Common conditions with low renin concentrations
Mineralocorticoid excess
Primary aldosteronism (see Chap. 1)
Cushing’s Syndrome (see Chap. 3)
Glucocorticoid/cortisol resistance (see Chap. 4)
Apparent mineralocorticoid excess syndrome
Licorice or carbenoxolone in excess
Congenital adrenal hyperplasia (11beta- and 17alpha-hydroxylase deficiencies)
11-Deoxycorticosterone (DOC), 18-hydroxy-DOC excess
Salt retention (Gordon and Liddle syndrome)
Geller syndrome
Salt loading (oral or intravenous)
Rare conditions leading to low renin levels
Increasing age
Low renin essential hypertension
Hyporeninemic hypoaldosteronism
Hyperkalemia
Therapy with beta-adrenergic blockers
Autonomic dysfunction
Decrease of renal tissue or being anephric
Most frequently, the underlying condition for syndromes of mineralocorticoid excess is autonomous secretion of aldosterone or primary aldosteronism (see Chap. 1 in this book and ref. [2]). Aldosterone mediates its action through the mineralocortoid receptor (MR) which regulates salt homeostasis in the kidneys and plays a range of other roles in the vasculature, heart, brain, and adipose tissue. To mediate transcription of target genes, the MR interacts with both mineralocorticoids and glucocorticoids (reviewed in [3]). The MR is able to exert tissue- and ligand-specific effects via its interactions with a range of binding partners. The MR also plays a major role in inducing glomerular podocyte injury and progression of chronic kidney disease which can be reduced by MR antagonists or selective MR inhibition [4].
Excessive MR activation can promote inflammation, fibrosis, and heart disease as well as psychiatric illness including anxiety and depression through key modulators including the glucocorticoid receptor (GR) and 11β-hydroxysteroid dehydrogenases (11βHSDs), which determine the amount of intracellular concentrations of active glucocorticoids [5, 6].
Aldosterone secretion is stimulated by angiotensin II, high potassium, vasopressin, and ACTH (transient), and inhibited by high levels of natriuretic peptides. Apart from aldosterone the MR is also activated by products of abnormal adrenal steroid biosynthesis (Fig. 2.1), dysregulated metabolism of cortisol in cells that are targets of mineralocorticoids, and activating mutations of the MR or of ion channels that are inducible by the MR to mediate its action.
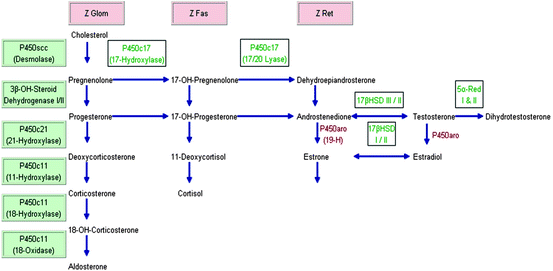
Fig. 2.1
Adrenal steroid synthesis. Modified from ref. [7]. Z Glom zona glomerulosa; Z Fas zona fasciculata; Z Ret zona reticularis; 19-H 19-Hydroxylase; HSD Hydroxysteroid dehydrogenase; P450aro aromatase; 5alpha-Red 5alpha-Reductase. The three adrenal cortex zones Z Glom, Z Fas, and Z Ret stand above the “column” of hormones that are produced in the respective zone. The steroidogenic enzymes on the left starting with P450scc (Desmolase) are listed in order for “vertical and horizontal reading,” i.e., Desmolase converts cholesterol to pregnenolone, 3beta-OH-Steroid Dehydrogenase I/II convert pregnenolone to progesterone, 17-OH-Pregnenolone to 17-OH-Progesterone, and P450c11 converts deoxycorticosterone to 18-OH-Corticosterone and 11-Deoxycortisol to cortisol, etc
SME and its inherited forms of mineralocorticoid hypertension have helped our understanding of normal physiology and of the pathogenesis of hypertension called “essential” which represents a major public health problem [8].
Hypertension is defined as a blood pressure exceeding 139/89 mmHg for adults aged 18 years or older based on the mean of two or more properly measured seated BP readings on each of two or more office visits. Hypertension affects approximately 31% of Americans; blood pressure control is suboptimal and is achieved in less than 1 in 3 [9, 10]. The prevalence of resistant hypertension varies from 34 to 53% in different large studies: ALLHAT (34%), NHANES (53%), or Framingham Heart Study (48%) [11]. The Joint National Committee (JNC VII) [12] classified hypertension for adults 18 years and older as follows:
Normal | <120 mmHg (systolic) | <80 mmHg (diastolic) |
Pre-HTN | 120–139 | 80–89 |
Stage 1 HTN | 140–159 | 90–99 |
Stage 2 HTN | ≥160 | ≥100 |
Clinical Findings and Clues to Identify Patients with Endocrine Hypertension Caused by Mineralocorticoid Excess
Individuals homozygous for an inherited condition of SME may develop hypertension very early in life and become refractory to antihypertensive therapy (failing triple-drug treatment). However, the clinical phenotype varies and some patients may not be found to have SME before they reach adulthood (Table 2.2).
