Visit the Endocrine Hypertension: From Basic Science to Clinical Practice , First Edition companion web site at: https://www.elsevier.com/books-and-journals/book-companion/9780323961202 .



Introduction
To the patient with hypertension, the detection of an underlying, specifically treatable endocrine cause can have a profound beneficial impact. Correction of the causative entity may lead to cure or at least improvement (often marked) in hypertension control, resulting in reduced pill burden, morbidity and even mortality and improved quality of life. Furthermore, adverse consequences of hormone excess which are not necessarily dependent on blood pressure elevation may also be averted. In addition, simply solving the mystery as to why a patient has had hypertension can be a considerable comfort and relief to them. For all these reasons, the majority of hypertensive patients who are offered the opportunity to undergo screening tests for secondary endocrine causes will readily consider it. Of course, this needs to be balanced against factors such as prevalence, costs of screening, and likelihood of beneficial outcomes. For this reason, the case for widespread screening among hypertensive patients is greater for some endocrine conditions than others. For example, a growing number of investigators have advocated screening for primary aldosteronism in the great majority, and possibly all newly diagnosed hypertensives as it is common, screening requires an inexpensive blood test (the plasma aldosterone/renin ratio; ARR) and specific management has well-documented highly beneficial sequelae. Thyroid disorders and hyperparathyroidism are also common and are routinely screened in most medical settings. Greater selectivity is advocated for renovascular hypertension, in which the prevalence is somewhat less, and screening involves more costly imaging approaches, including renal artery duplex ultrasonography (which, in addition, requires substantial expertise to obtain accurate results)
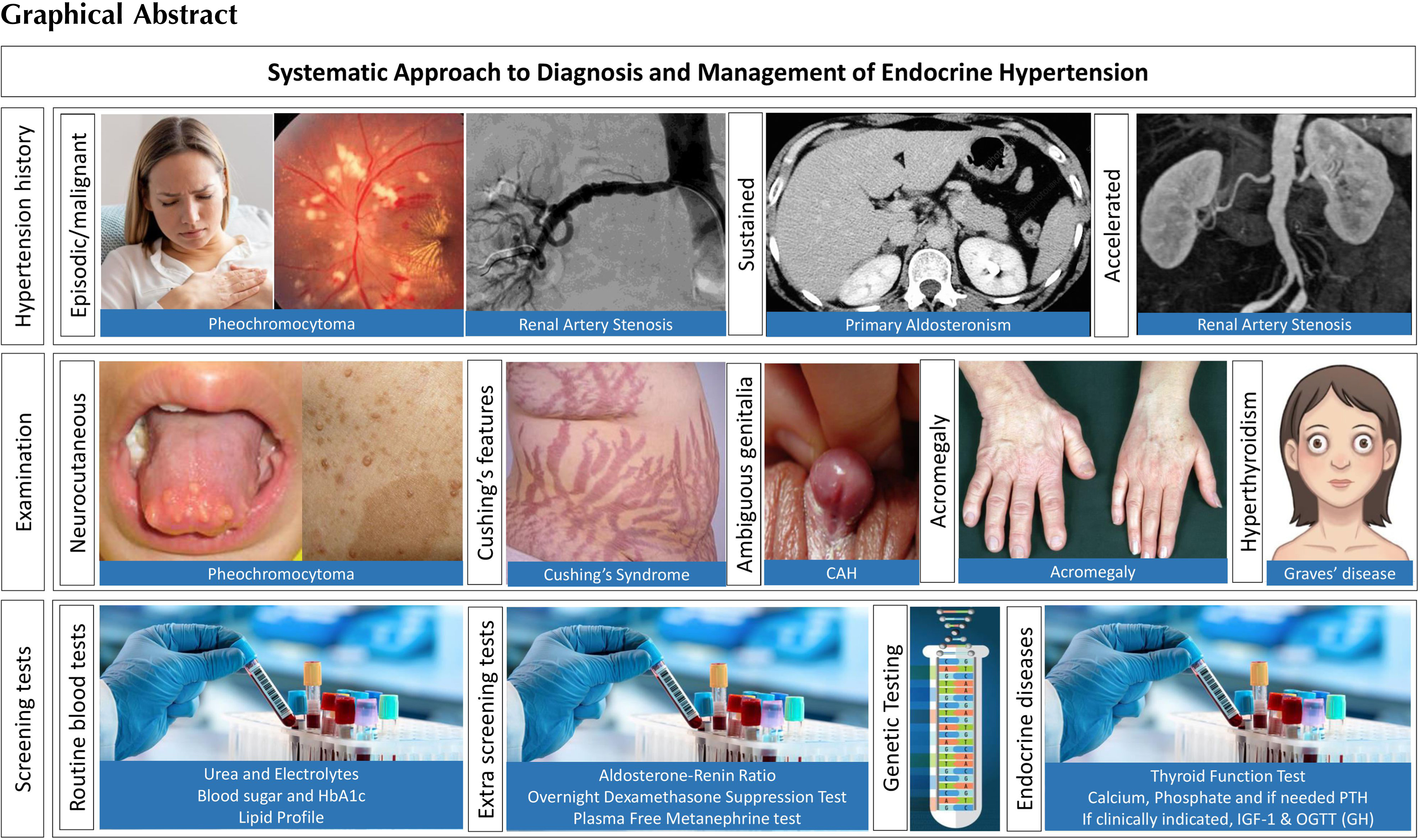
This chapter will attempt to provide an approach toward the diagnosis and management of various forms of endocrine hypertension with a focus on practical clinical aspects.
Primary aldosteronism (PA)
Clinical diagnosis
Symptoms and signs of PA
Almost all patients with PA are hypertensive. Rare exceptions include occasional normotensive (yet genetically positive) members of some families with inheritable forms [ , ], and others in whom PA has been heralded by hypokalemia or adrenal incidentaloma rather than hypertension [ ]. Hypokalemia is present in approximately 20%–25% of patients [ , ] and may be accompanied by muscle weakness, cramping, tetany, paresthesias, palpitation, polyuria, and nocturia, the latter of which often occurs even when plasma potassium is normal. Other common symptoms in patients with PA include lethargy, impaired mental concentration, and mood disturbances [ ].
Although uncommon, malignant hypertension can occur in PA, and the accompanying intrarenal ischemia may lead to renin levels in the unsuppressed range, which could result in the diagnosis of PA being missed [ ].
During pregnancy, hypertension and biochemical manifestations of PA may be masked [ , ] due to the high circulating levels of placental progesterone (a mineralocorticoid receptor [MR] antagonist). Conversely, in other women, PA due to aldosterone producing adenoma (APA) may develop during pregnancy possibly due to (i) the presence of somatic activating mutations in CTNNB1 (encodes β-catenin in the Wnt cell-differentiation pathway and may explain tumor development) and (ii) aberrant receptor activation of LHCGR (encoding lutotropin-chorionogonadotropic hormone receptor) and GNRHR (encoding gonadotropin releasing hormone receptor) within these APAs by high levels of human chorionic gonadotropin hormone, leading to stimulation of aldosterone production [ ].
The importance of accurate assays in the diagnostic workup of PA
Highly reproducible assays are essential for all stages of the diagnostic workup of PA, including screening, confirmatory suppression testing, and adrenal venous sampling (AVS).
Concerns have been raised regarding the accuracy and reproducibility of immunoassays for serum or plasma aldosterone (which circulates at low, picomolar concentrations) and especially about the degree of specificity of the antibodies used and the need for meticulous laboratory technique. Because of this lack of specificity, newer highly accurate and reproducible methods of measuring aldosterone using high-performance liquid chromatography and tandem mass spectrometry (LC-MS/MS) generate aldosterone levels within paired samples that are substantially lower than those measured by immunoassays [ , ], necessitating adjustment of diagnostic thresholds such as the ARR and confirmatory suppression testing [ , ]. The risk of overestimation of serum or plasma aldosterone with immunoassay techniques becomes greater when concentrations are at the lower end of the reference range, presumably because other steroids which cross-react with the aldosterone antibody would be expected to make up a greater proportion of the total amount of steroid that is bound. This is particularly relevant for confirmatory suppression testing, during which “true” aldosterone concentrations in patients without PA fall to very low values, but when measured by an immunoassay with poor specificity performance characteristics, may demonstrate apparent failure to suppress (i.e., a false-positive test). The use of semiautomated technology allows high sample throughput, rapid generation of assay results, and relatively low running costs, making them suitable for the clinical setting. The initial cost outlay associated with purchasing the equipment continues to represent a barrier to wide application of LC-MS/MS but given the savings that are likely to occur through more accurate diagnosis of PA (i.e., with lower rates of false positive ARR and confirmatory suppression testing results leading to fewer unnecessary confirmatory tests and AVS procedures respectively), this situation may gradually change with time.
Accurate measurement of renin is critical for detection of PA as it has a profound influence on the ARR with small changes in absolute renin concentrations resulting in potentially large variations in the ratio [ ]. Renin is most commonly measured as plasma renin activity (PRA) or of direct (or active) renin concentration (DRC). While PRA measures the amount of angiotensin I generated by the action of renin on its substrate, angiotensinogen, in a defined volume of plasma per unit of time and hence is a measure of renin enzyme activity, DRC measures the plasma concentration of the active (cleaved) form of renin. The main advantage of DRC is that it can be undertaken by automated immunometric assay [ ], which is much faster and less labor intensive than PRA (which uses immunoassay or occasionally LC-MS/MS to measure angiotensin I). However, as with aldosterone assays, DRC assays tend to lack reliability and reproducibility at the lower end of the reference range, which is especially pertinent for PA in which renin levels are suppressed [ ]. In the case of PRA, on the other hand, for samples with levels less than 1 ng/ml/min, reproducibility of the assay can be maintained by extending the incubation time from the recommended 90 min to 18 h [ ]. Use of DRC (but not PRA) is associated with higher ARR values with the potential for false-positives during the luteal phase of the menstrual cycle or in women receiving estrogen-containing oral contraceptive or hormone replacement preparations, possibly because the concentration of angiotensinogen (substrate) increases in response to higher estrogen, and DRC falls to maintain angiotensin II concentrations in the normal range, whereas PRA remains relatively unaffected [ , ]. Automated immunometric renin assays (DRC) have, however, been reported to have better interlaboratory agreement than PRA [ ].
A promising alternative to measuring renin is the mass spectrometric measurement of angiotensin II (during a period of equilibrium of formation vs. metabolism) [ ] which is arguably more relevant in clinically assessing patients for the presence of angiotensin II-independent aldosterone overproduction given it is the major direct physiological hormonal modulator of adrenal cortical aldosterone synthesis (unlike renin).
Screening for PA
Screening should be considered in most patients with hypertension, and especially those with moderate to severe forms, spontaneous or diuretic induced hypokalemia, adrenal incidentaloma, sleep apnea, or a family history of early onset hypertension or cerebrovascular accident at a young age (<40 years), and all hypertensive first degree relatives of patients with PA [ ]. It is no longer deemed appropriate to restrict screening to hypertensive patients with hypokalemia as this would result in up to 80% of patients with PA being missed [ ]. Some investigators suggest screening of all hypertensives [ ], ideally at the time of first diagnosis. Screening before commencement of antihypertensive medications avoids potentially confounding effects of these agents on renin and aldosterone levels and permits earlier institution of specific treatment with better outcomes [ ].
At present, the most popular method of screening for PA involves measurement of the ARR. Demonstration of frankly elevated plasma aldosterone levels lacks sensitivity [ , ]. “Normal” aldosterone levels are commonly seen in patients with PA and should be viewed as “inappropriately normal” in the face of renin suppression which, in individuals without PA, should be accompanied by low aldosterone levels. Elevated aldosterone levels in isolation (i.e., without knowledge of the renin level) also lack specificity as they may be secondary to activation of the renin-angiotensin system (secondary hyperaldosteronism) due, for example, to treatment with diuretics or renovascular hypertension. Suppressed renin levels lack specificity, in that they are also observed in patients receiving beta-adrenergic or nonsteroidal antiinflammatory agents, ingesting a high dietary salt intake, of advanced age, or with chronic kidney disease or other varieties of low renin hypertension such as Liddle syndrome, hypertensive forms of congenital adrenal hyperplasia, familial hypertension and hyperkalemia (Gordon syndrome), the syndrome of apparent mineralocorticoid excess, ectopic adrenocorticotropin (ACTH) syndrome, deoxycortisone-secreting tumor or glucocorticoid resistance states. The ARR is much more sensitive for detection of PA than plasma potassium or aldosterone measured in isolation, and more specific than isolated renin, in that aldosterone is also suppressed is most other salt-dependent low-renin forms of hypertension and hence the ARR is not elevated.
False-positive and false-negative ARRs can result from a variety of physiological and pharmacological factors ( Table 21.1 ), an appreciation of which aids substantially in the optimal use and interpretation of the ARR. The most common of these are antihypertensive medications, with beta-adrenoceptor blockers, clonidine, and alpha-methyldopa having the potential to cause false-positives, and diuretics (including potassium-sparing), angiotensin converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARBs) and dihydropyridine-type calcium channel blockers (CCBs) prone to cause false-negatives [ , ].
| Category | False positives | False negatives |
|---|---|---|
| Antihypertensive medications | Beta-adrenoceptor blockers Clonidine α-Methyldopa | Diuretics (including potassium-sparing) ACE inhibitors Angiotensin receptor blockers Dihydropyridine CCBs |
| Other medications | NSAIDs Estrogen-containing OCPs a Estrogen-containing HRT a | SSRI inhibitors |
| Clinical conditions | Chronic kidney disease Familial hyperkalemic hypertension | Malignant hypertension Renovascular hypertension Vomiting and diarrhea Hypokalemia |
| Physiological factors | High dietary sodium intake Luteal phase of menstrual cycle a Advanced age | Low dietary sodium intake |
a Only if renin is measured as direct renin concentration and not as plasma renin activity.
Where possible, antihypertensives known to affect the ARR should be withdrawn (for at least 4 weeks for diuretics and at least 2 weeks for other agents) prior to screening. Antihypertensive medications which appear to have minimal, if any, effects on the ARR and can be used to control hypertension (where necessary) during screening and subsequent steps in the diagnostic workup of PA include [ ]:
- –
verapamil slow release: suggested starting dose (SSD) ½ × 240 mg tablet twice daily
- –
hydralazine (best used in combination with verapamil SR to avoid reflex tachycardia): SSD ½ × 25 mg tablet twice daily
- –
prazosin: SSD ½ × 1 mg tablet twice daily
- –
moxonidine: SSD 200 μg one table at night.
ARR testing while patients are still on interfering medications can still be informative. For example, a normal ARR in a patient on a beta-blocker (without other interfering medications) would make PA highly unlikely, whereas a raised ARR with suppressed renin in a patient receiving an ACE inhibitor, ARB or diuretic (but not a beta-blocker) would be highly suggestive of PA.
