Abstract
Lipids serve as metabolic substrates, structural components of cellular membranes, and signaling molecules. The steroidogenic machinery is compartmentalized at the organ, cellular, and subcellular level. Steroidogenic cells do not store premanufactured steroid hormones, and the rate-limiting step in the production of steroid hormones is the movement of substrate cholesterol to the inner mitochondrial membrane, where the first committed step in steroidogenesis takes place, which is the cholesterol side-chain cleavage reaction in which cholesterol is converted into pregnenolone. Several families of enzymes are involved in the synthesis of steroid hormones, the most important being the cytochrome P450 family and the hydroxysteroid dehydrogenases and reductases. In addition to steroid secreting glands, bioactive steroids are produced in small quantities in many cell types.
Eicosanoids are derived enzymatically from 20-carbon polyunsaturated fatty acids (e.g., arachidonic acid). They are produced by almost all cells in response to hormonal stimulation or trauma, acting as paracrine or autocrine modulators. Specificity of eicosanoid action is afforded by selective expression of biosynthetic enzymes and their receptors. Eicosanoids include products of the cyclooxygenase (COX), lipoxygenase (LOX), and P450 enzyme acting on arachidonic acid and other polyunsaturated fatty acids. Prostanoids are formed by the COX enzymes. They are essential for ovulation and have been implicated in the processes of implantation, embryogenesis, and parturition.
Keywords
Aromatase, cholesterol, cholesterol side-chain cleavage enzyme, cyclooxygenase, cytochrome P450, disorder of sexual development, eicosanoids, hydroxysteroid dehydrogenase, leukotriene, lipoproteins, lipoxygenase, lysophosphatidic acid, prostanoid, prostaglandin, prostacyclin, reductase, sex hormone binding globulin, sphingosine-1-phosphate, steroidogenic acute regulatory protein, steroidogenic factor-1, thromboxane, vitamin D
Lipids serve as metabolic substrates, structural components of cellular membranes, and signaling molecules. This chapter reviews the diversity of lipid molecules that participate in signal transduction related to human reproduction, including the nomenclature, the general features of their synthesis and metabolism, the ways in which these processes are controlled physiologically, the ways in which they can be modified by pharmacologic intervention, and some disorders that interfere with normal synthesis and metabolism. The chapter emphasizes steroid hormones and eicosanoids because of their prominent roles in regulating sexual differentiation, the normal function of tissues involved in reproduction, and the pathophysiology of disorders affecting those tissues.
Steroid Hormones: Structure and Nomenclature
- ◆
The steroid hormone backbone name is not synonymous with biological activity.
- ◆
The location of hydroxyl groups and whether they reside in the α (below the plane of the steroid nucleus) or β (above) configuration have major impact on the biological activity of steroid hormones.
- ◆
Synthetic “bioidentical” steroid hormones may contain a mixture of stereoisomers that affect potency.
Steroid hormones and the secosteroid prohormone, vitamin D, belong to an ancient family of signaling molecules with diverse functions, including central roles in the regulation of female and male reproductive processes. The human steroid hormones are derived from cholesterol, an abundant plasma lipid and a structural component of plasma membranes, and other organelles. Seemingly subtle modifications of the four fused rings of the sterol skeleton and side chain result in molecules with different activities.
Steroid hormones share a cyclopentanoperhydrophenanthrene backbone. Each carbon in this fused-ring structure is assigned a number identifier, and each ring is assigned a letter ( Fig. 4.1 ). The families of naturally occurring steroid hormones are named according to the saturated ring structures of the parent compound: cholestanes, of which cholesterol (5-cholesten-3β-ol) is a representative, have 27 carbons; pregnanes have 21 carbons (e.g., 4-pregnen-3-20-dione, also known by its trivial name, progesterone ); androstanes have 19 carbons (e.g., 17β-hydroxy-4-androsten-3-one, or testosterone ); and estranes have 18 carbons (e.g., 1,2,5[10]-estratriene-17β-ol, or estradiol ). Gonanes contain 17 carbons (the cyclopentanoperhydrophenanthrene backbone) represented by synthetic progestins (e.g., desogestrel, norgestimate, gestodene). The backbone name is not synonymous with biological activity, which is determined mainly (but not exclusively) by which members of a family of nuclear transcription factors (the classical steroid hormone receptors) the molecules activate. For example, cortisol and progesterone are members of the pregnane family, yet they have different biological activities and act through different receptors. Synthetic molecules that activate the progesterone receptor are derived from androgens containing 18 carbons (19-nortestosterone derivatives) and are, therefore, members of the estrane family. 27-Hydroxycholesterol, a member of the cholestane family, is a selective estrogen receptor modulator.
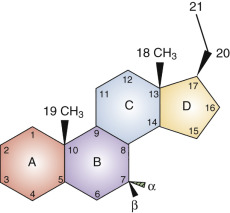
The locations of substituents in the steroid backbone are indicated by the carbon number to which they are attached. Substituents at several positions have a significant effect on metabolism and biological activity of steroid hormones, including carbons 3, 7, 11, and 17. Atoms attached to asymmetric centers are, by convention, given the designation α if they project below the plane of the ring structure (in figures of structures, a dashed line or hatched triangle indicates the α configuration). The designation β (a solid line or filled triangle) is given to atoms that project above the plane. Hormone receptors and steroid binding proteins (e.g., sex hormone binding globulin [SHBG]) distinguish between stereoisomers. In the case of estrogen receptors, 17β-estradiol is active, but 17α-estradiol is essentially inert. In the case of androgen receptors, testosterone with a 17β-hydroxyl configuration is active, but epitestosterone with a 17α-hydroxyl configuration has little activity.
Different enzymes catalyze the oxidation or reduction of α and β hydroxyl groups and reduce the Δ 4 double bond in the steroid A ring to form 5α or 5β molecules. Such 5α-reduced steroids can be active (e.g., 5α-dihydrotestosterone, which activates androgen receptors), or inactive (e.g., 5α-dihydroprogesterone) with respect to classical steroid hormone receptor function. However, the notion that pregnane 5α-reduced compounds are inactive has been challenged by the revelation that 5α-reduced glucocorticoids may have antiinflammatory activity acting through glucocorticoid receptors. Although 5β-reduced steroids are usually not capable of activating classical steroid hormone receptors, these steroids and some 5α-reduced molecules exert biological effects outside of the family of steroid hormone receptors, including modulation of neurotransmitter receptor function.
The naturally occurring steroid hormones are rarely referred to in the medical literature by their systematic names, which designate the parent structure and the number, location, and (if appropriate) orientation of substituents; instead, the trivial names are preferred (e.g., testosterone rather than 17β-hydroxy-4-androsten-3-one).
Steroid hormones used in clinical practice are derived from biological sources (e.g., conjugated equine urinary estrogens); semisynthetic, produced usually from a plant sterol starting material (e.g., stigmasterol or diosgenin) that can be chemically modified to yield a structure that is identical to the naturally occurring molecules; totally synthetic, which may result in the production of hormone isomers with different biological activity; or steroid drugs that are molecules not found in humans and animals (e.g., medroxyprogesterone acetate and norgestrel). “Bioidentical” steroid hormones produced by chemical synthesis may contain isomers that are not natural and may result in biological activities that differ from the steroid hormones produced in humans. Because of this concern, it has been argued that the term bioidentical steroid hormones can be misleading.
Organization of Steroidogenic Organs and Cells
- ◆
The steroidogenic machinery is compartmentalized at the organ, cellular, and subcellular level.
- ◆
The biosynthesis of estrogens usually requires the concerted effort of two different organs or cell types.
The steroidogenic machinery is compartmentalized at the organ, cellular, and subcellular levels, which has important implications for the control of steroid hormone production. Steroid synthesis involves a series of sequential modifications of cholesterol that result in the clipping of the side chain; alterations in olefinic bonds; and the addition of hydroxyl functions, proceeding invariably (although some have argued that shortcuts do exist) from cholesterol through the pregnane, androstane, and finally, estrane families.
Specific cell types can accomplish several of these sequential steps, but rarely can they generate an estrogen from cholesterol. The requirement for cooperative efforts by two different tissues or cell types is a characteristic of estrogen biosynthesis. This joint effort enables the modulation of both androgen and estrogen production by factors that independently influence the cells involved in precursor synthesis, in addition to the cell type in which the final step, aromatization, occurs. This cooperation is exemplified by estradiol synthesis in the ovarian follicle, where luteinizing hormone (LH) acts on the theca cells to stimulate production of androgen precursors and follicle-stimulating hormone (FSH) acts on granulosa cells to stimulate aromatization of these androgens into estrogens. Placental estrogen synthesis likewise requires precursors from another tissue, the fetal adrenal gland, that is under the control of fetal pituitary adrenocorticotropic hormone (ACTH). The sulfated dehydroepiandrosterone (DHEA) secreted from the fetal zone of the adrenal cortex has negligible androgenic activity in the fetus and increased solubility in plasma, so it can be efficiently transported to the placenta where cleavage of the sulfate group, followed by aromatization, occurs in the syncytiotrophoblast. Cooperative interaction among different cell types is also important in regulating the production of steroid hormones in the brain, and bioactive hormone production in endometriotic lesions, and breast and endometrial cancers.
Another example of compartmentalization of the steroidogenic machinery at the organ level is the adrenal cortex, which has histologically and functionally distinct zones that determine the relative production rates of mineralocorticoids, glucocorticoids, and adrenal androgens. The zona glomerulosa synthesizes mineralocorticoids; the zona fasciculata, glucocorticoids; and the zona reticularis and fetal zone of the fetal adrenal cortex produce androgens. One major functional distinction between the zona glomerulosa and the zonae fasciculata and reticularis is that aldosterone synthase is exclusively expressed in the zona glomerulosa, but 17α-hydroxylase/17,20 desmolase (CYP17A1, also referred to as P450c17) is not. However, CYP17A1 is abundant in the zonae fasciculata and reticularis. The zona reticularis expresses lower levels of type 2,3β-hydroxysteroid dehydrogenase (encoded by HSD3B2 ). Because the lyase activity of human CYP17A1 does not efficiently act on progesterone, reduced conversion of pregnenolone to progesterone resulting from the low 3β-hydroxysteroid dehydrogenase activity facilitates the conversion of pregnenolone into DHEA in the zona reticularis. Higher levels of cytochrome b 5 in the zona reticularis, which increase the lyase activity of human CYP17A1 by an allosteric mechanism, further augment the capacity to produce DHEA. The zona reticularis also has high levels of sulfotransferase. This constellation of enzymatic activities favors synthesis of DHEA sulfate.