Table 2.2
Clinical findings in patients with mineralocorticoid excess
Condition | Clinical presentation |
|---|---|
CAH: 11beta-hydroxylase deficiency | Early growth spurt initially, then short adult stature, advanced bone age, premature adrenarche, acne, precocious puberty in males, amenorrhea/hirsutism/virilism in females |
CAH: 17alpha-hydroxylase deficiency | Pseudohermaphroditism (male), sexual infantilism (female) |
Apparent mineralocorticoid excess | Growth retardation/short stature, nephrocalcinosis |
Liddle syndrome | Severe hypertension, hypokalemia, and metabolic alkalosis, muscle weakness |
Geller syndrome | Early onset hypertension (before age 20 year), exacerbated in pregnancy |
Glucocorticoid remediable aldosteronism (GRA) | Early onset of hypertension, presence of family history of mortality or morbidity from early hemorrhagic stroke |
Pseudohypoaldosteronism type 2 | Short stature, hyperkalemic and hyperchloremic metabolic acidosis, borderline blood pressure |
Glucocorticoid resistance (children)(Chrousos syndrome) | Ambiguous genitalia, precocious puberty. Women may have acne, excessive hair, oligo/anovulation, infertility |
Pathogenesis
The pathophysiological mechanisms of mineralocorticoid excess syndromes are better understood at present with the advent of new techniques in molecular biology and genetics. The increasing availability of affordable genetic/molecular testing may elucidate further the pathogenesis of these rather rare genetic conditions and their implication in hypertension seen in the general public. Excessive production of aldosterone (primary: familial hypoaldosteronism, aldosterone-producing adenoma/carcinoma/hyperplasia [13–16] or secondary: reninoma [17–20]) and sometimes of deoxycorticosterone (DOC) (primary: DOC-secreting adrenocortical adenoma/carcinoma [21–24] or secondary: 11beta-hydroxylase deficiency [25–27], 17alpha-hydroxylase deficiency [28–30], substances with high mineralocorticoid activity can lead to salt and water retention and consequently hypertension without causing hypernatremia.
11Beta-hydroxylase is responsible for the conversion of DOC to corticosterone (precursor of aldosterone) and 11-deoxycortisol to cortisol. In approximately 2/3 of individuals affected by a deficiency of this enzyme, monogenic low renin hypertension with low aldosterone levels ensues caused by accumulation of 11-deoxycortisol and DOC. Also, adrenal androgens are produced in excess in individuals with 11beta-hydroxylase deficiency compared with 17alpha-hydroxylase deficiency where they are deficient [25–30]. In patients with 17alpha-hydroxylase deficiency a persistent elevation of ACTH increases DOC and corticosterone levels and as a consequence affected patients will develop hypertension, hypokalemia, low aldosterone, and suppressed renin [28–30].
AME is caused by deficiency of the enzyme 11βHSD2) [31–33]. This enzyme is responsible for the conversion of cortisol to the inactive cortisone in renal tubular cells. Cortisol and aldosterone have equal affinity for the mineralocorticoid receptor but normal circulating concentrations of cortisol are 100–1,000-fold higher than those of aldosterone [34]. If 11βHSD2 is oversaturated or defective, more cortisol will be available to bind to the mineralocorticoid receptor [35]. Diminished 11βHSD2 activity may be hereditary or acquired. Acquired deficiency of this enzyme may result from its inhibition by glycyrrhetinic acid, the active metabolite generated when using licorice, certain brands of chewing tobacco, and carbenoloxone. The mutations in the 11βHSD2 gene are responsible for the reduced activity of the enzyme, which causes Na retention, low K, renin, and aldosterone.
In Liddle syndrome, “gain of function” mutations in the genes coding for the beta- or gamma-subunit of the renal epithelial sodium channel, located on chromosome 16p13, lead to constitutive activation of renal sodium and water resorption, hypertension, low renin, and aldosterone [36].