The ARR has greater sensitivity if measured in the morning in an upright (e.g., seated) posture [ ]. Cut-off values vary from laboratory to laboratory and depend, at least in part, on the assays used and the units in which aldosterone and renin are expressed. Because the ARR is mathematically highly renin-dependent, then at extremely low levels of renin (e.g., 1.5 mU/L for DRC or 0.1 ng/mL/h for PRA) the ratio may be elevated even if plasma aldosterone is very low (e.g., 110 pmol/L or 4.0 ng/dL) and clearly not compatible with PA. Under those circumstances, it is prudent to take the absolute aldosterone level into consideration (rather than simply the ratio alone) when deciding if to proceed to confirmatory suppression testing. The cut-off aldosterone level in this situation is a matter of debate. The figure used by the Endocrine Hypertension Center in Brisbane is the cut-off used for normal suppression at the conclusion of the seated saline suppression test (162 pmol/L, when measured by LC-MS/MS).
Controlling for factors that can affect the ARR or at least taking them into account enhances its usefulness in helping select patients for further diagnostic workup. The ARR also shows good within-patient reproducibility under such circumstances [ ] but not when potentially interfering factors are poorly controlled [ ]. Either way, the ratio should still be regarded as a screening test only and should be measured more than once before deciding whether or not to go on to confirmatory testing for PA.
Confirmatory testing for PA
The main purpose of confirmatory testing is to identify patients who, by testing positive, are definitively confirmed as having PA and are therefore candidates for undergoing AVS to determine the subtype; and conversely, to spare patients who test negative (and in whom the diagnosis of PA has therefore been excluded) from having to undergo this invasive and relatively expensive and difficult procedure.
In most cases (apart from the captopril challenge test), confirmatory testing involves measuring aldosterone during dynamic procedures designed to bring about complete suppression of circulating renin, and therefore of angiotensin II, aldosterone’s normal chronic regulator. Failure to suppress aldosterone confirms autonomous aldosterone production that is relatively autonomous of angiotensin II, which is PA by definition.
The most common confirmatory tests currently in use are oral sodium loading, saline infusion, and captopril challenge testing. Fludrocortisone suppression testing (FST), although regarded by some investigators to be the most reliable, is relatively labor-intensive and requires a five-day admission to hospital and hence, is rarely performed nowadays. During FST, plasma aldosterone is measured during 4 days oral administration of fludrocortisone (0.1 mg 6 hourly) and oral salt loading (by high sodium diet and administration of slow-release sodium chloride tablets, 1800 mg three times daily with meals) [ ]. Plasma potassium is maintained in the normal range by adequate slow release KCl supplementation (given 6 hourly). The FST is considered diagnostic of PA if upright (seated) 10:00 h plasma aldosterone fails to suppress to less than 6 ng/dL (165 pmol/L) when measured by immunoassay (4.8 ng/100 mL or 133 pmol/L by LC-MS/MS) at the conclusion of the test, provided that renin has been sufficiently suppressed (a prerequisite of all forms of suppression testing that involve salt loading).
For the oral salt loading test, patients are instructed to consume sufficient dietary salt to achieve a urine sodium excretion of over 200 mmol/day and are given adequate KCl supplementation to maintain normokalemia. A 24 h urinary aldosterone concentration of over 12 μg/day (33 nmol/day) on the third day is regarded as diagnostic [ ].
The saline infusion test (or saline suppression test, SST) involves intravenous administration of 0.9% saline (usually 2 L over 4 h) with measurement of plasma aldosterone at the end of the infusion. Originally, this was performed while patients were maintained in the recumbent position (unlike FST). Concentrations regarded as diagnostic for PA have varied from >5 to >10 ng/dL (>140 to >280 pmol/L) [ , ]. A major advantage of this approach over FST is that it can be performed as an outpatient procedure. However, recumbent SST lacks sensitivity, especially in patients in whom plasma aldosterone demonstrates responsiveness to the assumption of upright posture as their recumbent aldosterone levels are lower than those in posture-unresponsive patients [ ]. Performing SST in the seated position has been reported to result in markedly improved sensitivity for detecting PA while retaining high specificity [ ]. The diagnostic cut-off for normal suppression of plasma aldosterone is < 5.8 ng/dL (<162 pmol/L) if measured by mass spectrometry [ ]. A gray zone of between <6.2 ng/dL (<171 pmol/L) and 7.8 ng/dL (217 pmol/L) has been reported for plasma aldosterone measured by immunoassay [ ]. However, a more recent study comparing aldosterone concentrations measured by mass spectrometry and immunoassay in the same individuals undergoing seated SST has revealed a concerning rate of overdiagnosis of PA by immunoassay using these cut-off values, most likely due to the lack of specificity of the assay for aldosterone, which becomes particularly problematic at the lower end of the reference range [ ]. This has intensified calls for replacing immunoassay with mass spectrometry in laboratories offering clinical endocrine hypertension diagnostic services.
Captopril challenge testing involves measuring plasma aldosterone and renin basally and 1 or 2 h after administering 25–50 mg oral captopril, an ACE inhibitor, with the patient remaining seated throughout [ ]. In subjects without PA, plasma aldosterone is suppressed (>30%) and PRA stimulated by captopril and, as a result, the ARR falls. In one commonly used protocol, the cut-off for normal suppression of plasma aldosterone is < 8.5 ng/dL (240 pmol/L) and for the ARR is < 30 (with aldosterone expressed as ng/dL and PRA as ng/mL/h) 2 h after receiving 50 mg of captopril. Because it does not involve salt loading, the captopril challenge testing avoids the potential for inducing both hypokalemia and fluid overload in patients at risk because of compromised cardiac or renal function. However, its discriminatory power has been questioned [ , ] and symptomatic hypotension can occur as a result of the blood pressure-lowering effect of captopril in upright patients.
Confirmatory testing involving salt loading may be contraindicated in patients with severe hypertension, compromised cardiac status (e.g., reduced left ventricular systolic function, marked left ventricular hypertrophy or high risk of atrial fibrillation) or renal dysfunction because of the enhanced risk of inducing potentially dangerously elevated blood pressure levels, fluid overload, or episodes of atrial fibrillation. In these circumstances, it may be reasonable to proceed directly to subtype differentiation (including AVS) or to consider a trial of medical treatment with an agent that blocks aldosterone action as an attempt to establish better hypertension control and improve cardiac function (either as a temporary measure to reduce the risk of confirmatory testing to a more acceptable level, or as long-term management of PA).
Subtype differentiation
While the morphology associated with PA is diverse, the aim of subtype differentiation from a clinical standpoint is not to attempt to distinguish the many pathological variants from each other, but to assist selection of the optimal management approach. This is achieved by:
- 1.
genetic testing when familial PA is suspected;
- 2.
imaging to exclude large adrenal lesions which may warrant consideration of removal because of their malignant potential and to localize adrenal veins in preparation for AVS;
- 3.
determining whether autonomous aldosterone production is confined to one adrenal, and therefore potentially curable by unilateral adrenalectomy, or bilateral, in which case management usually involves treatment with medications which antagonize aldosterone action.
Of the various familial forms of PA, the strongest case for genetic testing can be made for FH-I (familial hyperaldosteronism type 1; glucocorticoid-remediable aldosteronism). This is because (i) it appears to be by far the most common in terms of familial forms for which a genetic basis has been found (a hybrid CYP11B1 / CYP11B2 mutation); (ii) affected individuals are at risk of severe hypertension and early death from hemorrhagic stroke unless detected and specifically treated; (iii) patients with FH-I demonstrate excellent clinical responses to glucocorticoid medications administered in small doses that do not cause Cushingoid side effects, making them the treatment of choice for this (but no other) subtype of PA; and (iv) detection heralds the need for genetic testing of other family members to identify additional individuals similarly at risk but who will also benefit greatly from timely specific glucocorticoid treatment. Genetic testing of peripheral blood DNA for FH-I involves the use of either Southern blotting or a faster PCR-based method [ , ]. The Endocrine Society recommends testing patients with onset of confirmed PA earlier than at 20 years of age and those who have a family history of PA or of strokes at young age (<40 years) [ ]. Germline mutations in CLCN2 (causing FH-II), KCNJ5 (FH-III), and CACNA1H (FH-IV) have so far been reported much less frequently than hybrid gene mutations and (unlike FH-I) no specific form of treatments are yet available for these conditions. Individuals found to have FH-III due to T158A, I151S, or G151R mutations in KCNJ5 tend to have more florid PA, often requiring bilateral adrenalectomy to control hypertension [ ]. Almost all reported individuals with FH-II, FH-III, and FH-IV have had early age of onset of hypertension, so it would seem reasonable to restrict genetic testing for CLCN2 , KCNJ5, and CANA1H mutations to very young patients with PA.
Adrenal imaging with CT or MRI permits detection of larger (e.g., >3 cm) mass lesions which have greater potential for malignant behavior and may warrant consideration for removal on that basis alone, or at least deserve careful follow-up. When performed with contrast, CT also allows localization of adrenal veins which can assist in successful cannulation at the time of subsequent AVS [ ].
Distinguishing unilateral from bilateral PA is an important aspect of the diagnostic workup of PA as the clinical responses to unilateral adrenalectomy in patients with unilateral forms of PA are generally quite gratifying, with cure of hypertension achieved in 40%–70%, marked reductions in pill burden, and better cardiovascular outcomes and improvements in quality of life compared to patients with PA treated medically with drugs that antagonize aldosterone action [ , ]. AVS remains the most reliable means of differentiating these two PA subtypes but is also the most invasive and expensive test and requires considerable expertise. Clinical prediction algorithms have relied on the general observation that unilateral forms (including APA) are generally associated with more florid biochemical manifestations of PA, but the considerable degree of overlap that exists has meant that algorithms incorporating such parameters as plasma sodium and potassium, total carbon dioxide, aldosterone, renin, and the ARR have shown variable (and not very reproducible) performance characteristics [ ]. Adrenal imaging by CT or MRI frequently misses APAs and can be frankly misleading by detecting nonfunctioning adrenal lesions [ , ]. Scintigraphy using labeled forms of cholesterol may detect large APAs [ ] but lacks sufficient resolution to detect smaller tumors. Nuclear imaging using PET-CT with 11 C-metomidate has been reported to be a viable alternative or adjunct to AVS (e.g., in difficult cases) [ , ]. However, the short half-life and lack of specificity of the isotope for CYP11B2 (being a ligand also for CYP11B1) limits the current widespread application and reliability of this technique though alternatives are being investigated and are awaited with great interest [ ]. Patients with APA more often show lack of responsiveness of plasma aldosterone to upright posture during posture stimulation studies and more often demonstrate elevated plasma or urinary levels of “hybrid steroids” (18-hydroxycortisol and 18-oxocortisol) compared with those with bilateral PA [ , ], but again separation is far from complete. To some extent, the phenotype regarding these two parameters among patients with APA appears to be genetically determined: APAs with somatic KCNJ5 mutations are usually associated with posture-unresponsiveness and elevated hybrid steroid levels and vice versa for somatic CACNA1D (and other) mutations [ ]. Although inferior to AVS, the above indicators can provide useful ancillary information, especially when AVS is unavailable, has been declined for some reason or results have been inconclusive (e.g., because of failure to cannulate one or both adrenal veins). For example, the presence of florid, hypokalemic, posture-unresponsive PA in a young patient with an obvious unilateral mass lesion on CT and elevated hybrid steroid levels (if available) would make APA highly likely.
To whom should AVS be offered and how should it be performed?
The Endocrine Society guideline [ ] recommends AVS for all patients with confirmed PA who desire adrenalectomy and for whom surgery is not contraindicated. The risk of adrenal hemorrhage associated with AVS is low (less than 2% of all procedures) in experienced hands [ ]. Performing AVS in the morning following overnight recumbency means an overnight stay but avoids the confounding effects of changes in posture on aldosterone concentrations and capitalizes on the higher endogenous ACTH levels (compared with later in the day), thereby ensuring that samples are collected during maximal ACTH-induced stimulation of aldosterone production. Arguably, however, these issues (and the need for overnight admission) may also be addressed by the use of exogenous ACTH stimulation, which aims to (i) maximize adrenal/peripheral venous cortisol gradients, (ii) reduce fluctuations in steroid secretion during AVS and (iii) stimulate aldosterone production by APAs and thus avoid sampling during a period of secretion “quiescence” [ ]. Successful cannulation of the adrenal veins (especially the right) is challenging in inexperienced hands but tends to improve with increasing throughput. Measures which can assist in attaining high success rates include performing contrast CT scanning prior to AVS to aid in localizing the adrenal veins [ ], limiting the procedure to a relatively small number of specialty radiology units within each region and radiologists within each unit to ensure high throughput for each proceduralist, collecting at least two samples from each side, and performing rapid point-of care cortisol testing to confirm correct catheter placement for collection [ ]. Cortisol ratios or “gradients” between adrenal and peripheral venous samples of at least two or three (or at least five if exogenous ACTH stimulation is used) are most commonly used to indicate adequacy of AVS. Calculating the aldosterone/cortisol ratio for each adrenal and peripheral venous sample corrects for differences in dilution with nonadrenal venous blood. Criteria for lateralization vary widely from one institution to another [ ] with some comparing adrenal venous aldosterone/cortisol ratios on each side with the simultaneous peripheral venous ratio (with lateralization defined as a the ratio being at least 2.5 times the peripheral on the affected side and no higher than peripheral on the other side), while others rely on the comparison of aldosterone/cortisol ratios on one side versus the other (the so-called “lateralization index”) with lateralization defined as the ratio on the higher side being at least 2–4 times, depending on the center, in comparison to that on the lower side.
When the patient is suspected of having an adrenal tumor that is concomitantly secreting cortisol, the use of cortisol to “normalize” aldosterone levels may bring about misleading results. This is because the higher cortisol levels on the APA side will lead to a lowering of the ipsilateral aldosterone/cortisol ratio, while feedback suppression of ACTH due to excessive cortisol secretion results in suppression of contralateral cortisol production and consequently a rise in the aldosterone/cortisol ratio on the contralateral side, ultimately potentially culminating in loss of lateralization. In these cases, a hormone other than cortisol (e.g., metanephrine) may be used for “normalization” of aldosterone levels, although criteria for lateralization are less well defined in those circumstances [ , ]. Performing an overnight 1 mg dexamethasone suppression test (ODST) helps to identify subjects who may be candidates for measurement of metanephrine in AVS samples and should therefore be considered as part of the routine diagnostic workup of PA.
Management
Although AVS results play a major role in guiding treatment in a patient with PA, management decisions should nevertheless be tailored to the particular characteristics and wishes of the individual patient. For occasional patients who lateralize on AVS, surgical treatment may not be appropriate. Likewise, in rare patients who do not lateralize but in whom medical management has proven unsuccessful for some reason, surgery may be a reasonable consideration. All management options and their possible outcomes should be fully explained and explored with the patient and, if appropriate, their family, before a treatment is chosen.