Acquisition, Storage, and Trafficking of Cholesterol
- ◆
Steroidogenic cells do not store premanufactured steroid hormones.
- ◆
Cholesterol is acquired for steroidogenesis from plasma lipoproteins, de novo synthesis, and hydrolysis of sterol esters stored in lipid droplets.
- ◆
Lipoprotein-derived cholesterol is the primary substrate for cells that elaborate large quantities of steroid hormone.
- ◆
The rate-limiting step in steroidogenesis is the movement of cholesterol to the inner mitochondrial membrane.
Steroidogenic cells have structural features that enhance their ability to obtain and store cholesterol for use in steroidogenesis ( Fig. 4.2 ). Unlike protein hormone–producing cells, steroid-producing cells do not store prefabricated hormone; they synthesize the hormones on demand from cholesterol that has been acquired from the plasma, synthesized de novo, or stored in membranes, or as sterol esters in lipid droplets. Because of cholesterol’s limited solubility in water (it forms aggregates at 25 to 40 nM [the critical micelle concentration]), multiple proteins are required to move the steroid hormone precursor efficiently from one cellular compartment to another.
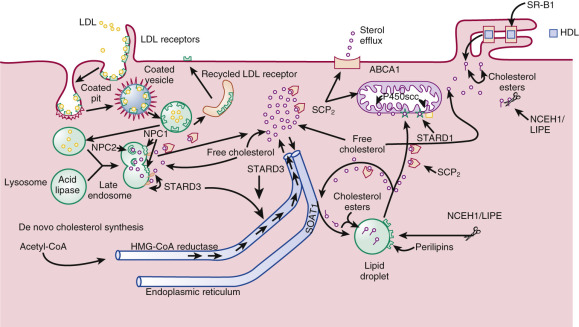
The plasma membrane has the highest content of free cholesterol, which is derived from plasma lipoproteins and de novo sterol synthesis. This sterol pool is not static; it exchanges with plasma free cholesterol, and regularly cycles through the cell and back to the plasma membrane. During this cycling process, sterols can be diverted for use in steroid hormone synthesis or esterified and deposited in lipid droplets.
Numerous microvilli project from the plasma membrane on which lipoprotein-gathering receptors of the low-density lipoprotein (LDL) receptor family are located (e.g., LDL receptors; LDL receptor–related protein, very–low-density lipoprotein [VLDL] receptors). These receptors mediate lipoprotein uptake by an endocytic mechanism that delivers the lipoproteins to the lysosomes where the apolipoproteins are degraded. The lipoprotein cholesterol esters are then hydrolyzed by acid lipase (lipase A encoded by the LIPA gene) to release free cholesterol. Severe acid lipase deficiency (Wolman disease) is associated with lysosomal accumulation of cholesterol esters and triglycerides, which can lead to damage of steroidogenic cells and compromised hormone production. Stimulation of steroidogenic cells by trophic hormones increases the number of LDL receptors on the cell surface and accelerates the rate of LDL internalization and degradation.
Free cholesterol is released from lysosomes through a system of sterol binding proteins encoded by genes (NPC1 , NPC2) that when mutated cause the cholesterol storage disorder, Neimann-Pick type C disease. NPC2, a soluble protein in the lysosome, delivers free cholesterol to NPC1, a membrane-associated cholesterol binding protein that controls sterol efflux from the lysosomes. Other sterol binding proteins, including sterol carrier protein-2 (SCP 2 ), STAR-related lipid transfer (START) domain protein 3 (STARD3), which is also known as metastatic lymph node 64 protein (MLN64), STARD4, and STARD5 may participate in the intracellular trafficking of cholesterol to various organelles including from endosomes to the endoplasmic reticulum, peroxisomes, and ultimately to the mitochondria. In steroidogenic glands regulated by tropic hormones (e.g., ovary, testis and adrenal cortex), the delivery of cholesterol substrate to the ultimate destination, the mitochondrial cholesterol side-chain cleavage enzyme, is effected by the steroidogenic acute regulatory protein (StAR or STARD1).
High-density lipoproteins (HDL) can also provide cholesterol for hormone synthesis by a pathway that differs from the “LDL pathway.” Receptors for HDL (scavenger receptor type B, class 1 [abbreviated SR-B1]) are located in closely apposed microvilli that form “microvillar channels” in which HDL particles are lodged. Endothelial lipases (hepatic lipase or endothelial cell–derived lipases) may facilitate uptake of the HDL-carried sterols by steroidogenic cells, including the selective uptake the HDL sterol esters. Similar to LDL receptor expression, SR-B1 expression is upregulated in response to trophic stimulation, facilitating the usage of HDL-delivered substrate.
HDL cholesterol esters are selectively internalized by SR-B1, leaving the apolipoproteins on the cell surface. The internalized unhydrolyzed HDL cholesterol esters are then cleaved by a cytosolic, neutral pH optimum sterol esterase (also referred to as hormone-stimulated lipase , encoded by LIPE , or neutral cholesterol ester hydrolase , encoded by NCEH1 ), thereby releasing free cholesterol.
De novo synthesis of cholesterol, a process that involves at least 17 enzymes, occurs primarily in the abundant smooth endoplasmic reticulum (SER). Some steroidogenic cells have up to 10-fold more SER by volume than rough endoplasmic reticulum. In certain cells, the SER takes on unique forms, exemplified by the whorls found in testicular Leydig cells. Enzymes involved in steroid hormone formation and metabolism are also embedded in the SER. Trophic hormones that stimulate steroidogenesis generally increase both cellular cholesterol synthesis and lipoprotein uptake.
Several studies have suggested that de novo cholesterol synthesis generates important local regulators of reproductive function in addition to the substrate for steroid hormone synthesis. The biosynthetic intermediates between lanosterol and cholesterol have been shown to stimulate oocyte maturation in in vitro assays and have also been proposed to play roles in spermatogenesis. These 4,4-dimethyl sterols, referred to as meiosis-activating sterols, contain 29 carbons and are found in the testis and follicular fluid in low micromolar concentrations. However, their physiological role in gamete maturation in vivo is uncertain based on results from germ cell-specific knockout mouse models (targeted deletion of cytochrome P450 lanosterol 14α-demethylase, Cyp51A1) that indicate that de novo synthesis of meiosis-activating sterols is probably not essential for reproduction. The potential use of meiosis-activating sterols in clinical settings of animal and human reproduction remain subjects of debate.
The quantitative importance of circulating cholesterol carried by LDL, HDL, and other lipoproteins as steroid hormone precursor in tissues that produce large amounts of steroid hormone (e.g., the corpus luteum and adrenal zonae fasciculata and reticularis), as opposed to de novo cholesterol synthesis, is demonstrated by the fact that radiolabeled plasma cholesterol in humans is almost fully equilibrated with the steroidogenic pool of cholesterol. Additional evidence for an important role of circulating lipoprotein cholesterol in steroidogenesis comes from the study of hypobetalipoproteinemia, a disorder in which there is virtually no circulating LDL. This rare metabolic disease is associated with reduced adrenocortical steroid production and diminished progesterone levels in the luteal phase and in pregnancy, although the lower levels of progesterone elaborated are still sufficient to achieve a term pregnancy. In the case of HDL, individuals with an SR-B1 missense variant (Pro297Ser) that reduces uptake function have increased HDL levels and attenuated adrenal steroidogenesis in response to ACTH stimulation.
Individuals with familial hypercholesterolemia due to inactivating mutations in the LDL receptor have modest impairment of steroidogenic gland function, reflecting the capacity of alternative sterol acquisition mechanisms to compensate for LDL receptor deficiency. The commonly used cholesterol-lowering statins (which inhibit 3-hydroxy-3-methylglutaryl coenzyme A reductase [HMG-CoA reductase], the rate-limiting enzyme in de novo cholesterol synthesis) do not impair adrenal, testicular, or luteal steroidogenesis in adult humans despite the lowering of plasma LDL levels.
Smith-Lemli-Opitz syndrome, an autosomal recessive disease, offers insight into the relationship of plasma cholesterol and de novo sterol synthesis for the supply of precursors for fetal steroidogenesis. The disease is caused by inactivating mutations in an enzyme involved in the terminal steps of cholesterol synthesis, 3β-hydroxysteroid Δ 7 -reductase (encoded by DHCR7 ). As a result, cholesterol levels are low and 7-dehydrocholesterol levels are elevated. Hypospadias or ambiguous genitalia are frequent findings in affected male neonates, reflecting diminished fetal testicular testosterone synthesis. Estrogen production during pregnancy is also reduced, due to impaired fetal adrenal hormone production. Adrenal insufficiency has been reported in some affected individuals, although others compensate with elevated ACTH secretion. B-ring unsaturated equine-like steroids (1,3,5[10],7-estratetrenes) are produced from the 7-dehydrocholesterol that accumulates, showing that the steroidogenic enzymes do not have an absolute requirement for cholesterol as a substrate. Desmosterolosis, a rare autosomal recessive disease caused by mutations in 3β-hydroxysterol Δ 24 -reductase (encoded in DHCR24 ) is also associated with ambiguous genitalia in affected males, presumably because of impaired fetal testicular testosterone synthesis.
