Syndromes of Extreme Insulin Resistance
Allison B. Goldfine
Alan C. Moses
Glucose homeostasis depends on a balance between the production of glucose predominantly by the liver and the utilization of glucose by insulin-dependent tissues (muscle and fat) and insulin-independent tissues (brain and kidney). Insulin controls glucose homeostasis primarily through the suppression of hepatic glucose production and stimulation of peripheral (and to a lesser degree, splanchnic) glucose uptake. Insulin resistance occurs when the ability of endogenous or exogenous insulin to promote glucose clearance is impaired (1). Several pathophysiologic conditions are associated with insulin resistance of varying severity (Table 28.1). Medical treatment of patients with these conditions may be difficult but often leads to improved insulin action.
TABLE 28.1. Conditions Frequently Associated with Clinical Insulin Resistance | ||||||||
|---|---|---|---|---|---|---|---|---|
|
Syndromes of extreme insulin resistance represent the far end of the spectrum of insulin resistance. In general, the syndromes of extreme insulin resistance are caused by genetic defects in the insulin action pathway or in pathways that intersect with the insulin action pathway, or by the presence of autoantibodies to the insulin receptor that affect receptor function, or by autoantibodies to insulin itself (Table 28.2).
TABLE 28.2. Classification of Syndromes of Severe Insulin Resistance | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
DEFINITION OF SEVERE INSULIN RESISTANCE
Patients with severe insulin resistance may be euglycemic if their β-cells are able to produce sufficient insulin to compensate for the defect in signaling, or they may have only impaired postprandial glucose tolerance, or they may have overt diabetes. Thus, the characterization of these patients differs, depending on the level of glycemia. For individuals with normal glucose levels, serum insulin levels are a good index of the severity of insulin resistance, because the ability of the β-cell to produce insulin, together with the magnitude of the peripheral insulin resistance, will determine the overall degree of hyperinsulinemia. Although there is no established value above which a fasting insulin level reflects severe insulin resistance, in the absence of obesity a fasting insulin value greater than 30 to 50 μU/mL and/or a postprandial insulin level or an insulin level following glucose challenge that is greater than 200 μU/mL should warrant further evaluation. However, identifiable genetic defects are less likely to be found when stimulated insulin levels are less than 1,000 μU/mL.
As patients progress to impaired glucose tolerance or overt hyperglycemia, the β-cell defect may progress and insulin levels may decline. For patients receiving insulin therapy, insulin resistance may be defined according to the insulin dose required to achieve glucose control. Because the normal adult secretes an average of 25 to 40 units of insulin daily (0.3 to 0.5 U/kg per day) and about 70% of exogenous insulin is bioavailable, any patient requiring more than 60 to 100 units of insulin every 24 hours has some degree of insulin resistance. However, for clinical purposes, the requirement of more than 1.5 to 2.0 U/kg body weight suggests more severe insulin resistance and the need for further evaluation. Behavioral problems or improper diabetes management
should be rigorously excluded as the cause of “insulin resistance.” The exclusion of these causes may require careful evaluation of the patient’s dietary and drug history and understanding of insulin therapy. Often hospitalization may be necessary to reveal a behavioral problem.
should be rigorously excluded as the cause of “insulin resistance.” The exclusion of these causes may require careful evaluation of the patient’s dietary and drug history and understanding of insulin therapy. Often hospitalization may be necessary to reveal a behavioral problem.
CLINICAL FINDINGS ASSOCIATED WITH EXTREME INSULIN RESISTANCE
The most common clinical features associated with severe insulin resistance include acanthosis nigricans, ovarian dysfunction with hyperandrogenism and hirsutism, and lipoatrophy.
Acanthosis Nigricans
Acanthosis nigricans is characterized by a velvety rash with papillomatosis, hyperkeratosis, and hyperpigmentation in the epidermis (Fig. 28.1). Lesions are commonly found at the neck and axillae and occasionally in the antecubital fossae and other areas. When severe, acanthosis nigricans can involve the entire face and much of the truncal skin. Acanthosis nigricans has been observed in a variety of clinical settings, including several endocrinopathies and internal malignancies, particularly gastrointestinal cancer (2). However, when present, acanthosis nigricans typically indicates at least a moderate degree of underlying insulin resistance.