Surgical management
In centers which use relatively strict definitions of lateralization, approximately 30% of patients with PA who undergo AVS demonstrate clear lateralization [ , , , ]. These patients are candidates for unilateral adrenalectomy (nowadays usually performed laparoscopically, either by an anterior or retroperitoneal approach), which results in cure of hypertension in 50%–60% and significant improvement in the remainder [ , ]. Centers which employ more lenient definitions report higher rates of lateralization but lower cure rates. In the Primary Aldosteronism Surgical Outcome (PASO) study, development of a consensus on criteria defining clinical and biochemical cure or improvement of PA following surgery [ ] permitted a standardized framework for robust comparisons of outcomes according to a variety of potential variables (which in that study varied from <20 to >60%), including the level of experience of the center, the approach used to diagnose PA and/or to differentiate unilateral from bilateral forms, the method used to reverse the cause of excessive aldosterone production (e.g., total vs. partial adrenalectomy, surgery vs. ablation) and so forth.
Other than in a few highly specialized centers that perform “selective” sampling of adrenal vein branches [ ], AVS only indicates which adrenal is excessively producing aldosterone in patients with unilateral PA, and not which part of the gland. It therefore cannot confirm with certainty whether a nodule visualized on CT scanning is actually an APA, rather than a nonsecreting nodule, which happens to be situated in the same gland as a smaller, nonvisualized autonomously secreting lesion. For this reason, the entire adrenal is almost always removed even when a discrete mass lesion is seen on CT.
In rare patients with bilateral forms of PA, inability to control hypertension at maximally tolerated doses of spironolactone (and/or amiloride) may lead to consideration of the option of unilateral adrenalectomy. In those instances, the decision as to which gland to remove should be guided by AVS (with the aim being to remove the gland that has the higher aldosterone/cortisol ratio) or CT (to identify the larger gland if the ratios are similar on both sides). Not surprisingly, the likelihood of benefit is less than with clearly lateralizing PA [ ]. Surgery may also be contemplated in patients with bilateral PA in whom one adrenal contains a large mass (e.g., greater than 3–4 cm) which may warrant removal based on malignant potential. Rarely, patients with marked, bilateral adrenal hyperplasia and severe, bilateral PA (including those with severe forms of FH-III), require bilateral adrenalectomy to control hypertension and biochemical manifestations of PA [ ].
Preoperative treatment for several weeks with an aldosterone antagonist (spironolactone or eplerenone), or epithelial sodium channel blocker (amiloride) is usually associated with a “smoother” peri-operative course in terms of blood pressure and plasma potassium control. Longer periods of such treatment may be advisable in patients with lateralizing PA whose hypertension has been particularly severe and caused substantial hypertensive heart disease in an effort to reduce operative risk by improving cardiovascular function through optimization of hypertension control, repletion of body potassium stores and blockade of direct adverse cardiovascular effects of aldosterone excess.
Immediately following unilateral adrenalectomy for APA, plasma potassium levels should be monitored closely (e.g., at least twice daily for the first 1–2 days and at least daily thereafter until discharge from hospital). Aldosterone production by the remaining adrenal gland may be markedly suppressed because of chronic suppression of renin/Angiotensin II (although this is less likely if the patient has received treatment with an agent that blocks aldosterone action in the weeks leading up to surgery). Because of this, potassium replacement should be withheld during the operation and in the first 24 h postoperatively unless concentrations drop to <3.0 mmol/L, in which case potassium replacement should be given only with extreme caution (at half the usual rate). Again, on the assumption of low aldosterone levels and thus a tendency to urinary salt wasting, postoperative fluids should be given as normal saline 1 L 12 hourly or 8 hourly. Potassium sparing diuretics (spironolactone, eplerenone and amiloride) are withheld just prior to surgery. Some or all other antihypertensives can generally be withheld, and reintroduced over the next few days if required to maintain normotension. In patients still receiving antihypertensives at discharge, gradual withdrawal of medications often continues to occur over the ensuing three to 12 months. In our Center, all operated patients have been cured of hypertension or have required substantially reduced medications following unilateral adrenalectomy.
Simultaneous autonomous overproduction of cortisol and aldosterone by an APA is being increasingly recognized [ ]. If a patient with unilateral PA is suspected of having concomitant autonomous adrenal overproduction of cortisol, not only may the AVS protocol need to be modified (by incorporating measurement of metanephrine or some other hormone other than cortisol as discussed above), but the requirement for steroid cover perioperatively and for a variable period following surgery should be anticipated. For these reasons, there is an argument for performing overnight dexamethasone suppression testing with basal (predexamethasone) ACTH measurement on all patients undergoing diagnostic workup for PA. Failure of serum cortisol to suppress to <1.8 μg/100 mL (<50 nmol/L), accompanied by a low basal ACTH suggests concomitant autonomous adrenal overproduction of cortisol.
When present preoperatively, hypokalemia almost always resolves within days to weeks following unilateral adrenalectomy for unilateral PA. Normalization of the ARR is another reassuring sign that PA has been biochemically “cured.” Blood pressure may take longer (sometimes a year or more) to return to normal but may remain elevated (but always in our experience to a lesser degree than preoperatively), even if PA has been biochemically cured, if the patient has reasons other than PA which are contributing to ongoing hypertension. Serum creatinine usually rises after surgery, and this reflects correction of the volume expanded state and glomerular hyperfiltration (caused by excessive sodium retention) that existed prior to adrenalectomy rather than a deterioration in renal function per se [ ]. Patients within our center are encouraged to undergo suppression testing (previously by FST and more recently by seated SST) several months postoperatively in order to detect any autonomous aldosterone production by the remaining adrenal. By this means of assessment, preoperative lateralization on AVS by our criteria has been associated with biochemical cure of PA in over 90% and significant improvement in all remaining patients postoperatively [ ]. Other reported benefits of surgery have included improvements in left ventricular mass index on echocardiography [ ] and urinary albumin excretion [ ], marked improvements in quality of life [ , , ], reductions in cardiovascular morbidity and reduced mortality [ ].
Medical management
In patients with PA who do not lateralize aldosterone production to one side, or who lateralize but prefer medical to surgical treatment or are unfit for surgery, treatment with specific drugs which block aldosterone action is indicated. These medications usually correct hypokalemia effectively, and potassium supplements should therefore usually be ceased when they are commenced. Overtreatment with these agents can cause volume contraction with prerenal failure, raising creatinine levels, and potentially life-threatening hyperkalaemia [ , ], which is more likely in patients who are taking other potassium-retaining agents such as ACE inhibitors, ARBs, or nonsteroidal antiinflammatory drugs (NSAIDs). They should be used with great caution in patients with existing renal impairment.
Because the onset of action of these agents on blood pressure is slow, several weeks should be allowed to elapse before assessing their full effect on blood pressure at each dose. Small doses (for example, spironolactone 12.5 mg once or twice daily, amiloride 2.5 mg once or twice daily and eplerenone 25 mg once or twice daily) should be initiated [ , ]. At these doses, these drugs are usually well tolerated. If necessary, other antihypertensive agents with more rapid onset of action could be employed during this period, and then later reduced or withdrawn.
Spironolactone, which has a steroidal structure, competitively inhibits aldosterone at the MR. Spironolactone is an inhibitor at the testosterone receptor and an agonist at the progesterone receptor (PR), and its use can therefore be associated with sex-steroid-related adverse effects, including gynecomastia and loss of libido, menstrual irregularities, and aggravation of breast fibrocystic change [ ], the incidence of which are dose-related. At 12.5–50 mg daily, the incidence of gynecomastia is approximately 10%–15% [ , ] while at doses of 150 mg daily or more it exceeds 50% [ ]. Combining spironolactone at lower dose with either amiloride or eplerenone can reduce the likelihood of gynaecomastia and other side effects.
Eplerenone is a newer MR antagonist that appears to be more selective for the receptor than spironolactone, and is, therefore, less likely to produce sex-steroid-related adverse effects [ ]. It may also be less likely to interfere with sexual development in children. Hence, it may be particularly suited for adult patients who have demonstrated intolerance to spironolactone because of sex-steroid-related adverse effects, or in children with PA (including those with FH-I). Eplerenone is already available and being used in clinical practice. However, indications for its use in different countries vary, and in some countries, it is not approved for government-subsidized use in PA. Other, nonsteroidal MR antagonists in varying stages of development and likelihood of entering the clinical arena for the management of PA include esaxerenone and finerenone [ ].
Amiloride (and the less commonly used triamterene) acts directly on the epithelial sodium channel where aldosterone exerts its effects. Because of its less potent aldosterone antagonist action and its much lower propensity to induce adverse effects, amiloride may be particularly suited to patients with milder forms of PA and to patients who are particularly concerned about the possibility of developing the sex steroid-related side effects of spironolactone.
In many patients with PA being treated medically with agents that antagonize aldosterone action, achievement of hypertension control requires the addition of other antihypertensive medications. Because of the propensity to induce or worsen hypokalemia, potassium-wasting diuretics should be generally avoided, other than in occasional patients with reduced glomerular function on aldosterone antagonists as they can help to prevent hyperkalemia in that situation. Otherwise, there is little evidence to favor one class of agents over another in this situation. Both ACE inhibitors [ , ] and calcium channel antagonists [ ] have been shown to lower blood pressure in patients with PA.
Glucocorticoids are highly effective for the long-term treatment of FH-I. In our experience, most patients are able to maintain control of hypertension on doses of dexamethasone as low as 0.125–0.25 mg per day [ ]. Hybrid gene expression is not completely suppressed at these doses, as evidenced by suppressed PRA, elevated ARR and elevated urinary 18-oxocortisol levels and tight correlation of circadian aldosterone with cortisol (rather than PRA) levels [ ]. Our Center’s approach is to use the lowest dose of glucocorticoid treatment required to maintain normal clinic, home and ambulatory blood pressures and normal left ventricular mass index and diastolic function on periodic echocardiographic assessments. Patients also undergo dual energy X-ray absorptiometry (DEXA) scans every 2–3 years to monitor for the development of glucocorticoid-induced osteoporosis. Spironolactone, eplerenone and amiloride are alternative options in the treatment of hypertension in FH-I. Amiloride or eplerenone may be preferable for affected children, since they avoid the potential problems of growth-retardation associated with glucocorticoids and androgen blockade with spironolactone.
Incorporation of dietary salt restriction into the management of patients with PA may help not only to lower blood pressure, but also to limit and even reverse target organ damage and morbidity through non-blood pressure-dependent means. Left ventricular mass [ ] and degree of proteinuria [ ] in patients with PA appear to be dependent on 24 h sodium excretion rate (as a marker of dietary salt intake).
It could be argued that treatment should strive for “complete” reversal of excessive aldosterone action rather than just normalization of potassium and blood pressure levels given that aldosterone excess is now known to have adverse cardiovascular effects that are not dependent on hypertension and hypokalemia alone. Normalization of renin levels may serve to be a better indicator in this sense, since it suggests that the dose of the MR antagonist is adequate to correct the sodium/volume expansion and is therefore providing better long-term protection against aldosterone-induced cardiovascular and renal injury (the development of which appears to be dependent on sodium balance). Indeed, recent studies have suggested better long-term clinical outcomes in medically treated patients with PA in whom renin levels have been maintained in the “unsuppressed” range [ ]. However, the usefulness of monitoring of renin levels is likely to depend on whether the patient is being coadministered other medications (such as ACE inhibitors, ARBs, or beta-blockers) which also affect renin levels. In some patients, tolerability will limit the degree to which the dose of spironolactone can be increased in order to achieve normalization of renin, since even at doses of 12.5–25 mg daily, side effects (such as gynecomastia in males and menstrual irregularity in females) are not uncommon. These are not seen during treatment with amiloride or eplerenone, which therefore represent options for alternative, or additive (spironolactone “dose-sparing”), treatment in such circumstances. The serious risk of developing hyperkalemia and uremia with any agent blocking aldosterone is dose-dependent, so that initial doses should be low and only gradually increased.
Measurement of renin can also be helpful in guiding changes in management when hypertension has not yet become optimally controlled on mineralocorticoid antagonist treatment (provided, again, that the patient is not receiving other medications that are affecting renin levels). For example, if renin levels have already become “unsuppressed,” it may be better to commence or increase the dose of other antihypertensive medications rather than increasing aldosterone blockade.
Aldosterone synthase inhibitors are in development and hold some promise as alternative treatment approaches for PA, but more information is required to confirm their efficacy and safety in treating PA. Remaining challenges include lack of specificity for aldosterone synthase (with evidence, for example, of reduced cortisol synthesizing capacity) and inferior treatment effect when compared to MR antagonism [ ].
(For further reading about PA: refer Chapter 7 ).
Other mineralocorticoid forms of hypertension
Common to the conditions described in this section is hypertension caused by excessive sodium resorption via the epithelial sodium channel (ENaC) in the distal tubule, due to either:
- •
excessive production of mineralocorticoids other than aldosterone, leading to excessive MR activation (as in congenital adrenal hyperplasia due to 11β-hydroxylase and 17α-hydroxylase deficiency, Chrousos syndrome, and deoxycorticosterone secreting tumours)
- •
excessive activation of the MR in the absence of mineralocorticoid excess (as in syndrome of apparent mineralocorticoid excess or Geller syndrome) or
- •
excessive activity of ENaC in the absence of MR overactivation (as in Liddle syndrome).
Those with ectopic ACTH syndrome would have excessive production of both deoxycorticosterone and cortisol causing MR activation (both 1 st and 2 nd mechanisms). In all these conditions, as in PA, circulating levels of renin (measured as PRA or DRC) are suppressed as a result of sodium retention and there is urinary potassium and hydrogen ion wasting which can lead to hypokalemia and alkalosis. However, unlike in PA, circulating levels of aldosterone are suppressed and the ARR is not elevated.
11β-hydroxylase deficiency
Clinical diagnosis
After 21-hydroxylase deficiency, 11β-hydroxylase deficiency is the second most frequent cause of congenital adrenal hyperplasia (5%–8%) but it is a rare cause of mineralocorticoid hypertension. 11β-hydroxylase deficiency is transmitted in an autosomal recessive fashion, being caused by loss-of-function mutations affecting both alleles of CYP11B1 which lead to partial to complete loss of 11β-hydroxylase enzyme activity [ , ]. The resulting failure of the normal conversion of 11-deoxycorticosterone (DOC) to corticosterone and of 11-deoxycortisol to cortisol [ ] leads to stimulation of ACTH release, which results in adrenocortical hyperplasia and an increase in DOC production causing mineralocorticoid hypertension and increased adrenal androgen production leading to hyperandrogenism [ , ]. Severity varies markedly among affected individuals, with virilization ranging from mild to marked and hypertension from absent to severe [ , , ].