Cytoplasmic lipid droplets represent another major depot of substrate in steroidogenic cells. As much as 80% of the total cholesterol content of steroidogenic cells is found esterified in these droplets. The sterol esters are synthesized in the endoplasmic reticulum from cholesterol acquired from lipoproteins or de novo synthesis by sterol O-acyltransferase 1 (SOAT1, previously named acyl-coenzyme A, cholesterol acyltransferase-1 or ACAT1), encoded by one of two related genes. The esters generated by SOAT1 accumulate within the SER, and subsequently bud off as lipid droplets (see Fig. 4.2 ). A targeted mutation in the Soat1 gene in mice results in markedly reduced sterol ester storage in the adrenal cortex without impairment of basal or ACTH-stimulated corticosterone production by adrenal cells. These findings suggest that transit through the sterol ester pool is not part of an obligatory itinerary for steroidogenic cholesterol.
The limiting membranes of the lipid droplets contain a family of proteins called perilipins. These proteins protect the droplet contents from hydrolysis in the basal state. They also serve as scaffolds, anchoring lipases to the lipid droplet surface, and mediating physical and functional interactions between lipid droplets and other organelles, including mitochondria. Mobilization of cholesterol ester stores occurs when cells are stimulated by trophic hormones. An adenosine 3′,5′ cyclic monophosphate (cAMP)-mediated process leads to phosphorylation of perilipins by protein kinase A and subsequent detachment of perilipins from the droplet surface, giving lipases access to the sterol esters.
The lipases that free cholesterol from lipid droplet sterol esters are hormone-sensitive lipase and neutral cholesterol esterases (see Fig. 4.2 ). Protein kinase A activates hormone-sensitive lipase by phosphorylation of serine residues, promoting binding of the sterol esterase to lipid droplets. This enzyme’s role in steroidogenesis was suggested by the reduced production of corticosterone under ACTH stimulation associated with an accumulation of lipid droplets in the adrenal cortex of mice deficient in Lipe . Targeted mutation of both the Lipe and Nceh1 genes results in adrenal enlargement and lipid accumulation but no impairment in ACTH-stimulated corticosterone production. These different mouse models suggest a role for both hormone-sensitive lipase and neutral cholesterol ester hydrolase activities in mobilization of sterol esters. The variable impact of enzyme deficiency on steroidogenesis in these murine models may be related to genetic background or the age of mice at the time of study since lipid accumulation may have secondary deleterious effects on cell function beyond the disruption of sterol ester hydrolysis.
The size and number of lipid droplets change as the ester pool expands or contracts. The quantity of sterol ester stored is determined by the availability of cholesterol to the cell through de novo synthesis, through accumulation of lipoprotein-carried cholesterol, and by the steroidogenic activity of the cell. Trophic stimulation promotes cholesterol ester hydrolysis and diverts cholesterol into the steroidogenic pool away from SOAT1, preventing re-esterification and resulting in a net depletion of cholesterol from the lipid droplets. Conversely, pharmacologic blockade of steroid hormone synthesis (e.g., with the cholesterol side-chain cleavage inhibitor, aminoglutethimide) or defects in cholesterol use for steroidogenesis (e.g., congenital lipoid adrenal hyperplasia) increase sterol ester storage by increasing the amount of cholesterol available to SOAT1.
The exact intracellular itinerary of lipoprotein-derived cholesterol, free cholesterol from the plasma membrane, or free cholesterol released from lipid droplets remains to be elucidated. Much is still unknown regarding the methods in which sterol is presented to the mitochondria, which is where the first step in steroidogenesis occurs. It is likely that sterol distribution to and from organelles occurs through a dynamic vesicular–tubular late endosomal compartment and through the assistance of lipid transfer proteins. The lipid transfer proteins involved in this process may include ATP binding cassette transporter G1 (ABCG1), proteins with a structure resembling StAR (STARD1), including its paralogues, STARD3, STARD4, STARD5 (see Fig. 4.2 ), and SCP 2 . The specific roles of these proteins remains to be clarified and their functions may be redundant, since knockout mouse models with the exception of STARD1 do not show strong steroidogenic phenotypes.
The mitochondria of steroidogenic cells are frequently found in close association with cytoplasmic lipid droplets, which may facilitate movement of substrate from these depots to the mitochondria. They have tubulovesicular cristae in contrast to the lamellar cristae that are characteristic of mitochondria in other cells. The remodeling of cristae in the human syncytiotrophoblast is believed to facilitate steroidogenesis. The inner mitochondrial membranes contain the cholesterol side-chain cleavage enzyme, which catalyzes the first step in cholesterol metabolism into steroid hormones leading to the formation of pregnenolone. The hydrophobic cholesterol substrate must move from the mitochondrial outer membrane across the aqueous intermembranous space to reach the inner membrane. This translocation process is the major rate-limiting step in steroidogenesis. The capacity to produce large amounts of steroid hormone in rapid response to trophic stimulation requires the action of STARD1, the prototypic member of the START domain family, which greatly enhances the flux of substrate to the side-chain cleavage system (see Fig. 4.2 ). The mitochondrial cholesterol side-chain cleavage system is also juxtaposed to downstream enzymes in the steroidogenic pathway on the endoplasmic reticulum, allowing for efficient metabolism of pregnenolone.
Regulation of Cellular Cholesterol Balance
- ◆
Cellular uptake of lipoprotein cholesterol and de novo synthesis are tightly regulated.
- ◆
Tropic hormones that stimulate steroidogenesis enhance lipoprotein cholesterol uptake and de novo cholesterol synthesis.
Cellular free (nonesterified) cholesterol balance is highly regulated by transcriptional, posttranscriptional, and posttranslational mechanisms. The expression of genes involved in cholesterol biosynthesis (e.g., the gene encoding 3-hydroxy-3-methylglutaryl coenzyme A reductase, HMGCR ) and the uptake of plasma cholesterol (LDLR) are controlled by master transcription factors, the sterol regulatory element-binding proteins (SREBF1, which encodes SREBP-1a and SREBPB-1c, and SREBF2, which encodes SREBP-2). SREBP-1a and SREBP-2 are the key transcription factors regulating cholesterol synthesis. The SREBPs are synthesized as inactive precursors that are bound to the endoplasmic reticulum. They interact with regulatory proteins, SREBP-cleavage activating protein (SCAP) and the insulin-induced genes 1 and 2, which inhibit SCAP. When cells are depleted of sterols, SCAP transports the SREBPs from the endoplasmic reticulum to the Golgi apparatus where SREBPs are cleaved through the action of two different proteases, releasing an NH 2 -terminal domain of the transcription factors that enter the nucleus where they activate genes controlling lipid synthesis and uptake. Cholesterol loading inhibits the movement of SREBPs to the Golgi and consequently the proteolytic processing, resulting in reduced transcription of the cholesterol synthesis and uptake genes. A micro ribonucleic acid, miR-33, derived from an intron located within the gene encoding SREBP-2, posttranscriptionally modulates expression of genes involved in cellular cholesterol balance.
Metabolites of cholesterol, the side-chain oxygenated sterols, including 22-, 24-, 25-, and 27-hydroxycholesterol, and 7-hydroxylated sterols, are endogenous regulators of cellular cholesterol metabolism acting through the liver X receptors (LXRalpha and LXRbeta, also known as NR1H3 and NR1H2, respectively). In addition to LXRs, steroidogenic factor 1 (SF-1, also known as NR5A1) and a related protein liver receptor homolog-1 (LRH-1, also known as NR5A2) play roles in regulating genes encoding cholesterol metabolizing enzymes including SOAT1 , LIPE , and STARD1 .
Posttranslational mechanisms influencing sterol synthesis and uptake include cholesterol-induced ubiquitination of HMG-CoA reductase, which tags the protein for degradation by proteasomes. Another posttranslational mechanism by which cells control sterol balance is the degradation of LDL receptors by proprotein convertase subtilisin/kexin 9 (PCSK9), a secreted serine protease that binds to LDL receptors on the cell surface and interferes with their recycling so that they are directed to lysosomes for degradation. Gain-of-function mutations in the PCSK9 gene cause autosomal dominant hypercholesterolemia, while loss-of-function mutations are associated with low LDL levels. Inhibition of PCSK9 reduces LDL cholesterol in hypercholestrolemic subjects.
Cells also control their sterol economy by reverse cholesterol transport (sterol efflux), mediated by members of the ATP-binding cassette subfamily A1 (ABCA1), which transfers cholesterol to plasma lipoproteins. In the primate corpus luteum undergoing functional luteolysis, a fall in STARD1 expression results in diminished progesterone production. To maintain free cholesterol balance, expression of lipoprotein receptors is reduced (reduced uptake) and expression of ABCA1 is increased (increased efflux), perhaps by hydroxysterol activation of LXR transcription factors. Excess cholesterol not available for steroidogenesis is esterified and deposited in cytoplasmic lipid droplets. These “homeostatic” adjustments reflect, in part, changes in gene transcription, posttranscriptional, and posttranslational processes.
Overview of Steroidogenesis
- ◆
Several families of enzymes are involved in the synthesis of steroid hormones—the most important being the cytochrome P450 family and the hydroxysteroid dehydrogenases and reductases
- ◆
The critical protein controlling steroidogenesis in cells that respond to tropic hormones is the STARD1.
- ◆
Mutations that inactivate key proteins involved in steroidogenesis cause a spectrum of phenotypes, many of them involving disorders of sexual development.
The cellular manufacture of steroid hormones involves the action of several classes of enzymes including the cytochromes P450, hemeprotein mixed-function oxidases (named because of their distinct absorption peak at 450 nm when reduced in the presence of carbon monoxide), hydroxysteroid dehydrogenases, and reductases.
Cytochrome P450s catalyze the major alterations in the sterol framework, cleavage of the side chain, hydroxylations, and aromatization. These hemeproteins require molecular oxygen and a source of reducing equivalents (i.e., electrons) to complete a catalytic cycle. Each member of the steroidogenic cytochrome P450 family of genes is designated “CYP,” followed by a unique identifying number that usually refers to the carbon atom at which the enzyme acts.