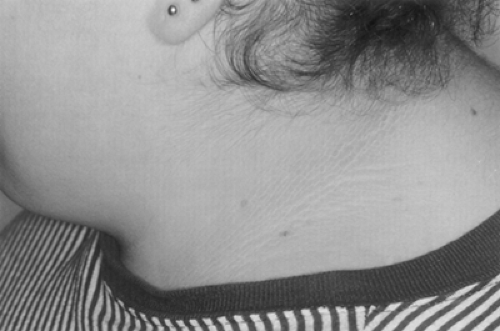 Figure 28.1. Acanthosis of the neck in a young woman with severe insulin resistance and obesity. (See also color plate.) |
When specifically searched for, the frequency of acanthosis nigricans may be higher than previously thought. In an unselected population of 1,412 schoolchildren in Texas, acanthosis nigricans was present equally among boys and girls, with a prevalence of 7.1% (3). The acanthosis nigricans was more frequently noted in obese children, and its severity was correlated with the fasting plasma insulin level. Acanthosis nigricans is even more common in selected groups of patients and was shown to affect up to 29% of women with hyperandrogenism referred to a subspecialty clinic and up to 50% of obese women with polycystic ovarian disease (4). Many of these patients have variants of the type A syndrome with moderate insulin resistance that at least partially reverts with weight loss (see below).
Pseudoacromegaly
Pseudoacromegaly, including frontal bossing, prominence of the jaw and malocclusion of the teeth, macroglossia, increased hand and foot size, skin tags, increased skin thickness, and median nerve entrapment at the carpal tunnel, has been described in some patients with severe insulin resistance (Fig. 28.2). The development of either acanthosis nigricans or an acromegalic appearance has been attributed to direct effects of hyperinsulinemia on skin tissue that result in epidermal hypertrophy. The presence of marked hyperinsulinemia in some patients is sufficient to bind to and activate the insulin-like growth factor 1 (IGF-1) receptor. IGF-1 receptors have a substantially lower affinity for insulin than do insulin receptors themselves, but when the ambient concentration of insulin is sufficiently high, IGF-1 receptors may become activated by a mechanism that has been called “specificity spillover” (5,6). Thus, increased stimulation of the IGF-1 receptor by hyperinsulinemia may be an important mechanism for the stimulation of epidermal growth in acanthosis nigricans. In addition, these mechanisms also may play an important role in the increased production of ovarian androgens in insulin-resistant states. In some patients, it is likely that insulin resistance is limited to insulin-receptor pathways involved in acute metabolic effects such as glucose transport or amino acid uptake and that “growth” effects of insulin are unrestrained and, in fact, accentuated by the hyperinsulinemia.
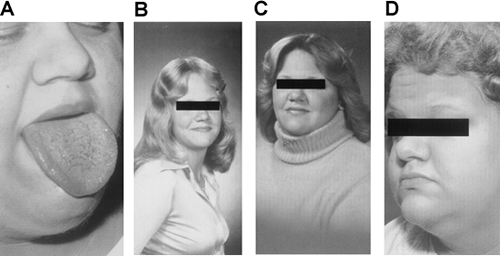 Figure 28.2. A: Tongue enlargement seen in the insulin-resistant patient with pseudoacromegaly. B–D: Progressive pseudoacromegaly in a young woman with severe insulin resistance. Note the progressive coarsening of facial features. (A, B from Flier JS, Moller DE, Moses AC, et al. Insulin-mediated pseudoacromegaly: clinical and biochemical characterization of a syndrome of selective insulin resistance. J Clin Endocrinol Metab 1993;76:1533–1541, with permission. Copyright 1993. The Endocrine Society.) |
Hyperandrogenism-Ovarian Dysfunction
Hyperandrogenism-ovarian dysfunction, presenting with primary or secondary amenorrhea or irregular menstrual cycles, is very common in female patients with syndromes of severe insulin resistance, with amenorrhea being more common in those with more severe resistance (7). The menstrual irregularity is associated with an increased production of ovarian androgens, leading to moderately elevated serum testosterone levels and various degrees of hirsutism and virilization (8). Moreover, free or bioavailable testosterone levels may be increased disproportionately in these patients owing to a decrease in sex hormone-binding globulin. The ovaries are typically polycystic and hyperthecotic. Although the exact pathophysiology of such ovarian dysfunction is not known, insulin may cause ovarian hyperstimulation via its own receptor or by cross-reacting with the receptors for IGF-1, which also are present in the ovary. Luteinizing hormone has also been shown to act synergistically with insulin to promote the production of androgen by the ovaries (9).