In females, the diagnosis may be suspected because of masculinization of the external genitalia at an early age [ , ], but normal internal reproductive organs. Acne, hirsutism, rapid somatic growth during childhood with precocious puberty and short adult stature due to early closure of the epiphyses is common in both sexes. Hypertension, present in about two-thirds of patients, is associated with suppression of plasma renin and sometimes hypokalemia. Plasma cortisol and corticosterone are low while basal or ACTH-stimulated levels of DOC and 11-deoxycortisol are elevated. Unlike in PA, aldosterone levels are also low, possibly because renin is suppressed and chronic ACTH excess leads to suppression (rather than stimulation) of aldosterone production [ , ]. Unlike 11β-hydroxylase deficiency, 21-hydroxylase deficiency (which also causes virilization) is associated with signs of mineralocorticoid deficiency, with salt wasting and potassium retention [ , ].
Management
Hypertension normally responds well to adequate glucocorticoid therapy (for example, oral hydrocortisone 10–20 mg/m 2 /day divided into 2–3 doses, or dexamethasone 20–30 μg/kg/day to a maximum of 2 mg/day [ ]), which suppresses ACTH and therefore lowers production of DOC. However, finding a dose that is sufficient to control androgen excess yet low enough to avoid Cushingoid side effects and growth suppression can be challenging and close clinical monitoring is required.
17α-hydroxylase deficiency
Clinical diagnosis
17α-hydroxylase deficiency is a rare cause of congenital adrenal hyperplasia (1% of patients) and mineralocorticoid hypertension [ , ] caused by loss-of-function mutations of both alleles of CYP17 . Cortisol biosynthesis is impaired, leading to increased ACTH secretion which results in excessive production of the mineralocorticoids, DOC and corticosterone and mineralocorticoid hypertension. A characteristic clinical feature of this condition is the absence of secondary sex characteristics which results from the lack of 17α-hydroxylase and 17,20-desmolase activity and consequently, deficient formation of sex steroids. The phenotype is transmitted in an autosomal recessive fashion.
The condition is frequently recognized at the time of puberty because the association of hypertension and hypokalemia with primary amenorrhea in females, and pseudohermaphroditism with hypospadias and small testes in males. Plasma levels of 17-hydroxyprogesterone, 11-deoxycortisol and cortisol are reduced, while gonadotropins (luteinizing hormone and follicle-stimulating hormone) are increased. As in 11β-hydroxylase deficiency, plasma aldosterone levels are usually low [ , ] but sometimes normal to mildly elevated [ ].
Management
Treatment consists of giving a glucocorticoid dosage sufficient to suppress ACTH secretion and hence DOC and corticosterone secretion, which usually results in control of hypertension. Supplemental sex hormone replacement therapy is usually also necessary in the young adult [ ].
For detailed reading on congenital adrenal hyperplasia, please refer Chapter 9 .
Primary glucocorticoid resistance (PGR) or Chrousos syndrome
Clinical diagnosis
The underlying defect in PGR is partial resistance of the human glucocorticoid receptor (hGR), resulting in chronic elevation of ACTH, adrenal hyperplasia and excessive production of ACTH-dependent steroids [ ]. The clinical spectrum is broad, ranging from asymptomatic to severe mineralocorticoid hypertension with hypokalemia and/or severe hyperandrogenism (due to excessive production of androgens). The mineralocorticoid hypertension probably results from the combined effects of (i) excessive production of ACTH-dependent DOC and corticosterone; and (ii) markedly elevated levels of cortisol which, although relatively inactive at the hGR (and therefore failing to induce signs of Cushing’s syndrome), appear to be normally active at the MR, where they exceed the capacity of the 11β-hydroxysteroid dehydrogenase type II enzyme (11β-HSD2) to render them ineffective by conversion to cortisone (see below). PGR can be sporadic or familial, with familial inheritance following either an autosomal dominant or an autosomal recessive pattern of transmission [ ].
PGR is characterized clinically by increased ACTH and cortisol levels and resistance of cortisol suppression by dexamethasone, in the absence of clinical stigmata of Cushing’s syndrome [ , ]. The circadian pattern of cortisol secretion is normal. The condition has been reported mainly in individuals who have presented with clinical manifestations of mineralocorticoid and/or androgen excess, and in family members who, although not necessarily symptomatic, have subsequently been found to have biochemical evidence of PGR. Plasma levels of DOC, corticosterone, and androgens such as androstenedione, dehydroepiandrosterone (DHEA), and DHEA-sulfate are variably elevated while renin (as PRA or DRC) and aldosterone levels are low [ , ].
The condition is caused by mutations within the gene encoding the glucocorticoid receptor ( NR3C1 ) which impair receptor function, thereby impairing glucocorticoid signal transduction, resulting in reduced tissue sensitivity to glucocorticoids [ , , , ].
Management
As in 11β- and 17α-hydroxylase deficiency, treatment involves ACTH suppression by administration of glucocorticoids, but doses required to do this are higher (for example, dexamethasone 1–3 mg/day) because of resistance at the level of the receptor. Treatment is not required for asymptomatic, normotensive individuals with PGR [ ].
Syndrome of apparent mineralocorticoid excess (SAME)
Clinical diagnosis
The underlying biochemical defect in SAME is reduced or absent activity of 11β-HSD2 which normally acts to “protect” the MR from activation by cortisol by converting it to cortisone, an inactive metabolite [ ]. Activation of the MR by cortisol in SAME induces manifestations of mineralocorticoid excess. Congenital SAME is caused by loss-of-function mutations in HSD11B2 , which encodes 11β-HSD2 [ ]. While classic congenital SAME is rare and characterized by childhood onset of severe mineralocorticoid hypertension (with hypokalemic alkalosis), milder forms with less severe or later onset hypertension, and without hypokalemia, have been described [ , ]. SAME may also be acquired through the ingestion of licorice, major constituents of which (glycyrrhizic acid and its hydrolytic product glycyrrhetinic acid) are potent inhibitors of 11β-HSD2 [ ]. Medical administration of carbenoxolone, the hemisuccinate derivative of glycyrrhetinic acid which was in the past used to treat peptic ulcer disease, was also associated with this syndrome [ ].
SAME should be suspected in patients presenting with mineralocorticoid hypertension in association with low levels of both aldosterone and renin, and confirmed by an increased ratio of urinary metabolites of cortisol (tetrahydrocortisol and 5α-tetrahydrocortisol) to tetrahydrocortisone (THF + allo-THF/THE), or of urinary free cortisol to urinary free cortisone [ , ], reflecting reduced 11β-HSD2 activity.
Once thought to be exclusively transmitted in an autosomal recessive manner, later reports of late-onset hypertension, variably suppressed aldosterone and renin, and high urinary free cortisol to cortisone ratios in heterozygote parents of more floridly affected homozygous patients have been consistent with codominant inheritance [ , ].
Management
Dexamethasone, which does not have significant affinity for the MR, is usually effective in correcting mineralocorticoid hypertension and hypokalemic alkalosis by suppressing ACTH and cortisol, but additional antihypertensives such as spironolactone and/or amiloride may be required [ , ].
DOC-secreting tumors
Clinical diagnosis
Primary deoxycorticosteronism may be caused by adrenal adenoma or carcinoma [ ]. Patients present with hypertension, hypokalemia and renin suppression, but unlike in PA, plasma aldosterone levels are low and plasma DOC levels are often very high. Adrenal carcinomas producing DOC may present because of the symptoms associated with a rapidly enlarging adrenal mass or metastases.
Management
Spironolactone is given before surgery. For DOC-producing adenoma, unilateral adrenalectomy has produced long-term cure [ , ]. As with other forms of adrenocortical carcinoma, the prognosis of DOC-producing carcinoma is poor, but long-term benefit from adrenalectomy has been reported [ ].
Ectopic ACTH syndrome
Clinical diagnosis
In Ectopic ACTH syndrome, due, for example, to production of ACTH by bronchial carcinoids or small cell cancers of the lung, excessive sodium reabsorption and volume expansion (due to very high levels of cortisol which overload 11βHSD2 and therefore gain access to the MR as in SAME and high levels of DOC acting on the MR) leads to hypokalemia and suppression of renin (and, as a consequence, suppression of aldosterone) [ , ]. Hypertension develops if the patient lives long enough and is not too ill from the effects of the neoplasm. As in SAME, these patients have elevated ratios of cortisol to cortisone metabolites (THF + allo-THF/THE) [ , ].
Management
Where possible, the optimal treatment is rapid and complete excision of the ACTH-secreting tumor. A period of preoperative cortisol-lowering drugs may be required. If surgery is not a viable option, approaches include combinations of pharmacological agents directed against cortisol excess, bilateral adrenalectomy and antitumoral interventions (see section below on Cushing syndrome for more detail) [ ].
Activating mutations of the MR or Geller syndrome
Clinical diagnosis and management
In 2000, Geller and coworkers [ ] described a kindred with severe hypertension associated with suppressed renin and aldosterone levels, transmitted in an autosomal dominant fashion. Affected family members were heterozygous for a gain-of-function mutation in the gene encoding the MR, resulting in sodium retention, urinary potassium wasting and suppression of renin and aldosterone. Most affected individuals were normokalemic. All pregnancies that occurred among female carriers were complicated by marked exacerbation of hypertension, accompanied by development of hypokalemia despite undetectable aldosterone levels. Spironolactone and progesterone, normally antagonists of wild-type MR, were found to be potent agonists of the mutated gene product, explaining exacerbation of the clinical phenotype during pregnancy and excluding spironolactone (and favoring amiloride) as treatment for this clinical entity.
Liddle syndrome
Clinical diagnosis
This syndrome results from constitutive activation of ENaC caused by mutations within the genes encoding its β or γ subunits which prevent its ubiquitination and degradation [ ]. The clinical outcomes are sodium retention and urinary potassium and hydrogen ion wasting, causing hypokalemic alkalosis with early onset severe hypertension and suppressed renin and aldosterone (due to low renin plus hypokalemia). Less florid varieties, with less severe hypertension and normal plasma potassium levels, have been described [ ]. The clinical manifestations respond to treatment with ENaC antagonists but not spironolactone. Both familial and sporadic cases have been described, the former following an autosomal dominant pattern of transmission [ , ]. The condition should be suspected in patients (especially children) who present with hypertension and hypokalemic alkalosis and are found to have low renin and aldosterone levels. It can be distinguished from hypertensive forms of congenital adrenal hyperplasia, PGR, and SAME by normal levels of 17α-hydroxyprogesterone, 11-deoxycortisol and cortisol, normal ratios of free cortisol to cortisone (or metabolites) in urine, and failure to respond to dexamethasone suppression of ACTH.
Management
The hypertension and hypokalemia in Liddle’s syndrome respond to amiloride or triamterene, but dietary sodium restriction is usually also necessary to maintain normotension. As with other forms of mineralocorticoid hypertension, some individuals, especially those who are older and have been hypertensive for longer periods of time, require additional antihypertensive medication to achieve control.
Familial hyperkalemic hypertension (Gordon syndrome)
Clinical diagnosis
Unlike all the above entities, Familial hyperkalemic hypertension (FHHt, Gordon Syndrome) has an autosomal dominant inheritance with salt-sensitive hypertension due to defective renal tubular function leading to sodium retention and reduced (rather than increased) potassium and hydrogen ion excretion (hyperkalemic acidosis) [ ]. It is unique in that chronic hyperkalemia exists in the absence of a reduced glomerular filtration rate (GFR). Despite renin suppression, plasma aldosterone levels can be low, normal or even raised due to stimulation by elevated plasma potassium. Because of this, the ARR can be elevated and FHHt is therefore one of the causes of a “false positive” ARR when screening for PA [ ]. FHHt is caused by mutations in regulators of the thiazide-sensitive Na + -Cl – Co-transporter, NCC, including ‘With No lysine (K)’ kinases ( WNK1 and WNK4 ), KeLcH-Like3 ( KLHL3 ) and CULlin3 ( CUL3 ) [ ], which lead to upregulation of NCC expression. As in Liddle syndrome, significant inter- and intrapedigree phenotypic variation is observed clinically [ , ].
Management
FHHt is effectively treated by thiazide diuretics and/or dietary salt restriction [ , ].
(For further reading on various familial causes of endocrine hypertension please refer Chapter 6, Chapter 8, Chapter 9, Chapter 10 )
Pheochromocytoma and paraganglioma
Prevalence, pathophysiology and clinical presentation
Pheochromocytomas and paragangliomas (PPGL) are catecholamine releasing tumors arising from adrenal or extra-adrenal chromaffin cells. Pheochromocytoma (around 80%–85% of PPGL cases) refers to a tumor originating from the adrenal medulla, whereas paragangliomas (15%–20% of PPGL cases) are derived from paravertebral sympathetic ganglia in the thorax, abdomen or pelvis, or parasympathetic ganglia in the neck and skull base. The latter group is generally hormonally inactive [ ]. PPGL is a rare diagnosis. A recent epidemiological study found a prevalence of 65 per 100,000 people [ ], although autopsy studies suggest that a significant number may remain undetected during life [ ]. Recent publications reported an incidence rate of 6.6 cases per million per year [ ], and similar figures when expressed as age-standardized incidence rate (SIR) which allows a comparison with historical data. This evaluation detected a rise in SIR from 1.4 per million person-years in 1977 to 6.6 in 2015 [ ]. This temporal change is most likely explained by an increase in early disease detection and the clinical presentation has shifted over time from the classical vignette of paroxysmal hypertension, to smaller and often asymptomatic lesions incidentally found on radiological imaging [ , ]. The pick-up rate depends on the background risk of the population that is screened. Of patients with an adrenal incidentaloma, as many as 7% is estimated to have a pheochromocytoma [ ]. On the other hand, in the general hypertensive population, PPGL remains a rare cause and accounts for less than 1% of all cases [ ]. Despite this, it is a diagnosis important not to miss because of the high risk of negative outcomes if the diagnosis is delayed or missed [ ].
Cardiovascular events are the most significant cause for morbidity and mortality. In a retrospective analysis of 145 patients with pheochromocytoma, the incidence of cardiovascular complications was found to be as high as 19.3%. Among these, commonly reported were arrhythmias, heart failure, ischemic heart disease, and stroke [ ]. The rise in blood pressure (BP) in PPGL is related to the cardiovascular actions of catecholamines, such as their vasoconstrictive effect to increase peripheral vascular resistance. The BP rise can be sustained, frequently seen in noradrenaline-dominant tumors, or paroxysmal. Orthostatic hypotension may occur with purely adrenaline-producing tumors. Dopamine-dominant tumors generally do not cause hypertension [ ]. Paroxysms can occur spontaneously but may also be provoked by numerous well-known triggers such as physical activity, surgery, emotional factors and direct tumor manipulation. Furthermore, multiple drugs are known for their ability to cause a surge in catecholamine release, most notably dopamine D2 receptor antagonists (such as metoclopramide), tricyclic antidepressants, monoamine oxidase inhibitors and corticosteroids. In addition to elevation of BP, PPGL patients also display other features of BP dysregulation including greater BP variability, loss of nocturnal dipping and a high incidence of orthostatic hypotension [ ].
