematologic toxicity is dose limiting, groups have explored dose intensification strategies using stem cell reinfusion after myeloablative doses of radiolabeled Mabs. Most have been phase I studies treating a limited number of patients. Stem cell–supported RIT studies primarily in breast cancer have reported objective responses.185,186 Other studies have reported fewer or no objective responses.132,187–190 Stem cell reinfusion has permitted higher myeloablative doses to be administered, but it appears that dose intensification alone will not result in the level of objective response rates initially hoped for and will need to be combined with other optimization strategies.
Fractionation of Administered Activity (see Table 22-3)
Fractionated RIT usually involves administration of the total activity through divided infusions spaced days to a week apart instead of as a single infusion and takes advantage of the differences in uptake and clearance kinetics of activity between tumor and normal tissues, such that with the appropriate fractionation schedule the ratio between tumor dose and marrow dose can be amplified. The optimum schedule therefore may be dependent on a complex interplay of multiple factors, including uptake and clearance kinetics of the antibody construct, decay kinetics of the radionuclide, tumor size, radiosensitivity of tumor and organs, and clearance kinetics of the individual patient. A number of groups have reported in animal models reduced hematopoietic toxicity and greater antitumor effects with fractionation.191–194 Meredith and colleagues141 used a fractionated RIT schedule with 131I-chimeric B72.3 in patients with colorectal cancer who received 28 or 36 mCi/m2 in 2 to 3 weekly fractions of 12 to 18 mCi/m2. There was a modest but statistically significant (p = .04) decrease in hematopoietic toxicity adjusted for whole body dose when compared with a similar population treated in an earlier trial195 who received similar doses of 131I-chimeric B72.3 as a single infusion. Divgi and colleagues196 also evaluated a fractionated schedule with the anti-TAG-72 antibody CC49 radiolabeled with 131I. Six patients with colorectal cancer received 15 mCi/m2 biweekly up to a maximum of four infusions. This schedule was well tolerated with no dose-limiting toxicities observed.
Engineered Immunoconstructs
Molecular engineering of antibodies with properties optimized for therapy continues to be pursued. Reducing antibody immunogenicity to permit multiple administrations has been successful in the clinic. Murine-derived Mabs have the disadvantage of being recognized as foreign by the patient’s immune system, which can lead to the formation of human antimouse antibodies (HAMA) in 50% to 100% of patients after a single administration,197–200 resulting in faster blood clearance of subsequently administered antibody and potentially compromised efficacy.199,201 Reduction in antibody immunogenicity has been observed with the use of immunosuppressive agents such as cyclosporine A.186,202,203 Others have reduced antibody immunogenicity through modification of the antibody molecule using recombinant DNA techniques to replace murine with human antibody domains (e.g., chimeric antibodies, where the murine variable domains are retained, and humanized antibodies, where only the murine complementarily determining region (CDR) antigen recognition sites are retained152,157,158,204–215 (Table 22-5).
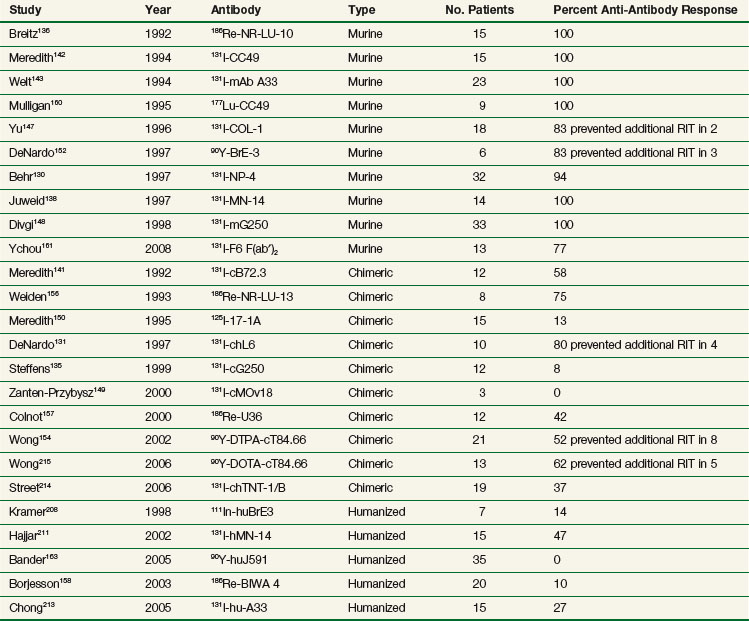
Recombinant antibodies can be designed with improved biodistribution and pharmacokinetic properties to optimize their use for therapy. Antibody fragments and other smaller-molecular-weight antibody constructs may ultimately prove superior for therapy because of their faster clearance, greater tumor penetration,216 more uniform distribution in tumor,217 higher initial dose rate in tumor,218 decreased immunogenicity,219 higher tumor to background ratios,115 and potential for higher administered activities and tumor doses for the same level of toxicity. Initial efforts evaluated radiolabeled Fab and F(ab′)2 fragments generated from whole immunoglobulin through enzymatic digestion132,136,139,146,220 (see Table 22-3). Unfortunately, antitumor effects appeared to be no better than with radiolabeled intact antibody. Although blood clearance may be more rapid with fragments, retention time in tumor is also reduced, resulting in no significant improvement in radiation dose to tumor over the intact antibody for equitoxic administered activities.