HIRSUTISM AND VIRILIZATION
Hirsutism, the development of excessive androgen-stimulated terminal hair in a female, may occur with severe insulin resistance. Virilization, including defeminizing signs such as loss of the female body contour or frank masculinization with clitoromegaly and deepening of the voice, increased muscle mass, temporal balding, and increased libido, is less common.
Lipoatrophy or Lipodystrophy
Lipoatrophy or lipodystrophy, a marked reduction in subcutaneous adipose tissue that occurs in a segmental or generalized distribution (Fig. 28.3), is an additional clinical sign associated with the insulin resistance syndromes of leprechaunism and lipoatrophic diabetes. Lipoatrophy can be classified as focal or generalized and congenital or acquired. If lipoatrophy is localized to sites of insulin injection, it can be the result of a form of insulin allergy in individuals without marked insulin resistance. Descriptions of various forms of lipodystrophy are provided later in this chapter.
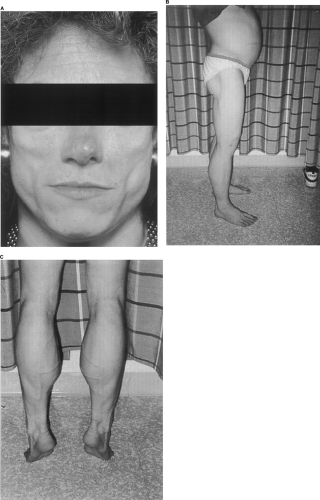 Figure 28.3. A: Lipoatrophy affecting the face in a patient with partial lipoatrophy. B, C: Lipoatrophy affecting the body and legs. Note the protuberant abdomen caused by marked hepatomegaly and the apparent muscular hypertrophy of the calves. |
CHARACTERIZATION OF INSULIN RESISTANCE
Patients suspected of having a syndrome of severe insulin resistance and who are not receiving exogenous insulin may be characterized by fasting and postprandial or post-glucose challenge serum insulin levels. The standard oral glucose tolerance test with both insulin and glucose levels is a rapid and reliable initial diagnostic evaluation. The more markedly elevated these levels, the more severe the resistance and the more likely that the evaluation will reveal an identifiable underlying defect, such as when the fasting insulin level is above 50 mIU/L and the postprandial levels are above 200 to 500 mIU/L, although in some affected patients they may exceed 1,000 mIU/L (with normal values between 2 and 20 mIU/L). The hyperinsulinemia commonly observed in these patients develops by two complementary mechanisms. One is an increase in insulin secretion by the pancreatic β-cell in response to persistent elevations in fasting or postprandial glucose levels, and the other results from defects in the peripheral action of insulin associated with a striking reduction in the metabolic clearance of circulating insulin (10).
Several additional procedures are used clinically to eval-uate the severity of insulin resistance. These tests vary in their complexity both in performance and in interpretation. They share the common element of administering exogenous insulin to rule out the unlikely possibility of an abnormal endogenous insulin molecule (insulin mutation) as the cause of hyperinsulinemia. The insulin tolerance test (ITT) assesses the glucose-lowering effect of an intravenous insulin bolus (6). After an overnight fast, in a normal individual, an intravenous bolus injection of a dose of regular insulin of 0.1 U/kg of body weight will reduce the blood glucose level to a value 50% or less of the initial value or may provoke frank hypoglycemia. A patient with peripheral insulin resistance will require a higher dose of injected insulin (≤0.2 U/kg) to produce this reduction in the fasting glucose level. If the patient requires an insulin bolus of 0.3 U/kg or more to induce hypoglycemia, the diagnosis of extreme insulin resistance is certain. Given the importance of the diagnosis, a serum insulin level should be obtained 10 minutes after insulin injection to confirm that insulin entered the vascular compartment.