The adverse cardiovascular risk extends beyond the hemodynamic effects of catecholamines. Catecholamine excess is characterized by dysglycemia as a result of insulin resistance, reduced insulin secretion and reduced peripheral glucose uptake [ ]. In further support of this concept, a direct correlation between noradrenaline and normetanephrine with HbA1c has been described [ ]. Although these observations are thought to be predominantly related to the direct metabolic effects of catecholamines, recent findings of excess glucocorticoid levels in pheochromocytoma patients suggest that glucocorticoid-mediated pathways may also play a role [ ].
Another important contributor to its negative outcomes is the potential of some PPGLs to become malignant. The overall risk of metastatic disease is estimated to be around 10%–17% [ ], but is significantly higher in particular genetic syndromes. The association with several clinical syndromes has been well described and over one third of PPGL cases have a germline mutation in one of the known susceptibility genes such as succinate dehydrogenase B ( SDHB) , SDHD , Von Hippel-Lindau ( VHL ), RET and Neurofibromatosis-1 ( NF-1 ). Another 30%–40% of sporadic cases may furthermore have somatic mutations in a relevant gene. The actual malignancy risk depends on the specific gene and gene mutation that is involved. Of these, SDHB mutations carry the highest risk of malignant disease [ ]. Many of the genetic syndromes are also associated with an increased risk of other tumors. An example of this is medullary thyroid cancer in multiple endocrine neoplasia type 2 (MEN2) [ ]. For these reasons it is important to consider genetic testing in all PPGL cases [ ]. With the advancement of next-generation sequencing technologies, the recommended pathway for genetic testing has shifted in recent years from a targeted approach based on clinical features and immunohistochemistry [ ], to application of a broad panel of relevant genes [ ].
Diagnosis
The initial step in diagnosis is to identify patients that require screening for PPGL. The 2014 Endocrine Society guidelines on PPGL recommend biochemical screening in patients with signs or symptoms of PPGL, especially if occurring paroxysmally, adrenal incidentaloma, a known or suspected predisposition to a genetic syndrome known to be associated with PPGL, and a previous history of PPGL [ ]. The classic symptoms of PPGL are headaches, excessive sweating and palpitations, often occurring in a paroxysmal manner. However, not infrequently other, often nonspecific, manifestations can occur in PPGL such as anxiety, pallor, chest pain, and abdominal symptoms [ , ]. Geroula et al. [ ] found that palpitations, hyperhidrosis, pallor, tremor and nausea, in combination with low body mass index (BMI) and elevated heart rate are key features to discriminate PPGL from non-PPGL. When combined into a clinical feature score, there was a positive correlation with urinary and plasma catecholamine levels and probability of disease, and this can be a useful tool for further risk stratification [ ]. Imaging characteristics may further help in risk stratification. Contrast washout on a CT scan is considered an unreliable marker to differentiate a pheochromocytoma from an adrenal adenoma [ ]. However, an attenuation value of 10 Hounsfield Units or less on an unenhanced CT scan may be sufficient to rule out a pheochromocytoma without the need for further biochemical testing [ ].
Key to the biochemical diagnosis is assessment of plasma or urinary catecholamines levels. The use of the metabolites like normetanephrine, metanephrine and 3-methxotyramine (3-MT) derived from their parent hormones noradrenaline, adrenaline, and dopamine, respectively, through the action of catecholamine O-methyltransferase is recommended due to their superior diagnostic accuracy [ , ]. When plasma or urinary metanephrines are more than twofold elevated the diagnosis is likely [ , ]. On the other hand, mild-to-moderate elevations can pose a diagnostic dilemma due to a high risk of false-positive results. Common causes for false-positive results need to be considered, such as body position at the time of blood sampling. It is recommended to draw blood after at least 30 min in the supine position to prevent a posture-related rise in catecholamine levels. Any other causes for increased sympathetic activity, such as an intercurrent acute condition, heart failure or obstructive sleep apnoea, need to be carefully considered [ ]. Furthermore, numerous pharmacological agents including tricyclic antidepressants, monoamine oxidase inhibitors, phenoxybenzamine, and sympathomimetics are known to cause false-positive results and should be discontinued if possible [ , , ]. In patients with mild-to-moderate elevations of (nor)metanephrine levels, a clonidine suppression test (CST) can be helpful to exclude false positive results. The CST is based on the premise that in patients without PPGL, the administration of clonidine – a centrally acting alpha-2-adrenergic receptor agonist that reduces sympathetic catecholamine release—would result in a decrease in catecholamine levels, whereas this suppression would not be seen in patients with autonomous catecholamine release [ ]. An elevated plasma normetanephrine level 3 h after an oral dose of clonidine and a less than 40% reduction from baseline is highly suggestive of PPGL [ , ]. The urinary CST can be a valuable alternative to the more widely adopted plasma CST [ ].
Once the biochemical diagnosis has been established, the final step is to localize the tumor. CT or magnetic resonance imaging (MRI) scanning are commonly used modalities for anatomical localization [ ]. Functional imaging such as 123 I-metaiodobenzylguanidine (MIBG) scintigraphy, 68 Ga-Dotatate Positron Emission Tomography (PET)/CT and 18 F-fluorodihydroxy-phenylalanine ( 18 F-FDOPA) PET scanning is of additional value, especially if the patient is at risk of multifocal or metastatic disease. This is particularly relevant when there is a (suspected) hereditary cause. The choice of modality is dependent on the context and availability. In sporadic pheochromocytoma either 18 F-FDOPA PET or 123 I-MIBG scintigraphy is recommended as first line, whereas in hereditary pheochromocytoma due to NF-1, RET, VHL, and MAX mutations 18 F-FDOPA PET is the modality of choice. In extraadrenal sympathetic PPGL, multifocal disease, suspected metastatic disease, SDH mutations, or in head-and-neck paragangliomas 68 Ga-Dotatate PET is the preferred imaging modality [ ].
Management
Surgical management
The treatment of choice is surgical resection of the tumor. A laparoscopic approach is suitable for most patients, but an open approach is recommended for large or invasive tumors and for most paragangliomas [ ]. In selected patients with bilateral pheochromocytomas who have already undergone a unilateral adrenalectomy, partial adrenalectomy or cortical sparing surgery may be considered to prevent adrenal insufficiency and the resultant need for long-term steroid replacement. This is particularly relevant for patients with hereditary PPGL such as MEN2, VHL and NF-1 who are likely to develop bilateral disease. The benefits of adrenal-sparing surgery need to be carefully balanced with the increased risk of disease recurrence and future need for reoperation and this requires an individualized and multidisciplinary approach to decision-making [ , ].
Perioperative management
Adequate blockade of catecholamines with alpha-adrenergic receptor blockers for a period of at least 7–14 days preoperatively is critical to prevent hypertensive crises, hemodynamic instability, and cardiovascular complications during surgery [ , ]. Phenoxybenzamine, a long-acting, noncompetitive, and nonselective alpha-1 and alpha-2 antagonist, is most widely used. However, short-acting, competitive, and selective alpha-1 antagonists such as doxazosin or prazosin can be used as an alternative. Studies comparing the efficacy of these agents have yielded conflicting results, but the interpretability has been limited by their retrospective and observational design [ ]. In a recent randomized controlled trial, Buitenwerf et al. [ ] found no difference in intraoperative duration of BP levels outside target range in PPGL patients treated preoperatively with either phenoxybenzamine or doxazosin but a score for intraoperative hemodynamic instability was significantly lower in the phenoxybenzamine group. Whether this has clinical significance is unclear. Although the authors observed no differences in perioperative cardiovascular outcomes, larger numbers are needed to answer this question with more certainty. Another point of contention has been whether preoperative alpha-blockade is indeed necessary or whether this could be safely omitted. Although a recent meta-analysis of the available literature to try and answer this question found no evidence for a benefit of alpha-blockade, the number of suitable studies was low and all were retrospective in design [ ]. Therefore, the recommendation that all patients with a hormonally active PPGL should receive preoperative alpha blockade remains unchanged [ ].
Beta-adrenergic receptor blockers can be used as second-line treatment but should only be initiated after adequate alpha-adrenergic blockade as inhibition of beta-2 adrenergic receptor mediated vasodilatation in the setting of ongoing alpha-adrenergic receptor mediated vasoconstriction may result in a life-threatening surge in BP [ ]. CCBs may be a useful addition when BP remains inadequately controlled, when side effects limit further titration of alpha-adrenergic receptor blockers, or when a patient suffers from catecholamine-induced coronary vasospasms [ ]. The tyrosine hydroxylase inhibitor alpha-methyl-paratyrosine, or metyrosine, which blocks the rate-limiting step in catecholamine synthesis, can be a further useful agent. It is not widely used due to limited availability and high costs, but can be considered in selected cases with high catecholamine levels due to large tumor size or metastatic disease and in preparation for high-risk procedures [ ].
In the postoperative phase, patients may develop hypotension due to the sudden drop in catecholamine levels after successful tumor removal. This may be exacerbated by the ongoing presence of alpha-adrenergic blockade, and a catecholamine-related contraction of circulating volume [ ]. A high sodium diet and an infusion of 1–2 L of normal saline in the 24 h prior to surgery are recommended to reduce the postoperative drop in BP [ ].
Follow-up
After resection of a functional PPGL, it is recommended to assess levels of (nor)metanephrine and 3-methoxytyramine 2–6 weeks after surgery to confirm complete removal. Long-term follow-up should include annual clinical review and biochemistry for a period up to 10 years. In patients with risk factors for recurrence or metastatic disease, such as a young age, a known hereditary factor or the presence of a paraganglioma, lifelong surveillance is indicated. In nonfunctional PPGLs, thoraco-abdomino-pelvic imaging every 1 to 2 years for up to 10 years is advised. Chromogranin-A may be an additional useful marker to monitor in patients who have normal metanephrine and 3-MT levels [ , ]. Surveillance of patients with hereditary PPGLs furthermore needs consideration of the risk of recurrence and of extra-adrenal malignancies that may occur as part of the specific syndrome and relevant clinical, biochemical and radiological surveillance recommendations need to be applied [ ].
For more detailed reading about PPGLs please refer to Chapter 11, Chapter 12, Chapter 2 .
Cushing’s syndrome
Prevalence, pathophysiology and clinical presentation
Cushing’s syndrome refers to a condition of excess glucocorticoids and is associated with a large number of adverse effects, increased morbidity and mortality [ ]. Since pharmacological doses of corticosteroids are widely used for the treatment of a broad range of conditions, exogenous steroid use is in fact the most common cause of Cushing’s syndrome [ ]. Endogenous Cushing’s syndrome on the other hand is rare with an estimated incidence between 0.2 and 5 per million population per year and a prevalence between 39 and 79 per million [ ].
Due to its role as a key metabolic hormone with a broad systemic action profile, cortisol excess can present with a wide variety of symptoms, signs and complications affecting almost every organ system. Well-known manifestations are weight gain and central obesity, characteristic skin changes, hirsutism and menstrual irregularities, neuropsychiatric symptoms, musculoskeletal complications (most notably osteoporosis and proximal myopathy), and multiple adverse metabolic risk factors including hypertension, glucose intolerance, and dyslipidemia [ , ]. Because many of these features are common in the general population, and the onset and progression of symptoms is often insidious, the diagnosis is frequently delayed [ ]. Violaceous striae, proximal myopathy, facial plethora, easy or spontaneous bruising and unexplained osteoporosis are characteristics with a higher discriminatory value and their presence, or the presence of unusual complications for age, or multiple and progressive features should trigger further evaluation [ , ].
Untreated Cushing’s syndrome is associated with a high mortality rate, and infections, cardiovascular complications, and malignancies are common causes of death [ ]. Early treatment significantly improves prognosis, but does not completely mitigate excess mortality [ , ]. This is partly explained by recurrent or residual disease in some patients [ ], but seems to persist even in patients with apparent successful treatment [ ]. The long-term increased mortality risk is particularly related to Cushing’s disease, and treated adrenal Cushing’s syndrome generally has a good prognosis [ , ]. In contrast, ectopic Cushing’s syndrome has a poor outcome in the majority of patients (see above within section on “other mineralocorticoid forms of hypertension”) [ ].
Hypertension is a very common complication of Cushing’s syndrome with an estimated prevalence of 70%–85% [ , ]. On the other hand, hypercortisolism remains an uncommon secondary cause of hypertension in the general population. The prevalence of Cushing’s syndrome in hypertensive patients evaluated in a general outpatient clinic was around 1% with a similar number found to have subclinical Cushing’s syndrome [ ]. The prevalence is likely to be higher in selected patients such as those with resistant hypertension, an unusually young age of onset, or patients with associated complications, reiterating the aforementioned principles of appropriate patient selection for screening [ ]. There appears to be a tendency toward higher BP in adrenal Cushing’s compared to pituitary causes [ ].
Several mechanisms contribute to the pathophysiology of hypertension in states of hypercortisolism, and both direct and indirect pathways appear to be involved [ ].
A key element of cortisol-induced hypertension is renal sodium and water reabsorption, and patients with Cushing’s syndrome have been found to display increased expression of different sodium transporters along the nephron in urinary exosome studies [ ]. Glucocorticoids influence renal sodium reabsorption through interaction with both glucocorticoid receptors (GR) and MR expressed in different parts along the nephron [ ]. Their ability to activate MRs in the aldosterone-sensitive parts of the tubular system is determined by activity of the enzymes 11-beta-hydroxysteroid dehydrogenase (11β-HSD) type 1 and type 2 [ , ]. Saturation of 11β-HSD type 2 will increase the availability of cortisol to activate MRs in mineralocorticoid target tissues such as the kidney [ ]. Furthermore, a reduction in plasma potassium levels can also play a contributory role to drive sodium reabsorption through activation of sodium-chloride-cotransporters (NCC) [ , ]. As a result of enhanced sodium reabsorption, renin levels are low in some, but can be inappropriately normal in others [ , ] implying that alterations at other levels of the renin-angiotensin-aldosterone system (RAAS) are involved. For instance, hypercortisolism is known to induce hepatic angiotensinogen synthesis [ , ]. In patients with nonsuppressed renin, other mechanisms may predominate, such as increased vascular responsiveness to vasopressors [ ]. Older studies have indeed shown an exaggerated BP response to vasopressors such as noradrenaline [ , ], and angiotensin II (Ang II) [ , ]. The latter observation may be mediated through a glucocorticoid-induced enhancement of vascular Ang II type 1 receptor expression [ ]. Elevated levels of the vasoconstrictor endothelin-1 were also postulated to be a contributing factor [ ], although this relation was not confirmed in other studies [ ]. Furthermore, a blunted response to different vasodilatory mediators such as atrial natriuretic peptide (ANP), nitric oxide and prostaglandins has also been suggested [ ]. More indirectly, metabolic changes including insulin resistance and hyperinsulinemia, obstructive sleep apnoea (a prevalent condition in Cushing’s syndrome), and vascular dysfunction and remodeling further contribute to the development and perpetuation of hypertension in Cushing’s syndrome [ ].