More recent efforts have used recombinant DNA technology to genetically engineer antibody constructs with properties to improve tumor to normal organ biodistribution, enhance in vivo stability, reduce immunogenicity, aid in conjugation and radiolabeling, and increase clearance kinetics. Constructs have ranged from approximately 25 to 160 kDa in size. The genes encoding the antibody’s variable light (VL) and variable heavy (VH) domains can be cloned and linked by a contiguously encoded peptide into a single-chain (sc) Fv construct. This small monovalent scFv, 28 kDa, clears rapidly from the circulation, increasing the tumor/blood ratio, making it attractive as a potential imaging agent.221,222 However, the absolute peak uptake and retention time in tumors are often reduced, limiting its use as a therapy agent.221–224 In addition, low- molecular-weight constructs can filter through the renal glomeruli, resulting in increased kidney uptake and radiation dose compared with higher-molecular-weight constructs. A more suitable construct for therapy might be bivalent, of intermediate molecular weight, and exhibit uptake and retention in tumor comparable to intact antibodies, but with faster clearance times than an intact Mab, resulting in an improved therapeutic ratio. Multivalent intermediate-molecular-weight constructs have been produced through covalent linking of monovalent fragments.193,223,225–233 Alternatively, single-chain monomers can be engineered with properties that promote spontaneous formation of dimeric and trimeric species. For example, Figure 22-1 shows an engineered series of recombinant fragments derived from the intact anti-CEA T84.66 Mab.234 This cognate family of anti-CEA constructs was radioiodinated and evaluated in animal biodistribution studies. The minibody (80 kDa), formed by the self-association of two scFv-CH3 single-chain monomers, was potentially suitable for therapy and resulted in peak tumor uptakes of over 20% to 30% injected dose per gram, which exceeded that of the diabody and F(ab′)2 and approached that of the intact antibody.235
A pilot imaging trial236 evaluating the biodistribution and tumor targeting properties of the 123I-cT84.66 minibody in 10 patients with colorectal cancer demonstrated faster blood clearance than intact cT84.66.237 To further evaluate its potential as a therapy agent, a follow-up pilot study evaluated 111In-DOTA-cT84.66 minibody, with four of five patients demonstrating tumor imaging. Predicted mean radiation dose to liver was 19.6 cGy, kidney 33.3 cGy, lung 1.5 cGy, and marrow 2.2 cGy per mCi of 90Y. Mean tumor dose was 12.4 cGy (6.8 to 25.1 cGy) per mCi of 90Y. Tumor dose to marrow dose ratio was 5.6, comparable to that seen in murine models, but less than the ratio of 10.5 seen for intact 90Y-DTPA-cT84.66154 and 11.6 for 90Y-DOTA-cT84.66215 seen in previous phase I trials.154,215
Other groups have investigated similar strategies.224,238–242 Forero and colleagues243 recently reported on a 153-kDa 131I-labeled humanized CH2 domain deleted construct (HuCC49ΔCH2) derived from CC49 anti-TAG72. Blood clearance 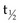 was 20 hours, comparable to cT84.66 minibody. A phase I dose escalation therapy trial in metastatic colorectal cancer was initiated, and preliminary results indicate reduced hematologic toxicity compared with intact murine 131I-CC49. At the initial dose level of 75 mCi/m2, only 25% of patients (one of four) developed hematopoietic toxicity higher than grade 3, compared with more than 60% of patients with 131I-murine CC49 in a previous trial.244
was 20 hours, comparable to cT84.66 minibody. A phase I dose escalation therapy trial in metastatic colorectal cancer was initiated, and preliminary results indicate reduced hematologic toxicity compared with intact murine 131I-CC49. At the initial dose level of 75 mCi/m2, only 25% of patients (one of four) developed hematopoietic toxicity higher than grade 3, compared with more than 60% of patients with 131I-murine CC49 in a previous trial.244
Modifying the Tumor Environment (see Table 22-3)
Up-regulation of tumor antigen expression as a means of increasing antibody targeting and radiation dose to tumor has been explored. Alpha and gamma interferon have been shown to increase CEA and TAG-72 expression,245–248 resulting in increased tumor antibody uptake and RIT efficacy in animal models.249,250 In the clinic, interferon has also increased tumor antigen expression and antibody uptake251–254 in breast, colorectal, and prostate cancer. However, antitumor effects remain modest with this strategy alone, leading investigators to now combine this approach with other optimization strategies.