In the steady-state plasma glucose test (SSPG), also known as the triple-infusion test, somatostatin is infused to suppress endogenous insulin production, and fixed doses of insulin and glucose are co-infused. At steady state, the higher the glucose level for a given insulin dose administered, the more insulin resistant the subject. The frequently sampled intravenous glucose tolerance test (FSIVGTT) provides a standardized method to assess both the pancreatic β-cell secretory capacity and peripheral
glucose uptake in response to a bolus intravenous administration of glucose (0.3 g/kg). Additional information on insulin sensitivity is gained by the administration of insulin (0.03 to 0.05 U/kg) 20 minutes after the glucose load. Euglycemic-hyperinsulinemic clamp studies are considered the gold standard for measurement of insulin action but are more difficult to perform. A dose-response curve to exogenous insulin is generated by measuring the variable infusion rate of glucose required to maintain euglycemia. The steady-state rate of peripheral glucose utilization (M value) is measured as milligrams of glucose used per kilogram of body weight per minute. Studies have demonstrated total-body glucose clearance to be reduced by 35% to 40% in subjects with type 2 diabetes as compared with that of age- and weight-matched healthy controls, with similar or more modest reduction in insulin sensitivity in persons with obesity, hypertension, and hyperlipidemia (1). More severe reductions are seen in persons with syndromes of extreme insulin resistance. In type 2 diabetes, the primary site of reduced insulin-mediated glucose uptake appears to be located in the peripheral (muscle) tissue (11), and both oxidative and nonoxidative (glycogen storage) components of glucose metabolism may be impaired (12).
glucose uptake in response to a bolus intravenous administration of glucose (0.3 g/kg). Additional information on insulin sensitivity is gained by the administration of insulin (0.03 to 0.05 U/kg) 20 minutes after the glucose load. Euglycemic-hyperinsulinemic clamp studies are considered the gold standard for measurement of insulin action but are more difficult to perform. A dose-response curve to exogenous insulin is generated by measuring the variable infusion rate of glucose required to maintain euglycemia. The steady-state rate of peripheral glucose utilization (M value) is measured as milligrams of glucose used per kilogram of body weight per minute. Studies have demonstrated total-body glucose clearance to be reduced by 35% to 40% in subjects with type 2 diabetes as compared with that of age- and weight-matched healthy controls, with similar or more modest reduction in insulin sensitivity in persons with obesity, hypertension, and hyperlipidemia (1). More severe reductions are seen in persons with syndromes of extreme insulin resistance. In type 2 diabetes, the primary site of reduced insulin-mediated glucose uptake appears to be located in the peripheral (muscle) tissue (11), and both oxidative and nonoxidative (glycogen storage) components of glucose metabolism may be impaired (12).
INSULIN RESISTANCE DUE TO MUTATIONS IN THE INSULIN-RECEPTOR GENE
Although more than 60 naturally occurring mutations in the insulin-receptor gene have been characterized, three distinct clinical syndromes are associated with mutations in this receptor. All share the clinical features of acanthosis nigricans and hyperandrogenism.
Leprechaunism
Leprechaunism (also known as Donohue syndrome), the most severe of the conditions associated with mutations in the insulin-receptor gene, is characterized additionally by intrauterine growth retardation, a characteristic bird-like facies, fasting hypoglycemia, and glucose intolerance despite insulin levels that may be more than 100 times the normal values (Fig. 28.4). Most patients die within the first year of life. The condition is associated with homozygous mutations in either the coding sequence or the regulatory domains of both alleles of the insulin-receptor gene. Rare patients have two null alleles and totally lack functional insulin receptors.
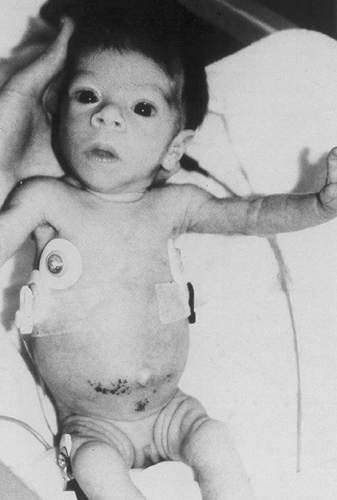 Figure 28.4. Infant affected with Donohue syndrome (leprechaunism). (From Kahn CR, Goldstein BJ, Reddy SSK. Hereditary and acquired syndromes of insulin resistance. In: Pickup J, Williams G, eds. Textbook of diabetes. Oxford: Blackwell Scientific Publications, 1991:276–285, with permission.) |
Rabson-Mendenhall Syndrome
Patients with Rabson-Mendenhall syndrome demonstrate abnormalities in the teeth and nails and pineal gland hyperplasia. These patients may survive until adolescence, and many have compound heterozygous mutations in the insulin receptor, producing severe impairment of receptor function.