The hydroxysteroid dehydrogenases reduce ketone groups or oxidize hydroxyl functions, employing pyridine nucleotide cofactors, usually with a stereospecific substrate preference and reaction direction. In addition to being involved in hormone biosynthesis in steroidogenic cells, this family of enzymes works with the reductases, steroid sulfotransferases, and steroid sulfatase to regulate the level of bioactive hormone in target tissues. The hydroxysteroid dehydrogenases are key determinants of the cellular response to endogenous steroid hormones and steroidal drugs.
The reductases, using the pyridine nucleotide nicotinamide adenine dinucleotide phosphate (NADPH) as a cofactor, produce saturated ring A steroids from Δ 4 -steroids (again, with stereospecificity). Table 4.1 lists the key steroidogenic enzymes by class and the respective gene designation. Fig. 4.3 outlines the pathways of steroid hormone synthesis indicating where specific enzymes act.
| Protein | Chromosomal | Major Activities | Known Deficiency States | ||
|---|---|---|---|---|---|
| Gene | Locus | Substrates * | |||
| StAR (STARD1) | STARD1 | 8p11.2 | Cholesterol flux within mitochondria | Sterol delivery to P450scc | Congenital lipoid adrenal hyperplasia |
| P450scc | CYP11A1 | 15q23-q24 | Cholesterol hydroxysterols | Cholesterol side chain cleavage | Side chain cleavage enzyme deficiency |
| P450c17 | CYP17A1 | 10q24.3 | Preg, 170H-PregProg, [170H-Prog]DHEA | 17α-Hydroxylase/16α-hydroxylase/17,20-lyase | 17α-Hydroxylase deficiency/isolated 17,20-lyase deficiency |
| P450c21 | CYP21A2 | 6p21.1 | Prog, 170H-Prog | 21-Hydroxylase | 21-Hydroxylase deficiency |
| P450c11β | CYP11B1 | 8q21-q22 | 11-Deoxycortisol | 11-Hydroxylase | 11-Hydroxylase deficiency |
| P450c11AS | CYP11B2 | 8q21-q22 | Corticosterone11-DOC18OH-Corticosterone | 11-Hydroxylase/18-hydroxylase/18-oxidase | CMO I deficiency/CMO II deficiency |
| P450arom | CYP19A1 | 15q21.1 | Androstenedione | 19-Hydroxylase | Aromatase deficiency |
| P450 oxido reductase | POR | 7q11.2 | Androgens/corticoids Testosterone | 17α-hydroxylase/17-20 lyase and 21-hydroxylase aromatization | Combined partial 17α-hydroxylase/17-20 desmolase and 21-hydroxylase deficiencies |
| 3β-HSDl | HSD3B1 | 1p13 | Preg, 170H-PregDHEA, Adiol | 3β-Dehydrogenase/Δ 5-4 -isomerase | |
| 3β-HSD2 | HSD3B2 | 1p13 | Preg, 170H-PregDHEA, Adiol | 3β-Dehydrogenase/Δ 5-4 -isomerase | 3β-HSD deficiency |
| 17β-HSD1 | HSD17B1 | 17q21 | Estrone, [DHEA] | 17β-Ketosteroid reductase | |
| 17β-HSD2 | HSD17B2 | 16q24 | Estradiol, testosterone DHT, 20α-OH-prog | 17β-Hydroxysteroid dehydrogenase/20α-hydroxysteroid dehydrogenase | |
| 17β-HSD3 | HSD17B3 | 9q22 | Androstenedione | 17β-Ketosteroid reductase | Male 17-ketosteroid reductase deficiency |
| 17β-HSD5 | HSD17B5 (AKR1C3) | 10p14-15 | Androstenedione, DHT, 3α-asdiol, 3α-androstanediol, asone, asdione | 17β-Ketosteroid reductase/3α-hydroxysteroid dehydrogenase | |
| 5α-Reductase type 1 | SRD5A1 | 5p15 | Testosterone, C21 steroids | 5α-Reductase | |
| 5α-Reductase type 2 | SRD5A2 | 2p23 | Testosterone, C21 steroids | 5α-Reductase | 5α-Reductase deficiency |
| 11β-HSDI | HSD11B1 | 1q32.2 | Cortisol, cortisone, corticosterone, 11-dehydrocorticosterone | 11β-Ketosteroid reductase | Cortisone reductase deficiency |
| 11β-HSDII | HSD11B2 | 16p22 | Cortisol, cortisterone | 11β-Hydroxysteroid dehydrogenase | Syndrome of apparent mineralocorticoid excess |
| Estrogen sulfotransferase | SULT1E1 | 4q13.1 | Estradiol, estrone | Sulfonation | |
| DHEA sulfotransferase | SULT2A1/SULT2B1 | 19q13.4 | DHEA/Preg, cholesterol | Sulfonation | |
| Steroid sulfatase | STS | Xp23.3 | DHEA sulfate, cholesterol sulfate | Sulfatase | Sulfatase deficiency |
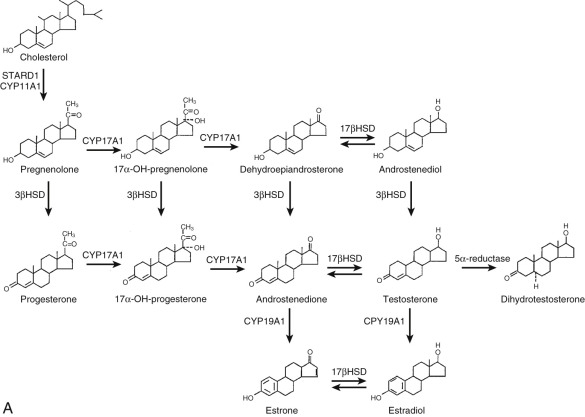
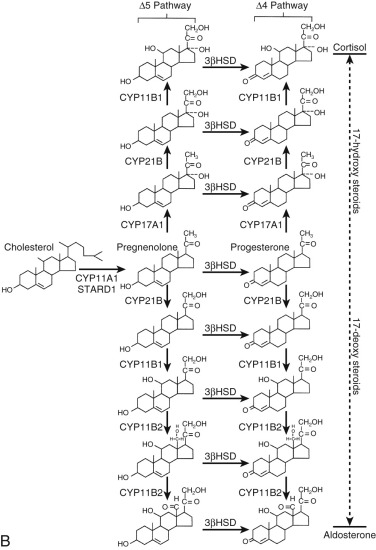
Key Proteins in the Biosynthesis and Metabolism of Steroid Hormones
Steroidogenic Acute Regulatory Protein: The Principal Regulator of Gonadal and Adrenal Steroidogenesis
Translocation of cholesterol from the outer mitochondrial membranes to the relatively sterol-poor inner membranes is the critical step in steroidogenesis. This translocation process occurs at modest rates in the absence of specific effectors. It is markedly enhanced by STARD1, a protein with a short biological half-life. The following evidence established STARD1 as the key mediator of substrate flux to the cholesterol side-chain cleavage system:
- 1.
Expression of STARD1 is directly correlated with steroidogenesis.
- 2.
Coexpression of STARD1 and the cholesterol side-chain cleavage enzyme system in cells that are not normally steroidogenic results in substantial pregnenolone synthesis above that produced by cells expressing the cholesterol side-chain cleavage enzyme system alone.
- 3.
Mutations that inactivate STARD1 cause congenital lipoid adrenal hyperplasia, a rare autosomal recessive disorder in which the synthesis of all adrenal and gonadal steroid hormones is severely impaired before the cholesterol side-chain cleavage step.
- 4.
Targeted deletion of the murine Stard1 gene results in a phenotype in nullizygous mice that mimics human congenital lipoid adrenal hyperplasia.
Human STARD1 is synthesized as a 285 amino acid protein. The N-terminus of STARD1 is characteristic of proteins synthesized in the cytoplasm and then imported into mitochondria, with the first 26 amino acid residues predicted to form an amphipathic helix. Newly synthesized STARD1 preprotein (37 kDa) is rapidly imported into mitochondria and processed to the “mature” 30-kDa form. The preprotein has a very short half-life (minutes), but the mature form is longer-lived (hours). The START domain consists of a unique structure that accommodates a cholesterol molecule. However, the binding of one sterol molecule is not sufficient to explain STARD1 action, and it is evident that STARD1 must be a catalyst for the transfer of multiple cholesterol molecules.
STARD1 contains two consensus sequences for cAMP-dependent protein kinase phosphorylation at Ser57 and Ser195. Ser195 of human STARD1 must be phosphorylated for maximal steroidogenic activity in model systems.
Tissues that express STARD1 at high levels perform trophic hormone–regulated mitochondrial sterol hydroxylations through the intermediacy of cAMP. STARD1 messenger RNA (mRNA) and protein are not present in the human placenta, an observation that is consistent with the fact that pregnancies hosting a fetus affected with congenital lipoid adrenal hyperplasia go to term. Although estrogen production is impaired in these pregnancies because of diminished fetal adrenal androgen production, placental progesterone synthesis is not significantly affected, indicating that the trophoblast cholesterol side-chain cleavage reaction is independent of STARD1.
The abundance of STARD1 protein in steroidogenic cells is determined primarily by the rate of STARD1 gene transcription, which is influenced by transcription factors that control other genes encoding proteins involved in cholesterol metabolism (e.g., SREBPs, NR5A1, NR5A2, LXRs). STARD1 mRNA stability and translational mechanisms (in some species determined by the 3′-untranslated region of the transcripts) may also contribute. In differentiated cells, the STARD1 gene is activated by the cAMP signal transduction cascade within 15 to 30 minutes. In differentiating cells (e.g., luteinizing granulosa cells), the induction of STARD1 transcription takes hours and requires ongoing protein synthesis.