Diagnosis
The diagnostic evaluation for Cushing’s syndrome has been described in detail in the Endocrine Society guidelines [ ], Chapter 13, Chapter 14 . The initial step in patients in whom a clinical suspicion exists, is to perform one of the following available screening tests: either a minimum of two measurements of 24-hour urinary free cortisol excretion, two late-night salivary cortisol measurements, or a low-dose DST. If adrenal Cushing’s syndrome is suspected, the use of either a low- dose DST or late-night salivary cortisol is preferred for the initial screening. Patients with an abnormal initial test require at least one additional positive test to confirm the diagnosis. Other tests such as the dexamethasone-corticotropin releasing hormone (CRH) test may further help in the process of confirmation [ ]. The next step in diagnosis is to determine whether the condition is adrenocorticotropic hormone (ACTH)-dependent or -independent. Patients with ACTH-independent Cushing’s syndrome (generally caused by a cortisol-producing adrenal adenoma) account for ∼20% of cases, whereas ∼80% of patients with Cushing’s syndrome have ACTH-dependent disease. The vast majority of these have an ACTH-producing pituitary lesion (i.e., Cushing’s disease), with a small group having an ectopic ACTH-producing tumor [ ]. An ACTH level below 10 pg/mL is suggestive of an adrenal origin and should be followed up by imaging of the adrenal glands. An ACTH level over 20 pg/mL indicates ACTH-dependent hypercortisolism. The high-dose DST and CRH stimulation test are dynamic tests that can assist to differentiate pituitary from ectopic ACTH production. Concordant dynamic test results in combination with a pituitary adenoma >6 mm on imaging confirms Cushing’s disease. In other cases, bilateral inferior petrosal sinus sampling may be required, as the gold standard test to discriminate pituitary from ectopic Cushing’s syndrome [ ].
Management
Resolution or improvement of hypercortisolism is the main treatment goal in Cushing’s syndrome and this is expected to also have a positive impact on BP. However, the degree of BP response is dependent on many factors including the underlying cause, treatment modality, treatment success and patient factors. If possible, surgical resection of the causative process is the preferred treatment with the highest chance of attaining cure or long-term remission. Especially in Cushing’s disease, however, additional treatment may be required, either as presurgical treatment, or after surgery in case of persistent or recurrent disease, and the possibilities include radiotherapy and pharmacotherapy. Pharmacotherapeutics can target the pituitary (e.g., pasireotide or cabergoline), adrenal steroidogenesis (e.g., metyrapone, ketoconazole or mitotane), or the GR (mifepristone) [ ].
Surgical management
Surgically treated patients with Cushing’s syndrome (both pituitary and adrenal) show an improvement in hypertension after treatment, but its prevalence continues to be higher than in aged-matched controls [ , ]. In a retrospective analysis, it was found that 92% of surgically treated patients achieved resolution or improvement of hypertension within 12 months, and 80% did so within the first 10 days of surgery. Forty-four percent of patients had complete remission of hypertension, whereas 56% of patients had residual hypertension [ ]. Furthermore, only a minority of patients with Cushing’s disease restored a normal BP dipping pattern even after curative surgery [ ]. Jha et al. [ ] found that younger age and lower BMI were predictors for a favourable BP response. In patients with adrenal Cushing’s syndrome, lower preoperative BP and shorter duration of hypertension predicted a favorable BP response 1 year after adrenalectomy [ ]. In subclinical Cushing’s syndrome, the cortisol level after a 1-mg DST may be another predictor of improvement in comorbid conditions including hypertension after adrenalectomy. Interestingly, a greater reduction of postoperative medication requirement was observed in patients with a post-DST cortisol between 1.8 and 3.0 mcg/dL (50 and 83 nmol/L) compared to patients with a lesser degree of cortisol suppression. It was proposed that these patients represent a milder group with a lower degree of comorbidities and possible associated irreversible target organ damage that may limit the long-term surgical outcome [ ]. Although Jha et al. [ ] did not find an association between duration of hypercortisolism and BP response, it is known that prolonged hypercortisolism contributes to excess long-term mortality risk after surgery. Persistence of metabolic risk factors, and long-term vascular dysfunction and remodeling are likely important factors to explain persistence of hypertension after management of hypercortisolism. Also, the alterations in the RAAS associated with hypercortisolism were found to persist long after normalization of cortisol levels [ ].
Pharmacological management
Pasireotide is a somatostatin analogue (SSA) with a high affinity for somatostatin receptor subtype 5, the predominant subtype expressed in corticotroph adenomas, and has emerged as a novel treatment option for medical management of Cushing’s disease [ ]. Both short-acting [ ], and long-acting pasireotide [ ] were shown to reduce BP, even without complete normalization of cortisol. A direct effect of pasireotide on BP is further supported by experimental studies which showed that in the presence of dexamethasone, pasireotide (and octreotide) reduced the Ang II-mediated vasoconstriction in iliac arteries derived from spontaneously hypertensive rats [ ]. The dopamine agonist cabergoline has also been found to be beneficial in the management of Cushing’s disease. In 20 patients who had uncontrolled Cushing’s disease after pituitary surgery, 75% showed biochemical responsiveness to cabergoline after 3 months, and 50% on longer term follow-up. This was associated with an improvement in hypertension, and although the reduction in BP was more marked in treatment responders, BP still improved in patients who showed treatment escape [ ].
Steroidogenesis inhibitors constitute a category of agents aimed to reduce adrenal steroid hormone synthesis. Ketoconazole is an imidazole derivative that blocks multiple adrenal steroidogenic enzymes including 11-beta-hydroxylase, 17-hydroxylase, and 18-hydroxylase. Metyrapone is an inhibitor of 11-beta-hydroxylase [ ]. These agents have been shown to reduce cortisol levels in a subset of patients with Cushing’s syndrome with an associated BP reduction in some [ ]. The lowering of cortisol by metyrapone may result in a compensatory rise in ACTH and consequently an increase in adrenal androgens and mineralocorticoid precursors, which could in fact exacerbate hypertension and hypokalemia [ ]. Other well-known steroidogenesis inhibitors are mitotane and etomidate. Mitotane is an adrenolytic agent which, due to its unfavorable side effect profile, is mainly limited to the treatment of adrenocortical carcinoma. Etomidate is another imidazole derivative with a rapid onset of action. Due to its parenteral route of administration, it is predominantly used in severe, life-threatening cortisol excess [ ].
Lastly, cortisol can also be blocked at the level of its main receptor GR. Mifepristone is a GR antagonist that has been shown to improve global clinical performance in patients with Cushing’s syndrome [ ]. In a 24-week, open-label trial, an improvement in diastolic BP (DBP) or reduction in antihypertensives was observed in over half of the hypertensive patients with Cushing’s syndrome after treatment with mifepristone, although no overall improvement in mean BP was seen. A subgroup of patients even developed a rise in BP which was associated with hypokalemia and peripheral edema [ ]. It is known that GR antagonists may lead to a compensatory rise in ACTH and cortisol further contributing to MR activation via the mechanisms described previously. Mifepristone is also a potent antagonist of the progesterone receptor (PR), and endometrial thickening and irregular vaginal bleeding has been observed in women treated with this agent [ ]. Relacorilant is a new selective GR modulator, which in a recent Phase II trial has shown potential in ameliorating BP in Cushing’s syndrome without causing associated antiprogesterone adverse effects. The compensatory rise in ACTH and cortisol was also substantially less than with mifepristone, which may therefore reduce the risk of developing of MR-mediated hypertension and hypokalemia [ ]. Further studies are awaited to determine its potential role in the treatment of hypertension in this setting.
In patients who fail to demonstrate a full response on monotherapy, combination treatment can be successful to achieve further improvement. When a step-wise approach was adopted in 17 patients with Cushing’s disease using pasireotide as initial treatment, the subsequent addition of cabergoline and ketoconazole in patients with inadequate control resulted in biochemical control in the majority. This was associated with a 12, and 8 mmHg reduction in systolic BP (SBP) and DBP, respectively [ ].
Multimodal treatment
The management of hypertension in Cushing’s syndrome requires a multimodal approach. To address the cause of hypercortisolism is key, but most treatment modalities take time to arrange (e.g., surgery), may not result in immediate or complete restoration of eucortisolism (e.g., radiotherapy and pharmacotherapy), or may fail over time. Also, hypertension may not fully resolve after treatment for the reasons explained above. Aggressive BP management to reduce cardiovascular risk is important in all stages of the disease, from diagnosis to long-term follow-up.
Isidori et al . [ ] describe a treatment algorithm for hypertension management in Cushing’s syndrome to target the different pathophysiological mechanisms that are in play. In light of the significant alterations in the RAAS at multiple levels, ACE inhibitors or ARBs are considered first-line treatment. An MR antagonist such as spironolactone can be considered as second line agent, especially if hypokalemia is present, or alternatively a CCB. If this fails to control BP, alpha-adrenergic receptor antagonists or nitric oxide donors can target the increased vasoconstrictive response. Thiazide diuretics, and beta-adrenergic receptor blockers are generally used with caution due to its potential negative effects on glucose tolerance, and especially in case of thiazides, their potential to exacerbate hypokalemia but can be used in selected cases [ ].
Acromegaly
Prevalence, pathophysiology, and clinical presentation
Acromegaly is a condition characterized by growth hormone (GH) excess. In the vast majority of cases this is related to a GH-producing pituitary adenoma, with GH releasing hormone (GHRH) producing tumors and ectopic GH producing tumors being extremely uncommon alternative causes [ ]. Acromegaly itself is a rare disease with a reported prevalence between 2.8 and 13.7 per 100,000 people. A significant delay in diagnosis with a median time-to-diagnosis of 4.5–5 years is common due to the often-insidious onset and progression of symptoms and signs, and the high prevalence of many of its associated complications in the general population [ ]. Excessive GH stimulates insulin-like growth factor 1 (IGF-1) synthesis predominantly in the liver which in turn acts on multiple target organs. Classical features such as acral enlargement, change in facial appearance, headaches and visual impairment due to a large pituitary lesion are the most frequently reported presenting symptoms, but these can be absent or subtle. When a patient presents with multiple acromegaly associated conditions, such as obstructive sleep apnoea, arthralgias, nerve entrapment syndromes, hyperhidrosis, and hypertension, this should trigger further investigations [ , ].
Acromegaly is associated with significant morbidity, mortality, and reduced quality of life due to its impact on multiple organ systems. Therefore, a rigorous assessment and management of associated comorbidities is warranted with a focus on cardiovascular, endocrine, metabolic and musculoskeletal disorders, as well as appropriate screening for malignancies [ , ]. Patients with acromegaly have an unfavorable cardiovascular risk profile due to the presence of traditional risk factors such as hypertension, type 2 diabetes mellitus and dyslipidemia [ ], and a higher incidence of atrial fibrillation and congestive heart failure [ ].
The relation between hypertension and acromegaly is well-known but the reported prevalence of hypertension varies widely ranging from 18% to 60% which is likely explained by differences in the populations from which the data were derived [ ]. In a retrospective comparison between acromegalic patients and carefully matched healthy controls, a nearly twofold higher prevalence of hypertension was found in the patient group (46% vs. 25%) and acromegaly preferentially raised DBP [ ]. Although the pathways have not been fully unraveled, multiple mechanisms are thought to be involved [ ]. Acromegaly causes expansion of extracellular volume through direct actions of GH and IGF-1 on renal sodium handling [ ] and indirectly through an inhibitory effect on ANP release. In addition, altered sympathetic activity, especially the loss of a normal circadian rhythm, may blunt the physiological nocturnal dip in BP. Also, acromegaly causes functional and structural cardiac and vascular changes resulting in increased vascular tone and resistance [ ]. Insulin resistance and hyperinsulinemia, common features of acromegaly, may further augment these pathophysiological and structural alterations [ ]. The presence of other risk factors, such as OSA, and concomitant hypopituitarism requiring exogenous steroid replacement further add to hypertension risk.
Diagnosis
When acromegaly is suspected, measurement of IGF-1 is the first step in diagnosis, and an elevated IGF-1 level in combination with typical symptoms is highly suggestive. A failure to suppress GH levels to <1 mcg/L (<1 ng/mL or <3 mIU/L) during an oral glucose tolerance test confirms the diagnosis. Pituitary imaging, preferably with a MRI scan, will usually visualize a pituitary lesion [ ].
Management
Management of acromegaly requires a multimodal approach. The main goals of treatment are to achieve biochemical control of GH excess, to control tumor mass, preserve or replace pituitary function, and to manage the associated long-term complications. Transsphenoidal surgery to remove the pituitary lesion is the preferred treatment as this is the best option to achieve cure if the whole lesion can be resected. Other available treatment modalities are somatostatin analogues (SSAs), dopamine agonists, the GH receptor antagonist (pegvisomant), and (stereotactic) radiotherapy, the latter if there is residual or recurrent disease after surgery, or if surgery cannot be performed. It is of great importance to adopt a multidisciplinary approach to decide on the most appropriate management for each individual patient [ ].
Although older studies had not yielded consistent evidence to show that disease control directly translates in improvements in BP [ ], this has become more apparent in recent publications. In retrospective studies, disease control was found to lower BP in acromegalic patients with hypertension at baseline [ ], but also in normotensive patients biochemical control of acromegaly was shown to prevent the incidence of hypertension [ ]. A prospective study of 45 new patients with acromegaly managed with SSAs as first-line treatment showed a significant reduction in the prevalence of hypertension after 5 years of follow-up [ ], further supporting the notion that disease control is directly associated with an improvement in BP. On the other hand, Gonzalez et al. [ ] found no change in the prevalence of hypertension in a cohort of 522 patients with acromegaly managed with a standardized multimodal approach after a median follow-up of 7.4 years. Part of the treatment effect on BP may have been masked by the influence of aging over the follow-up period, but notably no differences were seen between those who achieved biochemical control and those who still had active disease at the completion of follow-up. Possible factors to explain these discrepant results are the influence of age, disease severity and time-to-diagnosis mediated through classical hypertension risk factors, and the accumulation of structural and irreversible target organ damage which may impact on the likelihood of complete BP remission.
When assessing the cardiovascular benefits of treatment, it is important to consider hypertension in the broader context of overall cardiovascular risk profile. In a long-term follow-up of a large cohort of acromegaly patients treated with pegvisomant, it was observed that although average BP improved on treatment, the presence of hypertension remained a significant predictor of excess mortality and associated with a higher prevalence of other risk factors such as diabetes mellitus, hyperlipidemia, and cardiovascular disease [ ]. There are also indications that different treatment modalities have differential effects on cardiovascular risk factors independent of IGF-1 normalization [ ]. These studies emphasize the need for continuing assessment and aggressive management of cardiovascular risk factors in long-term follow-up of treated acromegaly patients.