Type A Syndrome
The type A syndrome of insulin resistance is the least severe of the receptor mutation disorders. Affected females generally also demonstrate significant hyperandrogenism. Patients are neither obese nor lipoatrophic. Although many use the term type A syndrome to identify patients with mutations in one allele of the insulin receptor, many use the term to describe patients who demonstrate the triad of hyperandrogenism, insulin resistance, and acanthosis nigricans (HAIR-AN). Because only a minority of the patients who manifest this triad have heterozygous mutations in the insulin receptor, it is likely that mutations exist in postreceptor insulin-signaling transduction proteins. Likewise, when this triad is seen in the setting of obesity, it is less likely to be associated with a mutation in the insulin receptor than with additional polygenic factors rendering the patient susceptible to the clinical syndrome.
Mutations of the insulin receptor have been described in the insulin-binding domain, the transmembrane domain, and at or near phosphotyrosine sites; not all of these defects are associated with severe insulin resistance and the acromegaloid and hyperandrogenic features. In addition, specific mutations in the insulin receptor are found in only a minority of patients with a severe insulin resistance syndrome.
Insulin action at the cellular level is complex (reviewed in Chapter 6). Postreceptor insulin signaling proteins have been assessed carefully in patients with severe insulin resistance in whom no receptor defect could be identified. There is a small increase in frequency in variations in the insulin-receptor substrate 1 (IRS-1) sequence in subjects with type 2 diabetes as compared with controls (∼15% vs. 5%, respectively), and some studies suggest that these may have some functional significance (13,14). IRS-1 variants may be more common among patients with severe insulin resistance; however, as these variants are also present in unaffected persons, they are not likely to account fully for the severe insulin resistance (15,16). Phosphatidylinositol 3-kinase (PI 3-kinase) is central in insulin-mediated glucose disposal and consists of a regulatory domain splice variants p85α and p85β and a catalytic domain p110. Variants of p85α Met326Ile, have been described in humans and do not appear to be associated with decreased activity; however, an Arg409Gln mutation has been identified in a patient with severe insulin resistance and is associated with lower activity in vitro and higher fasting glucose levels in the carriers within the proband’s family (17). In addition, impaired insulin activation of PI 3-kinase has been demonstrated in fibroblasts from severely insulin-resistant patients (18,19). Expression levels of genes for other proteins appear altered in some patients with type 2 diabetes, such as the gene belonging to the Ras-guanosine triphosphate superfamily, RAD (ras-associated with diabetes), which has been found to be overexpressed in muscle tissue of patients with type 2 diabetes (20), although the clinical significance of this finding remains uncertain and has not been demonstrated in conditions of extreme resistance.
The role of peroxisome proliferator-activated receptors (PPARs) in modulating insulin sensitivity has only recently been appreciated with the discovery of the thiazolidinedione class of drugs. These agents have been demonstrated to be potent insulin sensitizers, although the molecular mechanisms of action are only partially understood (see Chapter 41). The drugs bind to and activate an orphan nuclear receptor PPARγ and regulate the transcription of a number of insulin-responsive genes, including glucose transporters, critical for the control of glucose metabolism. PPARs exist as heterodimers with another nuclear receptor, RXR, and effects are dependent on the presence of separate insulin-regulated transcription factors. Two dominant negative heterozygote mutations in the ligand-binding domain of the PPARγ receptor have been identified in severely insulin-resistant, hypertensive patients with overt diabetes (21).
At the molecular level, multiple alterations in the insulin action cascade have been shown to be present and to contribute to the insulin resistance of type 2 diabetes and the insulin resistance syndromes; however, the relative importance of these alterations remains controversial. These changes are regulatory and include downregulation of the insulin receptor, decreased receptor kinase activity, decreased phosphorylation of receptor substrates, and defects in glucose transporter translocation (22,23,24,25,26). In addition to these signaling defects, several inhibitors of insulin action, such as cytokines, may be involved in the pathophysiology
of diabetes. For example, tumor necrosis factor-α (TNF-α), which is expressed by adipose tissue and increased in human obesity, inhibits insulin-receptor kinase activity, and neutralization of TNF-α improves muscle insulin sensitivity in insulin-resistant rats (27). Similarly, the transmembrane glycoprotein PC-1, a threonine-specific protein kinase, nonspecifically inhibits the activity of insulin-receptor tyrosine kinase and has been isolated from fibroblasts of a patient with severe insulin resistance (28), although a primary role for this kinase in the etiology of insulin resistance remains unproven. Although such regulatory involvement may be present in or contribute to syndromes of severe insulin resistance, none has been clearly demonstrated to date.