STARD1 was initially thought to stimulate cholesterol movement from the outer to the inner mitochondrial membrane as it was imported into the mitochondria. However, STARD1 protein lacking the mitochondrial targeting sequence is as effective as native STARD1 in stimulating steroidogenesis. Other STARD1 constructs engineered for prolonged tethering to the surface of the mitochondria were very active in stimulating pregnenolone production, suggesting that the residency time of the protein on the mitochondrial surface determines the duration of the steroidogenic stimulus. Recombinant human STARD1 lacking the mitochondrial targeting sequence enhanced pregnenolone production by isolated ovarian mitochondria in a dose- and time-dependent fashion, with significant increases in steroid production observed within minutes. Collectively, these findings strongly suggest that STARD1 acts on the outer mitochondrial membrane to promote cholesterol translocation. This perspective implies that import of the protein into the mitochondrial matrix, rather than being the trigger to steroid production, is actually the “off” mechanism, because it removes STARD1 from its site of action ( Fig. 4.4 ). Consequently, ongoing production of STARD1 preprotein is required to sustain steroidogenesis.
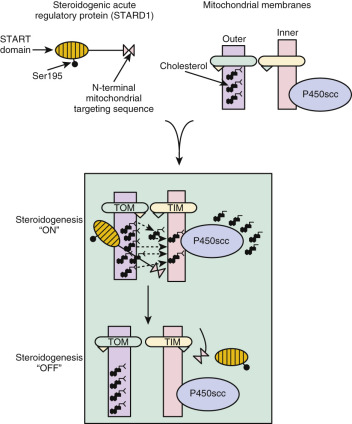
The findings of the previously described experiments are most consistent with the idea that STARD1 enhances desorption of cholesterol from the sterol-rich outer mitochondrial membrane to the relatively sterol-poor inner membranes. The desorption process may involve a pH-dependent conformational change (molten globule transition). Even though STARD1 contains a hydrophobic pocket that binds cholesterol, sterol binding is not required for steroidogenic activity.
The molecule or structure on the mitochondrial outer membrane that STARD1 acts on to promote cholesterol movement to the inner membrane has not been definitively identified. The STARD1 “receptor,” which could be a protein or a lipid, is not specific for mitochondria of steroidogenic cells, since STARD1 works in the context of COS-1 cells, which are not normally steroid hormone-producing cells. Thus the specificity of the mechanism of mitochondrial cholesterol translocation is determined by whether STARD1 is expressed. Initially the translocator protein (TSPO; also known as the peripheral benzodiazepine receptor ), which is found in the outer mitochondrial membrane of many cell types, was postulated to be a target of STARD1 because molecules that bind to TSPO stimulate steroidogenesis and knockdown of TSPO expression in cultured cells reduced steroid synthesis in the presence of STARD1. However, the subsequent phenotyping of TSPO-deficient mice revealed that TSPO is dispensable for steroidogenesis. Despite ongoing debate in the literature over the latter findings, the weight of evidence indicates that TSPO is not required for STARD1 action. Another potential candidate for the STARD1 effector molecule is the mitochondrial voltage-dependent anion channel 2 (VDAC2), which is also expressed by many different cell types and has been proposed to be a cholesterol “pore” through which outer mitochondrial membrane cholesterol could flow to the inner membrane.
As noted above, mutations in the STARD1 gene cause congenital lipoid adrenal hyperplasia, a rare autosomal recessive disease. Exceptions occur in Japan and Korea, however, where the mutation accounts for at least 5% of all cases of congenital adrenal hyperplasia. The pathophysiology of the disease entails a two-step process in which impaired use of cholesterol for steroidogenesis leads to accumulation of sterol esters in lipid droplets. These droplets ultimately compress cellular organelles, causing damage through the formation of lipid peroxides. This damage occurs prominently in the adrenal cortex and Leydig cells. Mutations inactivating NR5A1 (SF-1) also cause cholesterol accumulation in Leydig cells because STARD1 and cholesterol side-chain cleavage enzyme expression are impaired.
Mutations found in the STARD1 gene, which is composed of seven exons, include frameshifts caused by deletions or insertions, splicing errors, and nonsense and missense mutations. These mutations lead to the absence of STARD1 protein or the production of functionally inactive protein. Several nonsense mutations were shown to result in C-terminus truncations of STARD1. One of these mutations, Gln258Stop, results in the deletion of the final 28 amino acids of the STARD1 protein and accounts for 80% of the known mutant alleles in the affected Japanese population. Known point mutations that produce amino acid substitutions occur in exons 5 to 7 of the gene, the exons that encode the C-terminus. Mutations that cause partial loss of STARD1 activity (usually 20% to 30% of normal) are associated with a milder disease phenotype or “nonclassical” disease.
Although affected XY subjects are pseudohermaphrodites (46,XY disorder of sexual development [DSD]) because of an inability to generate sufficient fetal testicular testosterone to masculinize the external genitalia, 46,XX subjects have normal external genitalia, develop female secondary sexual characteristics, and experience menarche. They are, however, anovulatory and unable to produce large amounts of estradiol and progesterone in a cyclic fashion. The fact that some ovarian estradiol synthesis occurs reflects the existence of STARD1-independent substrate movement to the cholesterol side-chain cleavage system.
Cholesterol Side-Chain Cleavage Enzyme (P450scc Encoded by CYP11A1 )
Cholesterol side-chain cleavage is catalyzed by cytochrome P450scc and its associated electron transport system, consisting of a flavoprotein reductase (ferredoxin or adrenodoxin reductase) and an iron sulfoprotein (ferredoxin or adrenodoxin), encoded by the FDX1 gene, which shuttles electrons to cytochrome P450scc. The side-chain cleavage reaction involves three catalytic cycles: the first two lead to the introduction of hydroxyl groups at positions C-22 and C-20, and the third results in scission of the side chain between these carbons. Each catalytic cycle requires one molecule of NADPH and one molecule of oxygen so that the formation of one mole of the cleavage products, pregnenolone and isocaproaldehyde, uses three moles of NADPH and three moles of oxygen.
The slowest step of the reaction is the binding of cholesterol to the hydrophobic pocket of P450scc where the heme resides. The sterol substrate remains bound to a single active site on cytochrome P450scc for all three cycles because of the tight binding of the reaction intermediates. The dissociation constant (K d ) for binding of cholesterol, a measure of the enzyme’s affinity for its substrate, is approximately 5000 nM, whereas the K d for the binding of the intermediate product 22-hydroxycholesterol is 4.9 nM; the K d for 20,22-dihydroxycholesterol is 81 nM. The estimated K d for pregnenolone, the end product, is 2900 nM, which permits its dissociation from the enzyme at the end of the reaction.
Reducing equivalents are shuttled to cytochrome P450scc by ferredoxin in cycles of reduction and oxidation, facilitated by differential affinities of the proteins, depending on their state of oxidation or reduction. Ferredoxin forms a 1 : 1 complex with ferredoxin reductase, which catalyzes reduction of the iron-sulfur protein. The reduced ferredoxin then dissociates and forms a 1 : 1 complex with cytochrome P450scc and is subsequently oxidized when it donates its electrons to P450scc. Oxidized ferredoxin returns to ferredoxin reductase for electron recharging. This recharging is facilitated by the fact that ferredoxin reductase has a greater affinity for oxidized over reduced ferredoxin. The binding of cholesterol to cytochrome P450scc increases its affinity for reduced ferredoxin, which enhances the shuttle of electrons to substrate-loaded enzyme.
The rate of formation of pregnenolone is determined by:
- 1.
Access of cholesterol to the inner mitochondrial membranes
- 2.
The quantity of cholesterol side-chain cleavage enzyme, and secondarily, its flavoprotein and iron-sulfur protein electron transport chain
- 3.
Catalytic activity of P450scc, which can be influenced by posttranslational modification
Acute alterations in steroidogenesis generally result from changes in the delivery of cholesterol to P450scc, whereas long-term alterations involve changes in the quantity of enzyme proteins as well as cholesterol delivery.
Mutations in the CYP11A1 gene that result in significantly diminished cholesterol side-chain cleavage activity have been reported in association with adrenal insufficiency and 46,XY DSD (sex reversal), phenotypes that are similar to those associated with inactivating mutations in the STARD1 gene.
The discovery of mutations causing severe P450scc deficiency in some humans born at term challenges the notion that absence of P450scc activity in the fetus and placenta is incompatible with pregnancy progressing beyond the usually limited period of luteal progesterone support. Therefore, compensatory mechanisms appear to exist to maintain sufficient progestational activity, perhaps sustained corpus luteum function, to support pregnancy and fetal viability in women hosting a fetus with CYP11A1 mutations. Another surprising and distinguishing feature of some subjects with CYP11A1 mutations is the absence of adrenal hypertrophy, as seen in STARD1 mutations.
17α-Hydroxylase/17,20-Lyase (P450c17; CYP17A1)
P450c17 is an endoplasmic reticulum enzyme that catalyzes two reactions: hydroxylation of pregnenolone and progesterone at carbon 17 and conversion of pregnenolone into C19 steroids (in the case of the human enzyme, progesterone is also converted but to a much lesser extent). The 17α-hydroxylation reaction requires one pair of electrons and molecular oxygen. The lyase reaction requires a second electron pair and molecular oxygen. The reducing equivalents are transferred to the P450c17 heme iron from NADPH by NADPH cytochrome P450 reductase (POR), an enzyme functioning in the endoplasmic reticulum. The hydroxylase and lyase reactions are both believed to proceed through a ferryl oxene mechanism with the substrate bound to the enzyme in the catalytic pocket in the same orientation. P450c17 also catalyzes 16α-hydroxylation of progesterone and DHEA.
The importance of POR in steroid metabolism catalyzed by endoplasmic reticulum cytochrome P450 enzymes has been illuminated by the phenotype of individuals with POR deficiency, which causes a form of congenital adrenal hyperplasia. The autosomal recessive disorder results in a steroid profile suggestive of combined 21-hydroxylase and 17-hydroxylase/17,20-lyase deficiency, which presents as a range of phenotypes, including adrenal insufficiency, ambiguous genitalia (DSD), and Antley-Bixler syndrome-like skeletal malformation syndrome.
Several factors determine whether substrates only undergo 17α-hydroxylation or subsequent scission of the 17,20 bond, including:
- 1.
The nature of the substrate
- 2.
