Fig. 3.1
Diagnostic workup for patients with Cushing’s syndrome. Modified from Melcescu, Koch. Testing for endocrine hypertension, Chapter 7, In: Leslie De Groot, editor, online http:// www.endotext.org; 2012
Ectopic ACTH secretion usually causes higher glucocorticoid excess than eutopic ACTH oversecretion by an ACTH-oma in the pituitary gland. This explains in part why patients with ectopic ACTH syndrome usually have a higher prevalence of hypertension and diabetes mellitus than those patients with Cushing’s disease [2, 6]. CS is well known to associate with significant morbidity and to increase the mortality rate. The last, assessed as standardized mortality ratio, ranges between 1.7 and 4.1 [3, 4, 14–16], and is driven by associated morbidities which include hypertension, cardio- and cerebrovascular disease, hypercoagulability, diabetes mellitus, and depression. Of significant concern is the fact that mortality remains increased even after patients with CS have successfully been treated [16]. Although hypertension is seen more frequently in patients with Cushing’s disease than in those with adrenal CS, the survival rates are better for the first group of patients [17, 18]. Ectopic CS is often caused by neuroendocrine neoplasms, including small cell lung cancer, and the survival rate is expectedly lower than for either of the above [17].
Pathogenesis of Hypertension in Patients with CS
Hypertension is a common clinical feature of CS (sensitivity 74–90%, specificity 83%) but is neither universal nor predictable to the degree of other clinical features of the disease [5]. It is seen in about 80% of adults and up to 60% of children with CS [6, 19, 20]. While after successful cure of CS, hypertension still persists in adults in as many as 30%, in children and adolescents this figure is much more favorable—around 3–4% [21, 22]. Clinical consequences of CS depend on several factors, among these the duration and level of pathologic increase of glucocorticoids. Hypertension will relate mostly to obesity and increased peripheral resistance and sympathetic drive. In patients with ectopic ACTH production by neuroendocrine tumors, marked hyperplasia of the adrenals and a severe increase in various steroids can occur, which will lead to signs of more severe disease with an overall shorter course, more features of mineralocorticoid excess and higher probability of fast overload of functional capacity of 11-beta-hydroxysteroid dehydrogenase (11-βHSD—see below).
In this case, hypertension is seen in up to 95% of cases, while with exogenous glucocorticoids, hypertension is of significantly lower frequency—20%—and correlates with the dose of steroid used [19]. Administration of exogenous glucocorticoids rapidly increases both systolic and, to lesser degree, diastolic blood pressure for hydrocortisone, dexamethasone or ACTH [23, 24]. The establishment of the pathogenesis of CS-related hypertension is based on several lines of evidence: assessment of the phenomenon in patients with CS or chronic treatment with exogenous steroids, studies in healthy volunteers taking ACTH or oral steroids for a limited period of time, and animal studies. The last two have several expected limitations—while short-term hypercortisolemia in healthy people might not represent a long-term multi-organ response, animal studies show significant species variability and might not represent findings relevant to human physiology. For this discussion, we will try to select the most representative data and review additional studies where appropriate.
Causes of hypertension in CS are numerous (Fig. 3.2) and relate to different pathogenesis pathways [6, 25, 26]:
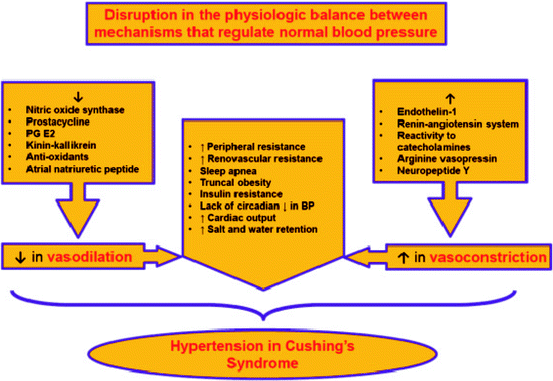
Fig. 3.2
Pathogenic pathways of hypertension development in CS
(a)
Signaling of glucocorticoids through the mineralocorticoid receptor (MCR)
(b)
Signaling of glucocorticoids through the GC receptor (GCR)
Inhibition of vasodilatation (decrease in nitric oxide synthase, prostacycline, kinin–kallikrein; increase in endothelin-1)
Activation of the renin–angiotensin system
Increase in vascular reactivity to catecholamines
Inhibition of norepinephrine metabolism
(c)
Indirect action of glucocorticoids
Increase in peripheral and renovascular resistance
Sleep apnea, truncal obesity, insulin resistance
Lack of circadian decrease in blood pressure
(d)
Unrelated to glucocorticoids—lack of hypertension resolution after cure from CS
Actions of Glucocorticoids Through the MCR: A Convenient Culprit
It is common knowledge that glucocorticoids can bind to and signal through the MCR. It is also a well-accepted dogma that the gate-keeping 11-beta-hydroxysteroid dehydrogenase type 2 (11-βHSD2) system is saturable and in case of significantly increased hypercorticolemia glucocorticoids, levels can reach its binding capacity. In case of significant hypercorticolemia, glucocorticoid levels can reach the MCR and act as pure mineralocorticoid (MC), which will expectedly associate with hypertension, as is the case for the syndrome of apparent mineralocorticoid excess (AME). The fact that glucocorticoid levels in patients with CS are significantly higher than normal, which are already at least 100 times higher than the physiologic level of MCs, would suggest that each and every case of CS should associate with clinical signs of severe hyperaldosteronism and appropriately resolve with spironolactone/eplerenone treatment. Unfortunately, this is not the case in most patients with CS and while treatment of CS or administration of GCR antagonist improves hypertension, spironolactone failed to do so [27, 28]. With this apparent conflict in mind, it seems reasonable to review the physiology of this system.