of diabetes. For example, tumor necrosis factor-α (TNF-α), which is expressed by adipose tissue and increased in human obesity, inhibits insulin-receptor kinase activity, and neutralization of TNF-α improves muscle insulin sensitivity in insulin-resistant rats (27). Similarly, the transmembrane glycoprotein PC-1, a threonine-specific protein kinase, nonspecifically inhibits the activity of insulin-receptor tyrosine kinase and has been isolated from fibroblasts of a patient with severe insulin resistance (28), although a primary role for this kinase in the etiology of insulin resistance remains unproven. Although such regulatory involvement may be present in or contribute to syndromes of severe insulin resistance, none has been clearly demonstrated to date.
LIPOATROPHIC DIABETES
Lipoatrophic diabetes encompasses a genetically heterogeneous group of rare syndromes characterized by insulin-resistant diabetes associated with a regional or complete absence of subcutaneous adipose tissue and presence of hypertriglyceridemia. Several forms of this condition have been reported according to mode of inheritance and extent and regional distribution of the lipoatrophy (29). These include congenital types of generalized lipoatrophic diabetes, which may exhibit either dominant inheritance (Köbberling-Dunnigan syndrome) or recessive inheritance (Berardinelli-Seip syndrome). In addition, syndromes of acquired total lipoatrophy (Lawrence syndrome) and various types of acquired partial lipoatrophy have been described.
Congenital Lipoatrophy
DOMINANT INHERITANCE (KöBBERLING-DUNNIGAN VARIETY)
By studying pedigrees of congenital lipoatrophy with dominant inheritance in Scotland and Germany, Köbberling and Dunnigan delineated two clinical subtypes with characteristic distributions of subcutaneous fat that appear to be consistent within each pedigree (30,31,32). In type 1 familial lipodystrophy, the loss of subcutaneous fat is confined to the limbs, with sparing of the face and trunk, and intracavity fat deposits. In type 2 congenital lipoatrophy syndrome, the trunk, with the exception of the vulva, also is affected, giving the impression of labial hypertrophy. Associated clinical features variably expressed in the pedigrees include severe hyperlipidemia, acanthosis nigricans, hepatosplenomegaly, tuberoeruptive xanthomata, elevated basal metabolic rate, and insulin-resistant diabetes without ketoacidosis. Genetic characterization of at least 10 kindreds with this condition demonstrates linkage to chromosome 1q, with evidence of a common founder haplotype (33,34). At least five different missense mutations have been identified in the LMNA gene associated with this condition. The protein product of the gene is lamin A/C, a component of the nuclear envelope. Lamins A and C are two alternatively transcribed products of the LMNA gene and are members of the intermediate filament family of proteins that form components of the nuclear lamina, a protein-rich fibrous area below the inner nuclear membrane. The defects in nuclear lamin A/C have been confirmed in additional affected kindreds, but not all patients with the phenotypic characteristics of the syndrome manifest mutations in this gene (35). The lipoatrophic disorder may be caused by a gene tightly linked to this site; however, evidence supports a direct involvement of the LMNA mutation in the disease, and there may be a series of genetic mutations responsible for the phenotype. Heterozygous mutations in LMNA, which are more extensive and involve several exons, have been associated with an autosomal dominant form of muscular dystrophy, the Emery-Dreifuss variant (36), characterized by muscle contractures and a cardiac conduction defect and with an age-related dilated cardiomyopathy and conduction system disease, CMD1A (37). Rodent models with a targeted deletion of this gene develop postnatal cardiac and skeletal muscle dystrophy and a loss of white adipose tissue (38). LMNA is expressed ubiquitously in cells; thus, the site-specific amino acid substitutions associated with these three distinct clinical syndromes reveal distinct functional domains of the lamin A/C protein to the different cell types. Further studies may reveal the role of lamin A/C in adipocyte function and insulin action.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