The amount of POR and flux of reducing equivalents
- 3.
Allosteric effectors such as cytochrome b 5
- 4.
Posttranslational modification of P450c17 ( Fig. 4.5 )
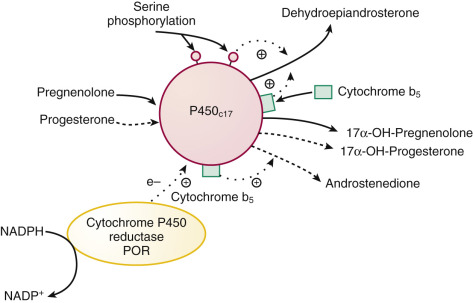
FIGURE 4.5
Factors modulating 17α-hydroxylase and 17,20-lyase activities of P450c17 (CYP17A1) .
Phosphorylation of serine/threonine residues and cytochrome b5 stimulate (designated by plus symbol) lyase activity, with pregnenolone being the preferred substrate for the lyase reaction. Dephosphorylation of the serine/threonine residues by protein phosphatase 2A reduces lyase activity. Flow of reducing equivalents ( e – , electrons) from cytochrome P450 reductase (POR) stimulates both 17α-hydroxylase and lyase activities. NADP , Nicotinamide adenine dinucleotide phosphate.
Collectively, these factors determine the nature of the products produced by the enzyme, which, in the gonads and zona reticularis, favor androgens through augmentation of lyase activity. In contrast, 17α-hydroxylation required for glucocorticoid and mineralocorticoid synthesis is favored in the zona fasciculata and glomerulosa.
Human P450c17 preferentially uses Δ 5 substrates for 17,20 bond cleavage. Cytochrome b 5 (CYB5A) promotes electron transfer via POR for the lyase reaction acting as an allosteric effector and not as an electron donor per se, because the apo b 5 protein is also effective. Cytochrome b 5 also increases the use of 17α-hydroxyprogesterone as a substrate for androstenedione synthesis. The distribution and regulation of cytochrome b 5 expression in the adrenal cortex, being greatest in the adult zona reticularis, supports a role for this protein in the regulation of lyase activity. The product of a second cytochrome b 5 gene (type 2 cytochrome b 5 , CYB5B), found in human testis and expressed in the adrenals, also increases lyase activity.
Phosphorylation of P450c17 at serine and threonine residues by a yet-to-be-identified protein kinase appears to be necessary for maximal 17,20-lyase activity. The phosphorylated P450c17 protein is evidently a substrate for protein phosphatase 2A (PP2A), since inhibitors of PP2A enhance lyase activity in cultured adrenal tumor cells.
Adrenarche, the increased production of adrenal androgens in the absence of increased production of cortisol or levels of ACTH, may result from enhanced P450c17 lyase activity due to increased expression of cytochrome b 5 or the state of P450c17 phosphorylation.
A P450c17-independent mechanism for conversion of pregnenolone into DHEA in glial tumor cell homogenates promoted by FeSO 4 has been described. This conversion presumably results from the fragmentation of tertiary hydroperoxides, which are probably derived from pregnenolone molecules oxygenated at carbons 17 and 20. The physiological significance of this pathway, if any, to the formation of C19 steroids in the brain or other tissues is unknown.
Mutations in the CYP17A1 gene cause combined or isolated deficiency states for each activity of P450c17. Individuals with combined deficiency have a marked diminution in the production of C19 and C18 steroids, low levels of cortisol that result in elevated ACTH secretion, and excess production of steroids proximal to the P40c17 reaction. Hypertension from sodium retention and hypokalemia are a consequence of the increased production of 11-deoxycorticosterone. The inability to produce sex steroid hormones prevents adrenarche, puberty in females, and results in incomplete or absent development of male genitalia (46,XY DSD). Isolated 17,20-lyase deficiency is very rare. The documented cases result from point mutations that permit pregnenolone or progesterone to be bound and undergo 17α-hydroxylation, but prohibit the efficient receipt or usage of a second pair of electrons provided by POR to support the 17,20-lyase reaction.
Aromatase (P450aro, CYP19A1)
Aromatase, an endoplasmic reticulum enzyme, catalyzes three sequential hydroxylations of a C19 substrate by using three molecules of NADPH and three molecules of molecular oxygen to produce one molecule of C18 steroid with a phenolic A ring. The first hydroxylation yields a C19 hydroxyl derivative, which is converted in a second hydroxylation to a gem diol that collapses to yield a C19 aldehyde. The final hydroxylation involves the formation of a 19-hydroxy-19-hydroperoxide intermediate that results in the elimination of the C19 methyl group as formic acid and concurrent aromatization. This sequence of reactions occurs at a single active site on the enzyme, with reducing equivalents transferred to P450aro by POR. The crystal structure of aromatase complexed with androstenedione revealed hydrogen bonding and tight packing of hydrophobic side chains that explain the enzyme’s specificity for androgen substrates.
The aromatase protein is encoded by a single large gene, CYP19A1 . This gene produces cell-specific transcripts from different promoters ( Fig. 4.6 ). The promoter driving ovarian aromatase expression lies adjacent to the exon encoding the translation start site (promoter IIa). In granulosa cells, FSH stimulates transcription of the genes encoding both aromatase and POR. A separate promoter lying approximately 100 kb upstream from the start of translation controls placental CYP19A1 transcription. Expression of aromatase in adipose tissue, skin, and brain is driven from other promoters. Cytokines (including interleukin-11, interleukin-6, oncostatin-M, and leukemia-inhibiting factor) increase P450arom expression in adipose tissue via the I.4 promoter.
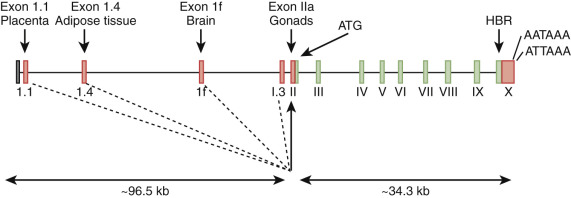
Several cases of aromatase deficiency have been described. Pregnancies in which the fetus is affected with aromatase deficiency are characterized by low maternal urinary estrogen excretion, maternal virilization, and ambiguous genitalia or female pseudohermaphroditism (46,XX DSD) in affected genetic females. The maternal and fetal virilization in the absence of placental aromatase activity highlights the importance and efficiency of the placenta in converting maternal and fetal androgens into estrogens.
Among the mutations identified in the CYP19A1 gene are an 87-bp insertion at the splice junction between exon 6 and intron 6, causing the addition of 29 in-frame amino acid residues, with the other mutations being mainly missense or nonsense mutations in exons 4, 9, and 10. The mutant protein with the 29 in-frame amino acid residues displayed less than 3% of the activity of normal aromatase. Compound heterozygous mutations in coding sequences found in patients with aromatase deficiency have also been shown to have minimal activity. When aromatase activity has been measured in placenta from offspring with CYP19A1 mutations, activities have been found to be markedly reduced.
The aromatase-deficient (ArKO) mice show many of the features of human aromatase deficiency, and the consequential lack of estrogens, including a profound bone phenotype with reductions in all indices of bone mineralization.
Families with autosomal dominant estrogen excess resulting from overexpression of aromatase have been reported. The phenotypes include severe prepubertal gynecomastia in males and macromastia and premature puberty in females. The aromatase overexpression is caused in some families by heterozygous genomic rearrangements. In some cases an inversion results in a constitutively active cryptic promoter placed in control of the CYP19A1 gene, driving excess aromatase production.
Inappropriate expression of aromatase in neoplastic and nonneoplastic tissue has also been found. In these pathological conditions, exemplified by breast cancer, there appears to be a shift in promoter use to favor the stronger gonadal (promoter IIa or I.3) over the weaker adipose tissue promoter (promoter I.4). This shift allows for activation of a cAMP-dependent signaling pathway, accounting for excessive aromatase expression and the resulting increase in estrogen synthesis.
11β-Hydroxylases (P450c11β and P450c11AS)
The human genome contains two genes that encode related mitochondrial enzymes involved in 11β-hydroxylation and aldosterone synthesis, respectively: P45011β, encoded by CYP11B1 , and P450c11AS (also referred to as “P450aldo,” “P450c18,” or “P450cmo”), encoded by CYP11B2. The encoded proteins differ in only 33 amino acid residues. Both enzymes display 11β-hydroxylase activity, but P450c11AS can also perform the two oxygenation steps at carbon 18 to produce aldosterone. They require molecular oxygen and reducing equivalents for catalysis, and the latter is shuttled to the enzymes by the adrenodoxin reductase–adrenodoxin system.
CYP11B1 , a gene whose transcription is stimulated by ACTH-triggered cAMP signaling pathways, is expressed in the zonae fasciculata and reticularis of the adrenal cortex. CYB11B1 is also expressed in human Leydig and theca cells and is responsible (along with type II 11β-hydroxysteroid dehydrogenase) for the production of 11-ketotestosterone, a molecule that activates androgen receptors. In contrast, CYP11B2 expression is restricted to the zona glomerulosa. Transcription of this gene is stimulated by protein kinase C signaling pathways, which are turned on by angiotensin II.
Mutations in the CYP11B1 gene cause 11β-hydroxylase deficiency, whereas mutations in CYP11B2 cause 18-hydroxylase or corticosterone methyl oxidase I deficiency and 18-oxidase or corticosterone methyl oxidase II deficiency. Unequal crossover of the adjacent CYP11B1 and CYP11B2 genes creates a third hybrid gene, in which the cAMP-regulated promoter of the CYP11B1 gene drives expression of a chimeric protein with aldosterone synthase activity. This leads to a state of glucocorticoid-suppressible aldosteronism.
11β-hydroxylase deficiency is characterized by high levels of 11-deoxycortisol and deoxycorticosterone, resulting in salt retention and hypertension. Affected females are virilized by the excess production of adrenal androgens driven by elevated ACTH levels. CYP11B1 mutations causing 11β-hydroxylase deficiency include those resulting in nonsynonymous amino acid substitutions and a premature stop codon.