Strategies to modify the tumor vasculature to increase blood flow or vascular leakage, using vasoactive agents, vasoactive immunoconjugates, radiation therapy, or hyperthermia, have been explored primarily in experimental models.255 Interest has focused on an emerging class of agents directed against the tumor neovasculature. Jain and colleagues256,257 and others have proposed using anti-angiogenic agents, such as anti-VEGF (vascular endothelial growth factor) antibodies, to “prune” or normalize the tumor vasculature, decrease tumor interstitial pressure, and increase tumor perfusion, which should increase macromolecule delivery. In some experimental models, tumor oxygenation is also increased,257 which can further enhance the antitumor effects of RIT. In a similar strategy, Burke and associates258 have looked at using cyclic RGD peptide, which recognizes the ανβ3 integrin receptor on tumor neovasculature. In a tumor-bearing breast cancer mouse model, the combination of RGD peptide and 90Y-chimeric L6 RIT resulted in higher tumor uptake of antibody and a greater percentage of mice cured than either therapy alone.258,259 In colorectal cancer tumor–bearing mice, Kinuya and colleagues260 combined the anti-angiogenic agent 2-methoxyestradiol with 131I-A7 RIT and demonstrated prolonged survival with the combination compared with either agent alone. Recently, Bodet-Milin and colleagues261 combined 131I-F6 anti-CEA RIT and bevacizumab (anti-VEGF) in medullary thyroid cancer–bearing mice and reported an increase in tumor growth delay with the combination compared with either agent alone.
Agents that increase vascular permeability also have been evaluated. A response-selective peptide agonist to human C5a resulted in an increase in 131I-B72.3 anti-TAG72 Mab and a twofold increase in colon cancer xenograft growth delay in vivo.151,262 The vascular disrupting agent combretastatin A-4-phosphate (CA4P) has been shown to increase tumor retention of 131I-AB7 anti-CEA antibody, resulting in an increased radiation dose to tumor263 and a significant increase in tumor growth suppression compared with 131I-AB7 RIT alone.264 A phase I/II trial was initiated and preliminary results recently reported.265 Twelve patients received 1311-A5B7 anti-CEA Mab given on day 1 and CA4P on days 2 and 3. Dynamic contrast-enhancing magnetic resonance imaging (MRI) demonstrated reduction in tumor vascular kinetics in 9 of 12 patients. Dose-limiting hematopoietic toxicity was observed at 131I-A5B7-administered activities of 43.2 mCi/m2 and CA4P of 54 mg/m2. Of 10 evaluable patients, 1 had stable disease.
Minimal Tumor Burden Setting (see Table 22-3)
Tumor size plays a dominant role in influencing antibody uptake, and clinically important outcomes are predicted if systemic RIT is applied in the subclinical or microscopic disease setting. Jain and colleagues216,266 have identified key physiologic factors that limit uptake of macromolecules. These factors include spatial heterogeneity of tumor vascularity, increased interstitial pressure in poorly vascularized areas, and limited diffusion distances of macromolecules, which work to significantly limit the ability of the antibody to reach all sites within the tumor. These factors are amplified as the tumor grows, resulting in an exponential decrease in tumor antibody uptake that has been demonstrated in vivo267–270 and in clinical trials.271,272 Given this inverse exponential relationship, a very modest reduction in tumor size will result in a substantial increase in antibody uptake.