Pseudohypoaldosteronism type 2 or Gordon syndrome is an autosomal dominantly inherited disorder with genes mapping to chromosomes 1, 12, and 17 [37, 38]. Mutations have been identified in WNK kinases WNK1 and WNK4 on chromosomes 12 and 17, respectively. More recently, mutations in the KLHL3, CUL3, and SPAK genes have been linked to Gordon syndrome. Published families with this condition (hypertension, hyperkalemia, metabolic acidosis, normal renal function, low/normal aldosterone levels) are predominantly from Australia [38] or the United States (Lifton and coworkers at Yale University, CT). Hypertension in these patients may develop as a consequence of increased renal salt reabsorption; and hyperkalemia ensues as a result of reduced renal K excretion despite normal glomerular filtration and aldosterone secretion [39–41]. The reduced renal secretion of potassium makes this condition look like an aldosterone-deficient state, thus the term “pseudohypoaldosteronism.”
Activating mutations in the amiloride-sensitive sodium channel of the distal renal tubule may explain the pathogenesis of this condition. Overactivation of the NaCl cotransporter results in Na and water retention and increased blood pressure.
Glucocorticoid resistance or Chrousos syndrome (see Chap. 4 in this book) is caused by inactivating mutations of the glucocorticoid receptor gene [42–44]. Chronic elevation of ACTH can lead to stimulation of adrenal compounds with mineralocorticoid activity (corticosterone, DOC), and elevation of cortisol may lead to stimulation of the mineralocorticoid receptor, resulting in hypertension and increasing androgenic activity (androstenedione, DHEA, DHEAS) in women.
Constitutive activation of the MR is an autosomal-dominant condition determined by “gain of function” mutations in the MC gene on chromosome 4q31 [45].
Figure 2.2 provides an overview of these pathogenic mechanisms involved in mineralocorticoid excess syndromes Fig. 2.2.
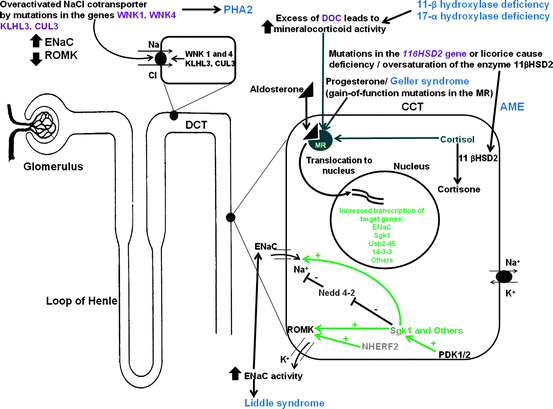
Fig. 2.2
Inherited endocrine conditions related to mineralocorticoid excess (modified after ref. [46, 47], and http://home.comcast.net/∼llpellegrini/urinarysystem.htm). Shown are the molecular pathways involved in dysregulation of NaCl homeostasis located in the distal renal tubules. AME – Apparent mineralocorticoid excess, GRA – Glucocorticoid-remediable aldosteronism, PHA2 – Pseudohypoaldosteronism type 2; MR – mineralocorticoid receptor; WNK – with-nolysine (K) kinase 1, 4; ROMK- renal outer medullary potassium channel; ENaC – epithelial sodium channel; KLHL3 – kelch-like 3; CUL 3 – cullin 3; Sgk1 – serum glucocorticoid kinase 1; Nedd4-2, ubiquitin-protein ligase; deubiquitylating enzyme Usp2-45; 14-3-3 proteins; NHERF2 – Na+/H+ exchange regulating factor 2; PDK1,2 – 3-phosphoinositide-dependent kinases 1 and 2; + activation; – inhibition
Diagnosis
As is true for measuring any hormone, when analyzing steroids, it is important to obtain accurate information. The most sensitive and specific method to detect a defect in adrenal steroid biosynthesis is urinary steroid profiling [47]. Particularly robust appears to be measuring steroid precursor/product ratios which are independent of 24-h excretion and usually not determined by commercial laboratories.
The diagnostic approach to syndromes of mineralocorticoid excess is based on the clinical presentation, laboratory, and genetic testing. The most important laboratory and genetic tests are described in Table 2.3.