Corticosterone methyl oxidase I deficiency results from the complete absence of P450c11AS activity. Aldosterone synthesis is absent, but the production of corticosterone and cortisol is retained. Corticosterone methyl oxidase II deficiency is caused by mutations that inactivate 18-methyl oxidase while retaining 18-hydroxylase activity. This causes elevated 18-hydroxycorticosterone and low aldosterone levels.
21-Hydroxylase (P450c21,CYP21A2)
P450c21 is an adrenal endoplasmic reticulum enzyme that catalyzes the 21-hydroxylation of progesterone and 17α-hydroxyprogesterone in the pathway of mineralocorticoid and glucocorticoid biosynthesis. The Michaelis constant (K m ) for 17α-hydroxyprogesterone (1.2 µM) is lower than that for progesterone (2.8 µM), and the apparent maximum velocity (V max ) for the former substrate is twice that for progesterone. The enzyme requires 1 mole of molecular oxygen and reducing equivalents (generated from NADPH through POR) to accomplish the hydroxylation of carbon 21. Mutations inactivating POR cause a partial deficiency in 21-hydroxylase activity and a partial deficiency in 17α-hydroxylase/17,20-lyase activity. The primary regulator of CYP21A2 expression in the zona fasciculata is ACTH.
The CYP21A2 gene is adjacent to a pseudogene (CYP21A1P) , separated by the complement C4B gene. These genes are embedded in the human leukocyte antigen region on band 6p21.1. Frequent unequal crossovers and gene conversions make 21-hydroxylase deficiency one of the most common autosomal recessive metabolic diseases, occurring in 1 : 10,000 to 1 : 15,000 births. Unequal crossover, with the complete loss of the C4B gene and a net deletion of CYP21A2 , along with gene conversion events in which mutations in the pseudogene are introduced into the expressed gene, result in reduced 21-hydroxylase enzyme levels or impaired catalytic activity ( Fig. 4.7 ). Large-scale deletions/gene conversions may extend into the adjacent gene encoding tenascin-X, which when mutated in both alleles, causes a form of Ehlers-Danlos syndrome.
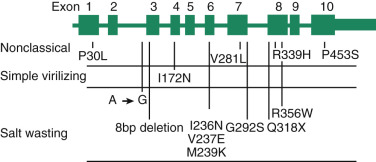
The signs and symptoms of congenital adrenal hyperplasia caused by 21-hydroxylase deficiency reflect deficits in cortisol (because of inability to convert 17α-hydroxyprogesterone into 11-deoxycortisol) and aldosterone (because of inability to convert progesterone into deoxycorticosterone). Another contributing factor is the accumulation of adrenal androgens that results from elevated ACTH levels due to the absence of cortisol-negative feedback on the hypothalamic–corticotrophin axis.
The clinical phenotypes are, however, variable and dependent on the severity of the 21-hydroxylase deficiency. The non–salt-wasting, salt-wasting, and nonclassic forms are associated with certain mutations that affect the amount of residual 21-hydroxylase activity, with the salt-wasting form being characteristic of severe enzyme deficiency (deletions and large gene conversions). The simple virilizing (non–salt-wasting) form is associated with mutations that substantially reduce activity (e.g., the missense mutation resulting in a nonsynonymous amino acid substitution Ile172Asp), and the nonclassic (or late-onset ) form is caused by mutations that do not severely impair the level of expression or activity of P450c21 (e.g., Val28Leu, Pro30Leu).
Hydroxysteroid Dehydrogenases and Reductases
Hydroxysteroid dehydrogenases (HSDs) or oxidoreductases catalyze the interconversion of alcohol and carbonyl functions in a position- and stereospecific manner on the steroid nucleus and side chain, using oxidized (+) or reduced (H) nicotinamide adenine dinucleotide, NAD(H), or NADP(H) as cofactors. In some instances, HSDs display bifunctionality (e.g., they oxidize or reduce 17β and 20α oxy functions), such as HSD17B2. Although the HSDs can catalyze both the oxidation and reduction reactions in vitro under different conditions (e.g., substrate, pH and cofactor), in vivo they catalyze reactions in one direction and can be classified as dehydrogenases or reductases. The enzymes are members of the short chain dehydrogenase/reductase (SDR) and aldo-keto reductase (AKR) superfamilies.
The importance of cofactor availability to HSDs is exemplified by inactivating mutations in the hexose-6-phosphate dehydrogenase (H6PDH) gene, which regenerates NADPH in the endoplasmic reticulum required for the HSD11B1 reaction. Individuals with these mutations have apparent cortisone reductase deficiency due to impaired HSD11B1 activity.
Multiple isoforms of HSDs exist, and, coupled with their tissue-specific expression, account for the ability of specific enzymes to act predominantly as reductases (ketone reduction) or dehydrogenases (alcohol oxidation). In steroidogenic tissues, HSDs catalyze the final steps in progestin, androgen, and estrogen biosynthesis. In steroid target tissues, HSDs can regulate the occupancy of steroid hormone receptors by converting active steroid hormones into inactive metabolites or relatively inactive steroids to molecules with greater binding activity.
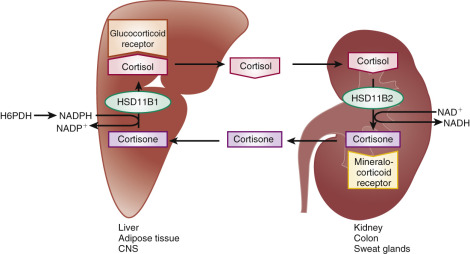
This is best exemplified by the human type 2 11β-HSD (HSD11B2), which controls mineralocorticoid activity in the kidney by converting cortisol (which has high affinity for both the glucocorticoid and mineralocorticoid receptors) to cortisone (which does not bind to the mineralocorticoid receptor). Thus the specificity of mineralocorticoid receptor activation is not determined by the receptor, but by activity of the HSD that removes the more abundant potential mineralocorticoid receptor ligand, leaving aldosterone as the controlling activator ( Fig. 4.8 ). Because of their tissue-specific roles in controlling the bioavailability of steroids, HSDs are interesting targets for pharmacologic manipulation.
3β-Hydroxysteroid Dehydrogenase/Δ 5-4 -Isomerases
The 3β-HSD/Δ 5-4 -isomerases are membrane-bound enzymes localized to the endoplasmic reticulum and mitochondria that use nicotinamide adenine dinucleotide (NAD+) as a cofactor. These enzymes catalyze dehydrogenation of the 3β-hydroxyl group and the subsequent isomerization of the Δ 5 olefinic bond to yield a Δ 4 ketone structure. They convert pregnenolone into progesterone, 3-, 17α-hydroxypregnenolone into 17α-hydroxyprogesterone, and DHEA into androstenedione.
The dehydrogenase and isomerase reactions are performed at a single bifunctional catalytic site that adopts different conformations for each activity. The 3β-hydroxysteroid dehydrogenase step is rate-limiting in the overall reaction sequence, and the NADH formed in this reaction is believed to alter the enzyme conformation to promote the isomerase reaction.
The human genome has two active 3β-HSD/Δ 5-4 -isomerase genes: one encodes a protein predominantly expressed in the placenta, liver, breast, and brain (HSD3B1), while the other encodes a protein expressed in the adrenal cortex and gonads (HSD3B2). These two genes, each consisting of four exons, lie 100 kb apart on band 1p13.1. The human genome also contains five unprocessed pseudogenes closely related to HSD3B1 and HSD3B2 on band 1p13.1, with two of them lying between the expressed genes. The DNA sequences of the exons of the two active genes are very similar, and the encoded proteins differ in only 23 amino acid residues. HSD3B1, however, has a lower K m for substrate than HSD3B2, which facilitates metabolism of lower concentrations of Δ substrate. Electron microscope cytochemistry has localized HSD3B2 activity to the perimitochondrial endoplasmic reticulum. In some cell types, the enzyme appears to be localized in the inner mitochondrial membrane, positioned to act on pregnenolone produced by the cholesterol side-chain cleavage system.
Because most steroidogenic cells have a large capacity to generate progesterone when presented with exogenous pregnenolone, the 3β-HSD/Δ 5-4 -isomerases are not believed to be rate-determining enzymes. However, mutations causing deficiency of HSD3B2 cause a form of congenital adrenal hyperplasia characterized by impaired adrenal and gonadal steroidogenesis with accumulation of Δ 5 steroids in the circulation. The presence of active HSD3B1 in extraadrenal and extragonadal tissues results in the formation of some Δ 4 steroids (e.g., 17α-hydroxyprogesterone).
In its severest form, HSD3B2 deficiency is associated with salt wasting because of insufficient mineralocorticoid production. Kinetic analysis of mutant proteins associated with the salt-wasting and non–salt-wasting forms of the disease showed a 4- to 40-fold reduction in catalytic efficiency for the conversion of pregnenolone into progesterone. The salt-wasting form of the disease is associated with frameshift mutations resulting in protein truncation and a variety of missense mutations that affect affinity for the cofactor and protein stability. The greater instability of the mutant proteins found in subjects with salt-wasting disease compared with those proteins found in the non–salt-wasting form appears to account, in part, for the different clinical phenotypes.
A so-called attenuated, or late-onset, form of 3β-HSD deficiency, diagnosed by steroid measurements, has been described in the literature. However, no mutations have yet been discovered in the genes encoding HSD3B1 and HSD3B2 in subjects with this clinical diagnosis. Mutations in the distal promoter or epigenetic factors that might alter enzyme expression cannot be excluded. The apparent reduced 3β-HSD activity could also be the result of alterations in the membrane environment that affect catalytic activity or posttranslational modifications to the enzyme that diminish its function. Mutations in HSD3B1 have not been detected, although several sequence variants of uncertain significance have been described.