Physiology of the GC/GCR/MCR System: Was it a Faulty Design?
Glucocorticoids are secreted from the zona fasciculata of the adrenal cortex as a part of the hypothalamic–pituitary–adrenal axis under tight control of both hypothalamus and pituitary. The last respond to both environmental and internal stressors, as well as show a robust feedback to cortisol levels. Glucocorticoids act as a ligand for two transcription factors—GR and MR, which belong to the nuclear receptor superfamily. While GR is specific for glucocorticoids, MR is equally affine to both GCs and MCs [29]. Both receptors reside in the cellular cytoplasm and upon activation by the ligand gain capability of nuclear translocation and action as transcription factors (Fig. 3.3).
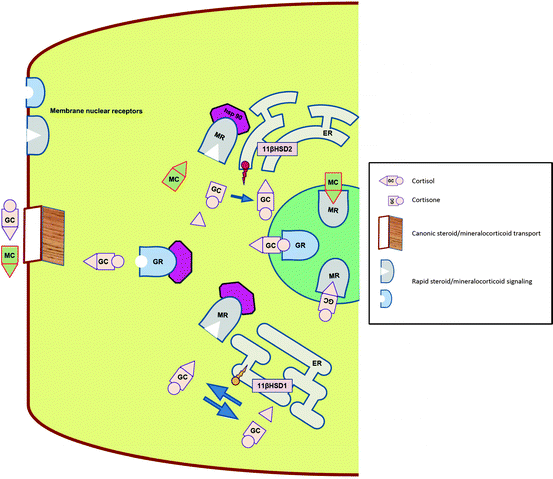
Fig. 3.3
Physiology of intracellular GC and MR transport, binding and signaling. GC glucocorticoid, MC mineralocorticoid, GR glucocorticoid receptor, MR mineralocorticoid receptor, Hsp 90 heat shock protein 90, ER endoplasmic reticulum, 11βHSD hydroxysteroid dehydrogenase
Although not part of this review, both have an exceedingly complicated co-activation/co-suppression complex, which further affect the action of these factors. There are several differences in physiology of MRs and GRs. The main two are: (a) while GRs are ubiquitously expressed throughout the body, MRs express selective distribution, providing thus “geographic” tissue selectivity—distal nephron of the kidney, colon, salivary, and sweat glands, (b) while GRs are accessible to the ligand at any given time, MRs use a pre-receptor gate keeper 11-βHSD2 (see below) to convert cortisol to cortisone, which lacks the ability to bind to the MR. This assures relative functional selectivity of the MR towards the true mineralocorticoid aldosterone. This process is especially important while keeping in mind the fact that circulating levels of cortisol are about 100–1,000 times higher than those of aldosterone.
Both MRs and GRs are ancient—both descended from an ancestral corticoid receptor (AncCR) by gene duplication over 450 million years ago. Original AncGR was affine to both 11-deoxycorticosterone (aldosterone appeared later in evolution) and cortisol, and could easily be called AncMR, which could probably be more deserving. Series of more prominent changes created GC-specific GCR, while whatever changes happened to the AncGR resulted in MR, which still binds both GRs and MRs with equivalent affinity. While it is hard to fault evolution for lack of imagination or time, specificity of MRs for MCs is maintained through principally different, enzymatic mechanisms rather than the logically expected specificity of the ligand-binding domain [30].
The binding promiscuity of MRs is extensive—it is functionally important for transcriptional activation by both mineralocorticoids (aldosterone and deoxycorticosterone [DOC]) and glucocorticoids (cortisol in humans and corticosterone in rodents), but can actually also bind estrogens. Both GRs and MRs are highly homologous, less at the steroid binding domain, though—56%—which seem not to be able to prevent similar high affinity binding of both MCs and GCs [31]. The canonic—mineralocorticoid action—of MRs is found in the classic MR target epithelial polarized cells—colon, distal nephron, and sweat glands. MR expression in non-epithelial tissues—hippocampus, endothelium, cardiomyocytes, and adipocytes—is the subject of intense interest and research [32].