Table 2.3
Laboratory testing protocols for some causes of mineralocorticoid excess syndromes
Disease | Laboratory testing | Genetic testing |
|---|---|---|
CAH: 17α-OH deficiency | ≠DOC, Ø 11-deoxycortisol ØØ Aldosterone, Ørenin, ØK ØPlasma 17-hydroxyprogesterone Øtestosterone ≠Urinary100*THDOC/(THE+THF+5αTHF) and (THA+THB+5αTHB)/(THE+THF+5αTF) | CYP17 gene |
CAH: 11β-OH deficiency | Potassium depletion (variable), Ørenin (The degree of hyporeninemia may vary widely) ØØAldosterone ØCortisol, ≠ACTH ≠11-Deoxycortisol, ≠DOC, ≠17-OHP ≠19-nor-DOC ≠≠Urinary 100*THS/(THE+THF+5αTHF) and 100*THDOC/(THE+THF+5αTHF) | CYP11B1 gene |
Apparent mineralocorticoid excess | Check the level of tritiated water in plasma samples when 11-tritiated cortisol is injected ≠24 h Urinary free cortisol/cortisone and ≠urinary (THF+5αTHF)/THE | 11βHSD2 gene |
Liddle syndrome | ØPlasma K, ≠urinary K, ØPRA and suppressed aldosterone secretion, metabolic alkalosis ØUrinary THALDO (<2 μg/24 h) Normal steroid profile (24-h urine cortisone/cortisol and other ratios) | ENaC gene |
Pseudohypo-aldosteronism type 2 | ≠≠K, hyperchloremic metabolic acidosis Øaldosterone, Ørenin ØSerum HCO3 (variable in children) Hypercalciuria (occasionally) ØUrinary THALDO | WNK1 and 4 gene KLHL3, CUL3, SPAK gene |
Geller syndrome | K, Øaldosterone, Ørenin ØUrinary THALDO | MCR gene |
Congenital Adrenal Hyperplasia: 17Alpha-Hydroxylase Deficiency
This enzyme deficiency is rare and affects both adrenal and gonadal steroid production.
Cytochrome P450c17 enzyme complex catalyzes both 17-hydroxylase and 17,20 lyase activity [25, 28–30].
Clinical findings: pseudohermaphroditism (46, XY male), sexual infantilism (46, XX female), varying degrees of hypertension and hypokalemic alkalosis.
Biochemical features: ≠DOC, Ø11-deoxycortisol, ØØ aldosterone, Ørenin, ØK.,
Ø Plasma 17alpha-hydroxyprogesterone, Øtestosterone, ≠urinary 100*THDOC/(THE+THF+5αTHF), and (THA+THB+5αTHB)/(THE+THF+5αTHF) ratios [47, 48].
Diagnosis of this autosomal recessive condition is suggested by delayed puberty, absent secondary sexual characteristics or amenorrhea combined with typical biochemical findings described above.
Genetic testing for mutations in the CYP17.
Cortisol is the first-line therapy and sometimes mineralocorticoid replacement, especially in cases of bilateral and complete (adrenal rest tumors) adrenalectomy. Oral hydrocortisone is often preferred for children to maximize their growth potential while minimizing the risk for developing iatrogenic Cushing’s syndrome.
Congenital Adrenal Hyperplasia: 11Beta-Hydroxylase Deficiency
11β-Hydroxylase deficiency is caused by several mutations in the CYP11B1 gene.
The inherited enzymatic defect (autosomal recessive) results in elevated serum levels of DOC, the principal steroid index of 11β-hydroxylase deficiency [49–53].
Accounts for approx. 5–8% of all CAH cases, approx. 1 in 200,000 births.
Chronic elevation of ACTH due to a deficient cortisol production stimulates the biosynthesis of other mineralocorticoids: 11-deoxycortisol, DOC, and others.
DOC is a weak mineralocorticoid, but at supraphysiologic levels promotes salt retention, volume expansion, and consequently hypertension.
Short stature (adult males), hirsutism, advanced bone age, amenorrhea, infertility are some of the most common findings in patients affected by this condition. Males may develop pseudoprecocious puberty (boys are tall as children).
Biochemical profile: potassium depletion (variable), Ørenin (variable), Øaldosterone, Øcortisol, ≠ACTH, ≠11-deoxycortisol, ≠DOC, ≠19-nor-DOC, ≠urinary 17-hydroxycorticosteroids and DOC, and ≠≠urinary 100*THS/(THE+THF+5αTHF) and 100*THDOC/(THE+THF+5αTHF) ratios [47, 48].
Other laboratory abnormalities: elevated serum level of 17alpha-hydroxy progesterone (17-OHP) and androstenedione and urinary pregnanetriol.