Limited data exist to suggest specific antihypertensive agents in this context, and the choice of agents is guided by the common comorbidities associated with acromegaly, such as diabetes mellitus and cardiomyopathy. It is therefore not surprising that ACE inhibitors and ARBs were reported to be most widely used by clinicians to treat hypertension in patients with acromegaly [ ].
For further reading on acromegaly and hypertension, please refer to Chapter 15 .
Thyroid disorders
Thyroid disorders are prevalent and frequently underrecognized in the general population [ ]. The effects of thyroid hormones on the cardiovascular system and the role of thyroid dysfunction as a risk factor for cardiovascular disease are widely recognized [ ]. Both hypo- and hyperthyroidism can lead to elevations in BP [ ]. The following section will discuss the prevalence, clinical relevance, diagnostic, and management approaches of hypo- and hyperthyroidism in relation to hypertension.
Hypothyroidism
Prevalence, pathophysiology, and clinical presentation
Hypothyroidism (either subclinical or overt) is a common condition. A meta-analysis of studies on the incidence and prevalence of thyroid dysfunction in different European populations found a combined prevalence of previously diagnosed and undiagnosed hypothyroidism of 3%. In this study, up to 85% of people with thyroid dysfunction had subclinical disease [ ]. There is a strong female preponderance in prevalence and incidence [ , ]. In the vast majority of patients, hypothyroidism is primary in nature. On a global scale, iodine deficiency remains the most significant causative factor. In iodine replete populations, chronic lymphocytic thyroiditis (Hashimoto’s thyroiditis) is the most common cause. Other well-known reasons for primary hypothyroidism are iatrogenic causes, such as after radioactive iodine (RAI) treatment, thyroidectomy, or external beam radiation to the neck region. Further, a broad range of pharmacotherapeutic agents can cause primary hypothyroidism. Central hypothyroidism, related to pituitary or hypothalamic pathology, and peripheral hypothyroidism, which includes consumptive hypothyroidism due to increased tissue expression of the inactivating deiodinase 3 enzyme, and thyroid hormone resistance, are rare but should be considered in the relevant clinical setting [ , ].
Hypothyroidism is widely recognized as a risk factor for (predominantly diastolic) hypertension [ ]. However, whether hypothyroidism is a significant cause of hypertension, especially in subclinical disease, remains a subject of debate [ ]. Most evidence is derived from observational studies, with some showing positive associations [ ], whereas others failed to substantiate this association [ ]. A recent large cross-sectional study which included 49,433 euthyroid patients and 7719 patients with subclinical hypothyroidism confirmed a higher prevalence of hypertension in patients with subclinical hypothyroidism, and this relation was stronger for females under the age of 65 years [ ]. Prospective and experimental studies further support the causative nature of this association. A systematic review and meta-analysis of nine studies showed a positive association between subclinical hypothyroidism and incident hypertension in women (OR 1.32). This association was significant only in women <65 years [ ]. Fommei and Iervasi (2002) studied 12 normotensive patients with a previous thyroidectomy and performed a 24-hour ambulatory BP measurement (ABPM) 6 weeks after thyroxine withdrawal, and 2 months after reinstatement of hormone replacement. They observed that the average daytime BP was significantly higher in the thyroxine withdrawal phase compared to the replacement phase (5.1 mmHg for SBP, and 8.2 mmHg for DBP) [ ].
Important factors to explain the BP rise are an increase in systemic vascular resistance (SVR) and increased arterial stiffness [ , ]. Contributors to the rise in SVR are the reduction in the physiological tri-iodothyronine (T3) mediated vasodilatory effect on vascular smooth muscle cells [ ], endothelial dysfunction [ ], and reduced vascular beta-adrenergic reactivity [ , ]. Hypothyroid patients were also found to have increased vascular stiffness [ ]. Altered vascular reactivity and endothelial dysfunction may further play a role, as well as structural vascular changes related to long-term hypertension and dyslipidemia. As such, the increased cardiovascular risk observed in hypothyroidism is likely related to a combination of risk factors which includes not only hypertension, but also an atherogenic lipid profile [ , ]. Case detection is important, as thyroid hormone replacement was found to improve BP [ , ], carotid intima-media thickness [ ], and indices for central arterial stiffness [ ]. However, there is a need for randomized controlled trials to better understand the benefits of thyroid replacement on cardiovascular risk factors and outcomes [ ].
Diagnosis
The diagnostic workup and management of hypothyroidism has been well-described in clinical practice guidelines [ ]. In most cases, measurement of thyroid stimulating hormone (TSH) provides a reliable initial screening test for thyroid dysfunction unless central hypothyroidism is suspected, and an elevated TSH level is indicative of hypothyroidism. Subsequent assessment of tetraiodothyonine (thyroxine or T4) and tri-iodothyronine (T3) can determine whether the hypothyroidism is subclinical or overt [ ].
Management
Thyroid hormone replacement with levothyroxine is the recommended treatment for hypothyroidism [ ], and BP is expected to improve on adequate replacement. When antihypertensives are required, dihydropyridine CCBs and ACE inhibitors have been suggested to be beneficial in this setting [ ], but the available evidence is limited. Furthermore, hypothyroidism causes a low renin, salt-sensitive form of hypertension which may respond to salt restriction and diuretics [ ].
Hyperthyroidism
Prevalence, pathophysiology, and clinical presentation
Hyperthyroidism is caused by an excess of thyroid hormones. The prevalence is highly variable and depends on the population and the geographical area where it is studied with numbers ranging from 0.1% to 2.9% [ ]. In a meta-analysis of epidemiological studies from Europe, a 0.75% prevalence of previously diagnosed and undiagnosed hyperthyroidism was found [ ]. Similar to hypothyroidism, hyperthyroidism is more common in women than in men [ , ]. The most common causes of hyperthyroidism are Graves’ disease and nodular thyroid disease with one or multiple autonomous nodules. Other well-known causes are thyroiditis (such as subacute thyroiditis and postpartum thyroiditis), and iodine-induced hyperthyroidism (Jod-Basedow effect). Primary hyperthyroidism constitutes the majority of cases and other causes such as central hyperthyroidism (TSH-oma) and ectopic thyroid hormone secretion (such as struma ovarii) are very rare [ , ].
The clinical picture of hyperthyroidism is characterized by adrenergic symptoms such as tremors, anxiety and tachycardia. Hyperthyroidism, especially if prolonged, is furthermore associated with multiple other adverse effects including weight loss, and osteoporosis [ ]. Hyperthyroidism is particularly associated with an elevated risk of cardiovascular complications and severe hyperthyroidism increases the risk of heart failure, atrial fibrillation, and stroke [ ]. The profound effects of hyperthyroidism on the cardiovascular system are well-known. It causes a hyperdynamic circulation with an increase in heart rate, cardiac preload, and contractility, and a decrease in SVR all contributing to an increase in cardiac output. Whether this leads to an increase in systolic BP depends on the balance between cardiac output and SVR [ ]. However, other hormonal factors may alter this balance toward an increase in BP. For instance, endothelin-1 levels may rise in hyperthyroidism. Furthermore, the reduction in SVR causes activation of the renin–angiotensin–aldosterone system (RAAS) resulting in salt and water retention and increased vascular reactivity in turn leading to a further increase in cardiac preload [ , ]. Although studies on this are relatively scarce, some investigators have found that management of hyperthyroidism can lead to improvement of hypertension [ ]. In subclinical disease, the evidence of an association with hypertension is more scarce and not as clear. One cross-sectional study found a higher prevalence of hypertension in subclinical hyperthyroidism (OR 2.8) [ ], whereas other studies, including one prospective study, failed to confirm this association [ ].
Diagnosis
The biochemical diagnosis of primary hyperthyroidism is confirmed when a suppressed TSH level is found with a normal or elevated T3 and/or T4 level indicating subclinical or overt hyperthyroidism. Additional investigations, including the measurement of TSH receptor antibodies, and a thyroid uptake scan may further help to establish the etiology [ ].
Management
The mainstay of management is to correct the hyperthyroid state. Treatment modalities that are available include pharmacotherapy with antithyroid drugs (ATDs or thionamides), RAI treatment and surgery, each to be used in the appropriate clinical setting [ ]. Antihypertensives may be indicated in situations where no specific treatment is available, such as in subacute thyroiditis, or where more rapid BP management is required. Beta-adrenergic receptor blockers are generally recommended to reduce the adrenergic effects of hyperthyroidism. The nonselective beta-adrenergic blocker propranolol has traditionally been the preferred agent of choice, due to its additional ability to block the conversion of T4 to T3. In patients with contraindications to nonselective betablockers, such as bronchospastic asthma, more selective beta1-adrenergic blockers such as atenolol or metoprolol can be used with caution [ ]. Although beta-adrenergic blocker treatment is largely given for symptom control, and to reduce tachycardia and its associated cardiovascular complications, this will also assist to improve BP. In a study of 30 patients with hyperthyroidism and hypertension, a three-month treatment with propylthiouracil in combination with either metoprolol or carvedilol, a nonselective beta-adrenergic blocker and alpha1-adrenergic receptor blocker, both resulted in a significant improvement in BP and heart rate with no differences between groups [ ]. When beta-blockers are contraindicated or not tolerated, nondihydropyridine CCBs such as verapamil or diltiazem can be considered [ ]. Furthermore, ACE inhibitors have been suggested as a useful agent in this context [ ].
Primary hyperparathyroidism
Prevalence, pathophysiology, and clinical presentation
Primary hyperparathyroidism (PHPT) is a condition defined by (semi-)autonomous secretion of parathyroid hormone (PTH) by one or multiple parathyroid glands. This generally results in elevated serum calcium levels, although cases of normocalcemic PHPT have been described. Patients can present with polyuria, polydipsia, constipation, nausea, vomiting and altered mental state, especially if calcium levels are significantly elevated. In the long run, PHPT is associated with a multitude of adverse sequelae. Well-known are the renal complications related to hypercalciuria, such as kidney stones, nephrocalcinosis, and renal impairment, and the skeletal complications, most notably osteoporosis [ ].
An estimated prevalence of up to 0.9% in the general population has been reported, with a significant proportion of that remains undetected [ ]. This number could be exceedingly higher when patients with normocalcemic PHPT are taken into account [ ]. In the majority of patients, the disease is caused by a single parathyroid adenoma, but multiglandular disease can also occur, usually due to parathyroid hyperplasia. Female sex and advanced age are important risk factors [ ]. Most cases are sporadic, but can occur as part of a familial syndrome, in particular one of the multiple endocrine neoplasia (MEN) syndromes. A familial cause should be considered in younger patients, males and multiglandular disease [ ].
In addition to the complications described above, studies have shown an association between PHPT and a high prevalence of cardiovascular risk factors, such as obesity, hypertension, hyperlipidemia and type 2 diabetes mellitus, compared to the general population [ ]. However, the significance of cardiovascular risk in PHPT has remained a subject of debate due to the heterogenous and observational nature of most studies, and cardiovascular risk mitigation is not currently considered an indication for surgical management of PHPT [ ]. With respect to hypertension, many studies have reported an association with PHPT [ , ]. Although the exact mechanisms are unknown, it is suggested that alterations in the RAAS, as well as a blunted vasodilatory and enhanced vasoconstrictive response in peripheral resistance vessels to pressor hormones such as norepinephrine play a role [ , ]. It also remains unclear whether the BP rise is related to hypercalcemia, or is caused by a direct effect of PTH per se [ ]. A meta-analysis of six prospective cohort studies has shown a positive correlation between PTH levels and hypertension risk [ ].
Importantly, there appears to be an intimate and bidirectional relation between PTH and the RAAS [ ]. Excessive aldosterone levels can cause secondary hyperparathyroidism due to induction of renal calcium and magnesium loss. Indeed, patients with primary aldosteronism are known to have higher PTH levels compared to patients with essential hypertension and PTH levels decrease after treatment of hyperaldosteronism [ ]. Conversely, PTH has been shown to stimulate aldosterone synthesis via direct and indirect pathways [ ]. Clinically, PHPT is associated with elevated aldosterone levels and this is ameliorated by parathyroidectomy [ ]. It is thought that this interplay between PTH and aldosterone exacerbates the target-organ damage caused by the excess of either hormone alone [ ].
Diagnosis
When establishing the diagnosis, an elevated serum calcium with an elevated or inappropriately normal PTH level is key. Other important biochemical measurements are serum phosphate, alkaline phosphatase activity, renal function, and vitamin D levels. Additional investigations into the presence of, or the risk for, complications should also be considered, such as a bone mineral density (DEXA) scan, a vertebral fracture assessment (VFA), a 24-hr urinary assessment for hypercalciuria, and abdominal imaging to look for renal calculi or nephrocalcinosis.
If surgery is contemplated, localization studies may aid to guide the surgical approach. Ultrasonography, a technetium-99m-labeled sestamibi scan ( 99m Tc-sestamibi), and a four-dimensional computed tomography scan are frequently used imaging modalities for this purpose [ ].
Management
Surgical management provides an opportunity for cure but is not warranted in all cases, and the indications for surgery are described in detail elsewhere [ ]. Whether parathyroidectomy is effective in reducing BP has remained unresolved, and available data on this has been inconsistent and heterogeneous [ ]. In a randomized controlled clinical trial, no beneficial effect on 24-hr ABPM was seen in patients with mild-to-moderate PHPT, 3 months after parathyroidectomy versus a 3-month observation period. In fact, in the treatment group, a small increase in ambulatory DBP was seen [ ]. Over a longer follow up of 3.5 years after parathyroidectomy, little change in hypertension prevalence was observed in 46 PHPT patients [ ]. Furthermore, Rydberg et al. [ ] reported on a 5 mmHg increase in average SBP on 24-hr ABPM, 6 months after parathyroidectomy in PHPT patients with pre-existent hypertension, whereas in normotensive patients no change in average 24-hr ABPM was found. It is likely that these disparate results may be explained by a variable degree of reversibility due to the presence of other cardiovascular risk factors, duration of hypertension, and irreversible target organ damage. In the absence of clinical prognosticators to predict the postoperative BP response after parathyroidectomy, hypertension per se should not be an indication for surgery, and further studies to identify patients who would benefit from the cardiovascular perspective are needed.