11-βHSD System: Too Smart to Fail
The existence of functional interconversion between active and inert forms of glucocorticoids (cortisol vs. cortisone in humans, and corticosterone vs. 11-dehydrocorticosterone in rodents, respectively) was characterized in the early 1950s [33]. It was soon discovered that functional activity of the compound was all about possession of a hydroxyl group at C11 [34]. Subsequently, two enzymes were characterized as having 11-βHSD properties—first was what is currently known as 11-βHSD1 and later 11-βHSD2 [35, 36]. Although the term “isoenzymes” is often applied to the pair, other than being 11-β hydroxysteroid dehydrogenases, these two enzymes have very little in common and their nomenclatural division into type 1 and 2 represents the sequence of discovery rather than anything else. Both enzymes are part of the short-chain dehydrogenase superfamily and share only 21% homology, mostly at sites homologous for the rest of the family. For the sake of continuous discussion related to the physiology of the MCR, we first discuss 11-βHSD2 and its relation to glucocorticoid signaling.
11-βHSD2 is a unidirectional dehydrogenase, expressed in mineralocorticoid target tissues—kidneys, colon, salivary gland, and placenta—and tightly co-expressed with MCR. The encoding gene is localized on chromosome 16 (16q22) and the protein consists of 405 amino acids. The enzyme uses nicotinamide adenine dinucleotide (NAD+) as a cofactor and has high (50 nM K m) affinity for cortisol [36]. The main functional role is dehydrogenation at C11 of species-specific active GC, while the main existential purpose is protection of MCR from same active GC. The functional importance of the enzyme is underlined by its inherited deficiency—syndrome of AME [37]. Clinically, AME presents with severe early onset hypertension associated with hypokalemia and low levels of both renin and aldosterone. Biochemically, deficiency of either 11-βHSD can be suggested based on the ratio between urinary excretion of A-ring reduced metabolites for both cortisol and cortisone (tetrahydrocortisol/tetrahydrocortisone), as well as the urinary free cortisol/cortisone ratio, the last is thought to be more specific for 11-βHSD2. The disease is caused by mutation in the 11-βHSD2 gene and shows genotype/phenotype correlation, based on the amount of functional enzyme. As a final proof that AME is caused by cortisol ability to signal through the MCR, it readily responds to treatment with spironolactone and/or low salt diet. Another phenomenon is lack of other clinical features of CS, despite increased half-life and thus levels of cortisol, which can be explained by decreased overall secretion through the negative feedback effect [38]. While it is thought that 11-βHSD2 protects MRs from being occupied by glucocorticoids, it has been shown that about 90% of epithelial MRs are occupied by glucocorticoids [39]. This phenomenon occurred in normal conditions—normal levels of endogenous glucocorticoids. In this case, transcriptional activity of the MR-GC complex maintained inactive by high levels of NADH, which activates the corepressor complex [40]. With change in redox that is associated with generation of reactive oxygen species, the MR-GC complex becomes active, changing the glucocorticoid action from antagonist to agonist [41]. The 11-βHSD2, as well as 11-βHSD1, is also expressed in the vascular endothelium, including coronary arteries. Functionally active MRs were described in hippocampus and hypothalamic neurons, as well as cardiomyocytes, adipocytes, and vasculature [32]. One can speculate that in case of significant oxidative stress and decrease in redox potential, the protective capacity of the enzyme will be severely diminished and, at least on the autocrine level, GC will signal through MRs.