RIT should therefore have its greatest impact in the adjuvant setting and in the treatment of minimal or microscopic disease. For example, Behr and colleagues133 and Vogel and associates194 successfully demonstrated improved survival rates and prevention of liver metastases in animal models with RIT delivered with adjuvant intent. Subsequent clinical trials evaluated this strategy in patients with small-volume disease. Behr and colleagues133 reported initial phase I trial results in 12 colorectal cancer patients with metastatic lesions of 2.5 cm or less. Patients received a single administration of 50 to 70 mCi/m2 131I-hMN14 anti-CEA RIT. Of 11 evaluable patients, 2 partial responses and 5 minor/mixed responses of up to 12 months’ duration were observed (see Table 22-3). This was followed by a phase II study of 30 patients with colorectal cancer and liver metastases who received a single administration of 131I-hMN14 anti-CEA RIT (60 mCi/m2).273 Of 19 evaluable patients who had small-volume disease of 3 cm or less, 3 patients experienced a partial response of 3 to 15 months’ duration and 8 patients had a minor response of 3 to 14 months’ duration. Five patients received a second cycle at time of progression at 8 to 16 months, which resulted in partial responses in two patients. In addition, nine patients received RIT for microscopic disease similar to the results in an adjuvant setting after a complete resection of hepatic metastases. Seven of nine remained disease free at more than 24 to 36 months, which compared favorably with historical controls at the same institution receiving 5-FU-based regimens following hepatic resection (see Table 22-3). These initial encouraging observations led to a phase II adjuvant RIT trial of 131I-hMN14 administered after resection of colorectal cancer liver metastases274,275 (see Table 22-3). In the most recent report, 23 patients who underwent R0 resection of liver metastases from colorectal cancer received a single administration of 40 to 60 mCi/m2 of 131I-hMN14 anti-CEA.276 Results were compared with those of a contemporaneous control group of 19 patients from the same institution treated following resection with 5-FU-based chemotherapy regimens. Median follow-up for the RIT group was 91 months and for the control group was 51 months. A statistically significant improvement in median overall survival rates in the RIT group (58 months vs. 31 months) was reported. Median disease-free survival time was greater for the RIT group (18 months vs. 12 months), but the difference did not reach statistical significance. The investigators concluded that a prospective randomized trial to evaluate 131I-hMN14 as adjuvant therapy in this patient population was warranted given the suggestion of a survival advantage. This same group recently reported preclinical results of an ongoing trial evaluating the feasibility of administering two courses of RIT after resection of liver metastases in patients with colorectal cancer.277 Thirty-two patients received a dose of 40 to 50 mCi/m2 131I-hMN14 anti-CEA, with 24 receiving a second RIT administration. Sixteen patients had no residual disease detectable after surgery and comprised the group that received adjuvant RIT. With follow-up of 21 months, disease-free survival time was 18 months for the adjuvant group and 6 months for the nonadjuvant group. The authors concluded that a second cycle of RIT is feasible with acceptable toxicity. A future randomized trial is planned in the same population that will compare two cycles of RIT versus RIT and chemotherapy as adjuvant therapy.
An adjuvant RIT trial in colorectal cancer was recently carried out by Ychou and colleagues,161 who administered a preoperative “diagnostic” dose of 131I-F6 F(ab′)2 anti-CEA (8 to 10 mCi) before planned surgery to 22 patients with one to four liver metastases. After complete resection (R0), 13 patients, including 10 who had tumor to liver uptake ratios of more than 5, received 180 to 200 mCi of 131I-F6 F(ab′)2 anti-CEA RIT. With a median follow-up of 127 months, median disease-free survival time was 12 months and median overall survival time was 50 months. One patient remained disease free at 93 months. The investigators concluded that RIT is feasible after surgery in this poor prognostic group and that RIT combined with chemotherapy should be further evaluated in this clinical setting.
At City of Hope, a recently completed phase I trial evaluated the feasibility and toxicities of 90Y-cT84.66 anti-CEA RIT combined with hepatic arterial fluorodeoxyuridine (FUdR) and systemic gemcitabine in colorectal cancer patients after resection and or radiofrequency ablation of liver metastases. No dose-limiting toxicities were observed in 13 patients with FUdR of 0.1 to 0.2 µg/kg/day for 14 days, gemcitabine 105 mg/kg, and RIT 16 .6 mCi/m2. Four patients remain progression free at over 4 to 37 months (see Table 22-3).