Normalization of DOC is an indicator of adequate glucocorticoid therapy which represents the most important therapeutic indication for this condition. Hydrocortisone is frequently used in children to maximize their growth potential while minimizing their risk for developing Cushing’s syndrome from overuse of glucocorticoid therapy (i.e., dexamethasone with its longer half-life and higher affinity for the glucocorticoid receptor) In selected patients, bilateral adrenalectomy may be safe and effective in managing blood pressure. In such cases, mineralocorticoid replacement (i.e., fludrocortisone) may be necessary.
Mutations in the CYP11B1 gene may not always be found.
Apparent Mineralocorticoid Excess
11β-hydroxysteroid dehydrogenase type 2 (11βHSD2) is highly expressed in the kidney where it metabolizes cortisol to cortisone. It is also expressed in all sodium-transporting epithelia.
A reduced enzymatic activity of 11βHSD2 caused by loss-of-function mutations explains the pathogenesis of the autosomal recessive syndrome of AME which was first described by Ulick et al. in 1979 [54–57].
Diminished 11βHSD2 activity may be hereditary or acquired.
Acquired deficiency of this enzyme may result from inhibition by glycyrrhetinic acid which may occur with use of licorice, chewing tobacco, carbenoloxone, and “asam boi” flavor (containing glycyrrhizic acid) if consumed on a daily basis in certain quantities [58].
Impaired 11βHSD2 activity alters the inactivation of cortisol in the target renal cell leading to above-normal cortisol activity, renal Na retention, and severe hypokalemia.
The severity of hypertension in patients with AME syndrome seems to correlate with the degree of loss of 11βHSD2 enzyme activity.
The clinical presentation of AME can include: growth retardation/short stature, hypertension, hypokalemia, and occasional nephrocalcinosis [59, 60].
A typical case of AME presents with: early hypertension (childhood), hypokalemia, suppressed renin, very low aldosterone levels, and metabolic alkalosis.
There is a large spectrum of clinical and biochemical features in patients with homozygous mutations of the 11βHSD2 gene; heterozygous individuals usually develop hypertension later in life, have milder biochemical profiles of AME and no phenotypic characteristics of AME [61].
The biochemical diagnosis can be made by detecting ≠ 24 h urinary-free cortisol/cortisone and ≠urinary (THF+5αTHF)/THE ratios [47, 48].
Patients diagnosed with AME syndrome respond well to low sodium diet and spironolactone, which blocks binding of both cortisol and aldosterone to the MCR.
Genetic testing for AME-associated loss-of-function mutations by DNA sequencing of the 11βHSD2 gene may further elucidate the diagnosis.
Licorice-Induced Hypertension
Licorice has been used for medicinal purposes for many years.
Licorice or its active component glycyrrhetinic acid may cause hypertension and hypokalemia due to cortisol-mediated activation of the MR by decreasing 11βHSD2 activity [62].
A detailed dietary history has to be obtained in each patient in the presence of resistant hypertension with associated hypokalemia to exclude the acquired forms of AME.
Various licorice containing foods, teas, or herbal products (like yokukansan) are available and their individual consumption varies considerably [63]. Possible adverse effects of yokukansan on blood pressure and electrolytes (low K) were reported recently [64]. A daily dose of 10 mg of glycyrrhetinic acid is considered unharmful for most healthy individuals [64]. Glycyrrhizic acid (a component of licorice) is used as a sweetener and also can be found in chewing tobacco products [65, 66].
Licorice seems to reduce serum testosterone by blocking 17-hydroxysteroid dehydrogenase and 17–20 lyase [67].
Licorice as a medicinal plant contains not only glycyrrhetinic acid but also compounds with estrogen-like or antiandrogen activity. PTH and calcium levels have been increased in healthy women after 2 months of licorice therapy reflecting its impact on calcium metabolism ([68], see Chap. 9 in this book on the topic Primary Hyperparathyroidism).Related posts:
 Congenital Adrenal Hyperplasia
Congenital Adrenal Hyperplasia
 Central Mineralocorticoid Receptors and Cardiovascular Disease
Insulin Resistance and Hypertension
Central Mineralocorticoid Receptors and Cardiovascular Disease
Insulin Resistance and Hypertension
 Testosterone Deficiency or Male Hypogonadism
Testosterone Deficiency or Male Hypogonadism
 Primary Generalized Familial and Sporadic Glucocorticoid Resistance (Chrousos Syndrome) and Hypersensitivity
Primary Generalized Familial and Sporadic Glucocorticoid Resistance (Chrousos Syndrome) and Hypersensitivity
 Primary Aldosteronism: Progress in Diagnosis, Therapy, and Genetics
Primary Aldosteronism: Progress in Diagnosis, Therapy, and Genetics

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