11-βHSD1 is a bidirectional microsomal enzyme, highly expressed in glucocorticoid target tissues—liver, lungs, central nervous system, gonads, and adipose tissue. The encoding gene is localized on chromosome 1 (1q32-41) and the protein consists of 292 amino acids. The enzyme depends on NADP(H) as a cofactor and acts as a low affinity both 11-oxo-reductase and dehydrogenase, although reductase seems to be the preferential activity in vivo [33]. The set point, as well as overall activity of 11-βHSD1 also depends on microsomal hexose-6-phosphate dehydrogenase (H6PD), which is responsible for generation and provision of NADPH to 11-βHSD1. The main purpose seems to be provision of active GC to GCR on as needed paracrine fashion. This speculation is based on the following facts [42, 43]:
(a)
Intrinsic features of cortisol
Circadian variability of levels
High binding to cortisol-binding protein
Relatively low affinity of GR to cortisol
(b)
Intrinsic features of cortisone
No circadian variability
No significant binding to cortisol-binding protein
Relatively constant, significant, and readily available to peripheral issues
(c)
Physiology of the paracrine GC system
Needed for acute and chronic inflammation
Needed for adipocyte maturation
Abnormal in obesity and metabolic syndrome
More Causes, Including Imbalance Between Vasoconstrictors and Vasodilators
Administration of synthetic glucocorticoids and ACTH were used extensively to assess the pathogenesis of hypertension in CS. However, the question remains about how much these represent pathophysiology of actual disease, because of relatively high doses, as well as baseline condition, which usually is normal health and acute mode of administration. These last may evoke acute compensatory responses that will significantly modify the process in comparison to actual chronic CS. Additionally, different synthetic glucocorticoids (prednisone, dexamethasone, triamcinolone) have different pharmacodynamics/pharmacokinetic profiles and intrinsic mineralocorticoid activity, which may affect severity and degree of hypertension through induction of additional mechanisms. It was found that oral dexamethasone causes an increase in total calculated peripheral resistance in humans without an increase in cardiac output or heart rate, while in dogs, a higher dose will also add decrease in cardiac output [44, 45]. Both hydrocortisone in humans and ACTH in rats caused hypertension, associated with an increase in both cardiac output and renal vascular resistance, but not total peripheral resistance or heart rate [44–46]. At least with hydrocortisone-induced hypertension in humans, neither β-blocker (atenolol), nor calcium channel blocker (felodipine) affected hypertension, which adds to reservations about clinical relevance of this model, as well as our understanding of this phenomenon [44, 45, 47]. One can argue that an increase in cardiac output and peripheral resistance can relate to an increase in plasma volume and/or fluid/salt retention, but further research shows that this is not the case [27, 28, 48].
Changes in the balance between vasoconstrictors and vasodilators can include different parameters—amount, expression, or sensitivity—and seem to play a significant role in the development of hypertension in patients with CS [6]. The group of vasoconstrictors/vasopressors includes the following: (a) endothelins, (b) angiotensin, (c) arginine vasopressin, (d) catecholamines, (e) neuropeptide Y, and others [6, 49].
Endothelins are potent direct vasoconstrictors and elevated endothelin-1 (Fig. 3.4) levels are found in patients with CS, as well as during treatment with prednisone [50, 51]. Synthetic glucocorticoids can induce catecholamine synthesis through activation of enzymes involved in the process, as well as increased expression of adrenergic receptors in vascular smooth muscles, thus promoting a vasoconstrictive response [49].
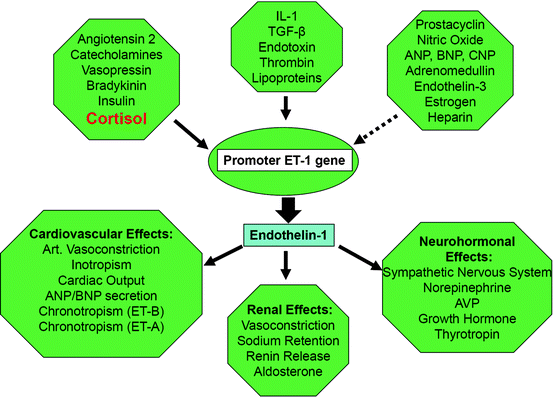
Fig. 3.4
Consequences of disease-related oxidative—stress regarding arteriolar tone. (Solid arrow: stimulation, non-solid arrow: inhibition), see http://www.endotext.org, Chapter 25. Hormones of the cardiovascular system by Pliquett, Buerke, Paschke, Koch CA. In: Chrousos GP, section editor. Adrenal disease and function; 2011
Metabolic Syndrome: An Easy Blame
Another way to look at comorbidities of CS, including hypertension, is based on a purely clinical rather than pathophysiologic approach. Metabolic syndrome which includes central obesity, hypertension, insulin resistance, dyslipidemia, closely associates with CS and its morbidity [3, 4]. As with other comorbidities, hypertension seems to persist as part of the metabolic syndrome after resolution of hypercortisolemia with seemingly no difference between different etiologies of CS [53, 54]. Weight gain, especially truncal obesity, obstructive sleep apnea, insulin resistance, loss of circadian BP variability—all by themselves associate with hypertension and do not fully resolve after the treatment of CS and thus can explain, at least in part, persistent hypertension after successful treatment. Differently from animal—especially rodent—models of obesity, obese humans show normal or decreased systemic cortisol levels [55]. Thus, while it is known that adiposity associates with local production of cytokines and that cortisol is needed for adipocyte maturation, it seems that it is a paracrine steroid delivery system, based on action of 11-beta-hydroxysteroid dehydrogenase type 1 that delivers the active hormone to the receptor, as both are closely co-expressed in these tissues. The mouse model with overexpression of 11-βHSD1 in white adipose tissue shows the phenotype of metabolic syndrome including hypertension, while obese humans show similar increased activity of 11-βHSD1 in their subcutaneous fat [43, 56].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