Are adjustments in pharmacotherapeutics warranted in patients with concomitant PHPT and hypertension? First of all, thiazide diuretics are known to cause hypercalcemia due to its ability to increase renal tubular calcium reabsorption (thereby reducing the urinary calcium excretion). Whether this exacerbates hypercalcemia in PHPT is unknown, but in a retrospective observational study, Griebeler et al. [ ] found that a high proportion of patients with thiazide-induced hypercalcemia had underlying PHPT, and thiazide use could unmask mild cases of PHPT. It was also suggested that thiazides may actually have beneficial effects on fracture risk due to reduction of hypercalciuria [ ]. In the prospective Nurses’ Health Study, use of loop diuretics, but none of the other antihypertensives including thiazide diuretics, was associated with a higher risk of incident PHPT [ ]. Furthermore, in the study by Griebeler, a small subgroup of PHPT patients safely continued their thiazide diuretic up till definitive surgical management [ ]. In the absence of any large-scale prospective data on the use of thiazide diuretics in the setting of PHPT, it is advisable to use these agents with caution.
In light of the aforementioned interaction between PTH and aldosterone, MR blockade may provide a targeted approach to reduce BP in PHPT. In a randomized clinical trial, an eight-week treatment with the MR antagonist eplerenone resulted in a significant improvement on 24-hr ABPM in patients with PHPT independent of changes in PTH [ ] and MR blockade may provide an exciting new strategy to reduce the cardiovascular risk associated with PHPT that deserves further investigation.
Please refer to Chapter 16 for detailed reading on hypertension in thyroid and parathyroid disease.
Renal artery stenosis and other rare forms of renin driven hypertension
Renal artery stenosis is a common condition which, in some patients, can cause secondary hypertension. In patients with established atherosclerosis the prevalence of renal artery stenosis is high, at up to 14%–42% depending on the study [ ], being more common in those with aortic or peripheral vascular disease and in patients with established chronic kidney disease. Renal artery stenosis due to fibromuscular dysplasia is typically found in younger patients, particularly females, though it can occur in other patient groups. It is important to recognize, however, that regardless of the underlying pathology, the detection of renal artery stenosis in a hypertensive patient is not sufficient in itself to establish it as the cause of the hypertension. While the coexistence of renal artery stenosis and hypertension is relatively common, cases of hypertension that are caused by renal artery stenosis (i.e. renovascular hypertension) and are therefore likely to be improved by vascular intervention are much less frequent. Other much rarer causes of renin driven hypertension include Page kidney and reninoma.
Clinical presentation
As implied above, the age and gender of the patient and presence or absence of vascular disease elsewhere or risk factors for cardiovascular disease can thus be a starting point when considering whether a patient may have renovascular hypertension. An unexplained acute kidney injury after the institution of an ACE inhibitor or ARB is commonly regarded as a potential indicator of significant renal artery disease. This typically occurs within days to weeks and is manifest by a greater than 30% rise in creatinine (often a much greater rise is seen). With significant renal arterial disease, GFR is at least partially dependent on angiotensin II mediated vasoconstriction of the efferent arteriole, and thus inhibition can cause a precipitous rise in creatinine. Severe hypertension with recurrent episodes of flash pulmonary edema is also regarded as a clinical clue suggesting bilateral renal arterial disease. In unilateral renal artery stenosis, the contralateral kidney can compensate with increased sodium excretion in the face of volume expansion (the pressure natriuresis phenomenon); however, in bilateral disease, this escape mechanism is not possible because of impaired perfusion to both kidneys.
Fibromuscular dysplasia can affect almost any arterial bed but the renal arteries are most commonly involved (75%–80% of cases) [ ]. Hypertension is the most common presenting sign but flank or abdominal pain (from ischemic events or dissection) can also be a presenting symptom of renal FMD, and headaches, tinnitus, transient ischemic attacks, and strokes can be an indicator of cerebrovascular FMD. Because FMD commonly affects multiple arterial beds in individual patients (up to 2/3 of patients in one recent registry study) [ ], modern guidelines suggest initial cross-sectional imaging from head to pelvis when FMD is found [ ].
Renal artery bruits may be found in renal artery stenosis, and careful examination for bruits of the carotid and subclavian arteries and more widely over the abdomen for other arterial bruits is useful especially in FMD. Evidence for peripheral vascular disease in the lower limbs is also helpful in determining the overall atherosclerotic burden.
Diagnostic approach
Renal duplex ultrasound is a useful noninvasive screening method for renal arterial disease. It is, however, highly operator-dependent and although sensitivity for significant renal artery stenosis is high (up to 98%) in high-volume referral centers [ ], it may not be the modality of choice in centers with less technical experience. CT angiography is less-operator dependent, has much greater spatial resolution and requires less technical expertise and in many settings is a better screening test but requires contrast administration with the risk of acute kidney injury or rarely allergic reaction. Magnetic resonance angiography (MRA) is an alternative imaging modality and can be considered where CT is contraindicated, for example because of contrast allergy. MRA however has lower sensitivity compared to CT because of lower spatial resolution and this can be important particularly in FMD which commonly affects the mid and distal renal arteries. Digital subtraction angiography is the reference standard test for diagnosis of renal artery stenosis and can be considered for diagnosis when there is a strong clinical suspicion of renal artery stenosis and particularly FMD where noninvasive imaging techniques have lower sensitivity [ ]. Angiography can also be followed by angioplasty in the same procedure. Other adjunctive imaging techniques including ACE inhibitor-augmented renography using nuclear medical techniques can give additional information (including differential renal function, which may assist in assessing management options, for example, unilateral nephrectomy in the case of nonfunctioning renin-producing kidney) and have reasonable sensitivity in the right setting, but have become less popular with greater availability of highly accurate CT scanners.
The most difficult aspect of renal artery stenosis is determining if a stenosis identified on imaging is hemodynamically or functionally significant, and if intervention will result in improvement in blood pressure control or renal outcomes. Illustrating this point, randomized trials to date have failed to demonstrate a benefit to angioplasty in atherosclerotic renal artery stenosis compared to standard medical therapy [ , ]. This reflects the difficulty in determining, from imaging studies alone, which patients have a hemodynamically significant lesion. In our Center’s experience, renal vein renin measurement has an important role in determining the significance of renovascular disease in terms of its contribution toward a patient’s hypertension and in planning intervention. Renin secretion from a stenotic kidney that is >1.5 times higher than the contralateral kidney, especially after the administration of an ACE inhibitor, is predictive of an improvement in blood pressure after intervention, though reports vary [ , ]. It is likely that the sensitivity and specificity of the test is improved with very strict control of factors influencing the renin-angiotensin system, including avoiding RAAS blockade and diuretics, a strict low salt diet prior to the test (to stimulate renin production), control of posture on the day of the test (subjects maintain recumbency overnight until the test is complete), and collection of pre- and post-ACE inhibitor stimulated samples [ ]. Performing this test in such circumstances is therefore difficult and requires an experienced unit with a strict protocol.
Management
Patients with renovascular hypertension due to FMD generally do well with angioplasty. Younger patients with a short duration of hypertension and focal FMD lesions have a particularly high chance of cure of hypertension after intervention of up to 50% in some studies [ , ]. Most FMD lesions respond well to angioplasty alone, with stent insertion appropriate for lesions which don’t respond to angioplasty or where a dissection has complicated angioplasty. Renal artery aneurysms or very complicated disease may be better treated with a surgical approach with bypass or resection. All patients should be treated with aspirin or an alternative antiplatelet agent. Patients with renal FMD should have ongoing follow-up with measurement of renal function and a renal artery duplex initially yearly seeking evidence of progression of stenosis or recurrence after intervention.
In contrast to FMD, multiple randomized trials of angioplasty in atherosclerotic renal artery stenosis have not demonstrated consistent benefit compared to optimal medical therapy [ , ]. Limitations of the intervention trials relate mainly to the selection criteria, with exclusion of patients which most guidelines suggest would be most likely to benefit from intervention, such as patients with acute pulmonary oedema, short duration of hypertension or drug resistant hypertension. Good quality observational data suggests that intervention in patients with specific indications such as acute pulmonary edema is more likely to result in benefit [ ]. Intervention for atherosclerotic disease should thus generally be reserved for those with a high likelihood of benefit, which might include patients with a short duration of hypertension, patients with drug resistant hypertension, patients with recurrent acute pulmonary edema and patients with very suggestive functional studies (such as ACE inhibitor augmented renograms or renal vein renin studies). Unlike FMD, atherosclerotic lesions generally require stent insertion due to elastic recoil, and stenting reduces rates of subsequent restenosis [ ]. In addition, patients with a small kidney poorly functioning kidney (<15% split function) with lateralizing renal vein renin studies might benefit from nephrectomy of a “pressor kidney.” All patients with atherosclerotic renal artery disease should have optimal cardiovascular risk factor reduction.
Page kidney
Page kidney is named after Irvine Page who demonstrated that wrapping an animal kidney in cellophane produced hypertension that responded to nephrectomy [ ]. The phenomenon occurs because of external kidney compression, which activates the renin-angiotensin system likely via an ischemic mechanism. The typical patient is a young male athlete presenting after sports trauma, however contemporary case series show that kidney biopsy and transplant allograft biopsy are common causes [ ]. Interestingly there can be a lag of many years between the trauma and the presentation with hypertension, though most subjects present acutely. Renin is typically high and renal vein renin studies invariably lateralize if they are performed. Patients usually respond well to decompression but nephrectomy may be required in some cases.
Reninoma
A reninoma is a benign tumor arising from juxtaglomerular cells. Cases have been sporadically reported in the literature with approximately only 100 reported to date. The typical presentation is of an adolescent or young adult with severe hypertension. There is a clear female predominance and patient presents with evidence of secondary hyperaldosteronism with normal or high renin, normal or high aldosterone, hypokalemia and a metabolic alkalosis. Ultrasound imaging may miss reninomas, but modern contrast CT will reveal most of them, which appear as well circumscribed low-density lesions. Renal vein renin studies typically lateralize strongly to the kidney in which a reninoma is present, and partial or total nephrectomy is very effective for treatment. Several case reports suggest that the direct renin inhibitor Aliskiren is also partially effective in treating reninoma though treatment breakthrough may occur [ , ]. Renal cell carcinoma and Wilm’s tumor can also secrete renin [ ] and histopathological examination of the tumor is important to confirm the diagnosis.
Conclusion
Searching for secondary endocrine causes of hypertension can be a highly rewarding experience for both patients and physicians but requires a high index of suspicion and a systematic approach. Fig. 21.1 (Panels A, B and C) summarizes the approach used within our Center at Princess Alexandra and Greenslopes Hospital for the initial diagnostic assessment of a patient referred with suspected endocrine hypertension.
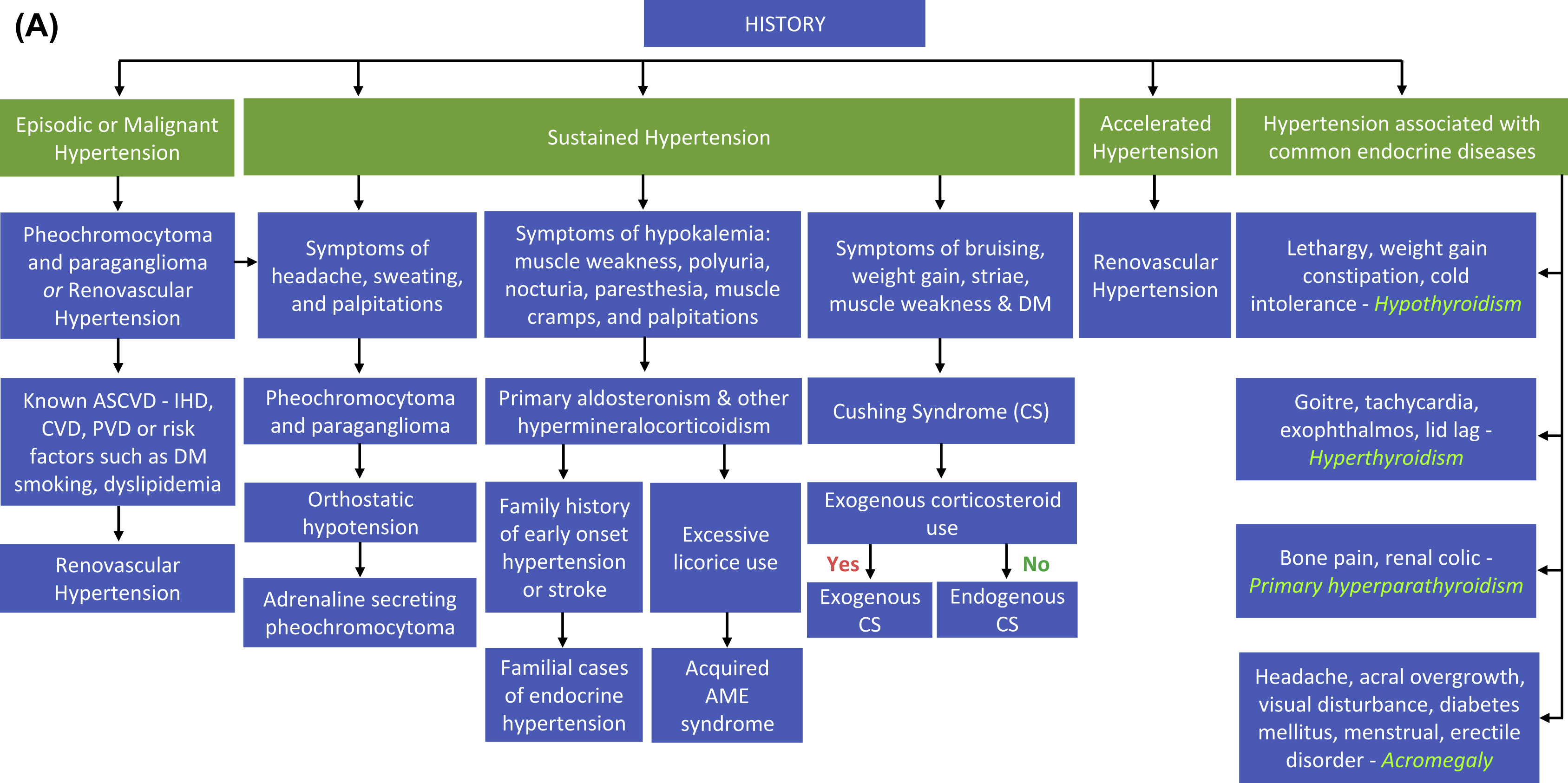
Related posts:
 Congenital Adrenal Hyperplasia
Congenital Adrenal Hyperplasia
 Syndromes of Mineralocorticoid Excess
Syndromes of Mineralocorticoid Excess
 Hypertension in Patients with Cushing’s Syndrome
Hypertension in Patients with Cushing’s Syndrome
 Renin–angiotensin–aldosterone system and blood pressure regulation
Renin–angiotensin–aldosterone system and blood pressure regulation
 Adrenal cortical hormones and blood pressure regulation
Adrenal cortical hormones and blood pressure regulation
 Hypertension in thyroid disease and primary hyperparathyroidism
Hypertension in thyroid disease and primary hyperparathyroidism
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