11β-Hydroxysteroid Dehydrogenases: Key Regulators of the Activity of Glucocorticoids
The biological activity of cortisol in target tissues is controlled by the action of two different 11β-hydroxysteroid dehydrogenases that are members of the short-chain dehydrogenase/reductase family (see Fig. 4.8 ). These enzymes catalyze the interconversion of active glucocorticoids and their inert 11-keto metabolites. The type 1 enzyme (HSD11B1) is an endoplasmic reticulum protein with reversible oxidoreductase activity in vitro but preferentially catalyzes the reduction of the 11-keto group, using NADPH as a cofactor in vivo. This enzyme is expressed in the liver, lung, adipose tissue, brain, vascular tissue, and gonads, where it regenerates cortisol from 11-ketosteroids. In the case of the placenta, the activity ensures transport of biologically active cortisol to the fetus in the first half of pregnancy.
Targeted deletion of the type 1 enzyme gene (Hsd11b1) in mice results in animals with lower blood glucose levels in response to overfeeding and stress, impaired activation of gluconeogenesis, and blunted sensitivity to natural glucocorticoids. This finding substantiates a role for HSD11B1 in the amplification of cortisol and corticosterone action. HSD11B1 also plays a key role in the pharmacology of glucocorticoids. Hepatic HSD11B1 converts cortisone and prednisone, inactive prohormones, into active cortisol and prednisolone.
As noted above, mutations in the H6PDH gene cause a syndrome characterized by high ratios of cortisone to cortisol; impaired cortisol negative feedback resulting in elevated ACTH secretion; and increased production of adrenal androgens leading to hyperandrogenism, sexual precocity, and a polycystic ovary syndrome-like phenotype. Mice with targeted mutations in H6pdh share many of the phenotypes of mice with Hsd11b1 mutations. Heterozygous mutations in the HSD11B1 gene cause a mild form of cortisone reductase deficiency.
HSD11B2, also an endoplasmic reticulum enzyme, has a higher affinity for its substrate than HSD11B1 and catalyzes the oxidation of cortisol with NAD + as a cofactor. It shares only modest amino acid sequence identity (21%) with HSD11B1. HSD11B2 is highly expressed in the kidney, colon, salivary glands, and placenta, all tissues that respond to aldosterone, or in the case of the placenta, tissues that act to separate the maternal and fetal endocrine systems (which is important in the third trimester). By converting cortisol and corticosterone to 11-keto compounds, HSD11B2 protects the renal mineralocorticoid receptors, which cannot distinguish cortisol or corticosterone from aldosterone, from inappropriate activation by the glucocorticoids.
Mutations that inactivate HSD11B2 produce a syndrome of apparent mineralocorticoid excess in humans, which is mimicked in the mice deficient in the enzyme that display hypertension, hypokalemia, and renal structural abnormalities. Glycyrrhizic acid, a component of licorice, and its metabolite carbenoxolone are competitive inhibitors of HSD11B2, but they also cause reduced expression of the HSD11B2 mRNA when administered in vivo. As a result, a drug-induced syndrome of apparent mineralocorticoid excess is produced.
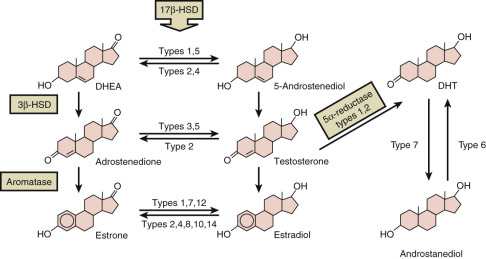
17β-Hydroxysteroid Dehydrogenases: Multiple Enzymes With Specific Biosynthetic and Catabolic Roles
The adrenals, gonads, and placenta reduce 17-ketosteroids into 17β-hydroxysteroids (which have greater biological potency), whereas target tissues usually oxidize 17β-hydroxysteroids, inactivating them. In humans, these metabolic processes are mediated by at least 7 of the 14 known mammalian 17β-HSDs, designated types 1 through 14, according to the chronological order in which they were identified ( Fig. 4.9 ). They are all members of the short-chain dehydrogenase/reductase family, except the type 5 enzyme, which is an AKR. They have different cofactor and substrate specificities, including molecules that are not steroids, subcellular locations, and tissue-specific patterns of expression.
The structures of the genes encoding the 17β-HSDs differ, and their nucleotide sequence homology is low. They can be grouped into enzymes that catalyze NAD + -dependent oxidation (types 2, 4, 6, 8, 9, 10, 11, and 14) and those that catalyze NADPH-dependent reduction (types 1, 3, 5, and 7). Because of the broad substrate specificities, the primary roles of several of these enzymes are in basic metabolic pathways unrelated to steroid metabolism, and deficiencies of these enzymes cause metabolic disease.
The type 1 enzyme (HSD17B1) is referred to as the estrogenic 17β-HSD, because it catalyzes the final step in estrogen biosynthesis by preferentially reducing the weak estrogen estrone to yield the potent estrogen 17β-estradiol. The enzyme is a cytosolic protein that uses either NADH or NADPH as a cofactor. It has 100-fold higher affinity for C18 steroids than for C19 steroids. It also converts 16α-hydroxyesterone into estriol and shows modest 20α-HSD activity.
The HSD17B1 gene is expressed in granulosa cells of the ovary and the placental syncytiotrophoblast. HSD17B1 is also expressed at higher levels in breast cancer cells relative to HSD17B2, which converts estradiol into estrone. Amplification of HSD17B1 in estrogen receptor-positive breast cancers is associated with lower survival compared with subjects without amplification. The crystal structure of HSD17B1 has been determined to a resolution of 2.2 Å with and without bound substrates, providing a molecular framework for the design of specific inhibitors which might be employed in chemotherapy of estrogen-responsive cancers.
HSD17B2 is an endoplasmic reticulum enzyme that inactivates hormones; it preferentially oxidizes testosterone into androstenedione, and it oxidizes estradiol into estrone using NAD+ as its cofactor. However, HSD17B2 can also convert 20α-hydroxyprogesterone into progesterone.
The gene encoding HSD17B2 is expressed in liver, secretory endometrium, and the fetal capillary endothelial cells of the placenta, as well as the endothelial cells of the larger vessels. This expression pattern is consistent with its role of inactivating testosterone and estradiol. HSD17B2 in the fetal capillary endothelium protects the fetal compartment from estradiol formed in the syncytiotrophoblast and from any testosterone escaping aromatization. Its expression in the secretory endometrium permits the conversion of estradiol to estrone, whereas 20α-hydroxyprogesterone is converted back into progesterone, resulting in progestational dominance. In normal breast tissue, HSD17B2 expression predominates over HSD17B1 expression.
HSD17B3 is referred to as the androgenic 17β-HSD, because it catalyzes the final step in testosterone biosynthesis in the Leydig cells, reducing androstenedione to testosterone using NADPH as cofactor. It also can reduce estrone to estradiol. HSD17B3, an endoplasmic reticulum enzyme, is not expressed in the ovary, requiring androgen-producing cells of the ovary to employ another enzyme, probably HSD17B5, to synthesize testosterone.
In the absence of HSD17B3 activity, the testes are unable to convert androstenedione into testosterone, resulting in male pseudohermaphroditism (46,XY DSD) associated with 10- to 15-fold elevations in the ratio of blood androstenedione to testosterone. Females with mutations in the HSD17B3 gene are asymptomatic and produce normal amounts of estrogens and androgens. Molecular analysis of the HSD17B3 gene in affected individuals revealed mutations that affect splicing, cause amino acid replacements in exons 9 and 10, and a small deletion leading to a frameshift. Many of the missense mutations result in proteins that are devoid of catalytic activity when expressed in eukaryotic cells.
HSD17B5, located on chromosome 10p15-14, is a member of the AKR family (AKR1C3). It has 3α-HSD and 20α-HSD activity in addition to 17β-HSD activity that produces testosterone from androstenedione. It is expressed in the adrenal cortex and may serve as the androgenic 17β-HSD of ovarian theca cells. HSD17B5 is also expressed in prostate, mammary gland, and Leydig cells.
HSD17B6 (also known as 11-cis retinol dehydrogenase, RODH) has 3α-HSD activity and catalyzes the conversion of androstanediol to dihydrotestosterone in the prostate. It is also a 3α,3β-epimerase.
Human HSD17B7 is involved in cholesterol synthesis but also thought to produce active estrogens and inactivate androgens. It transforms estrone into estradiol and has 3-keto reductase activity for dihydrotestosterone. The gene is expressed in the ovary, breast, placenta, testes, prostate, and liver cells.
3α- and 20α-Hydroxysteroid Dehydrogenase Activities
A number of HSDs catalyze reactions at both the 3 and 20 positions of steroid hormones (e.g., AKR1C1). The enzymes that are 3α-reductases (e.g., AKR1C2 and AKR1C4) are encoded by tandemly duplicated genes on chromosome 10p14-p15. They are expressed in liver, prostate, breast, and uterus, but some are also expressed in the testis and adrenals. Recessive mutations in one of these genes, AKR1C2, were discovered in 46,XY individuals with DSD, implicating this enzyme in a pathway of bioactive androgen biosynthesis through conversion of androsterone to androstanediol, followed by HSD17B6-mediated conversion of androstanediol to dihydrotestosterone. This finding suggests that two pathways are needed for testicular androgen synthesis to support normal male sexual differentiation.
The enzymes with 20α-HSD activity that are members of the AKR family reduce progesterone to yield the inactive steroid, 20α-hydroxyprogesterone (AKR1C1). They are cytosolic proteins with a molecular weight of approximately 34 kDa, exemplified by genes expressed in human keratinocytes and cells of the liver, prostate, testis, adrenal gland, brain, uterus, and mammary gland. These enzymes prefer NADPH to NADH as a cofactor.
Enzymes with 3α-hydroxysteroid oxidative activity that are members of the short-chain dehydrogenase/reductase family also have 20α-HSD activity and preferentially oxidize 20α-hydroxyprogesterone into progesterone.
Δ 4-5 -Reductases
The Δ 4-5 -reductases are membrane-associated enzymes that reduce the Δ 5-4 double bond in steroid hormones by catalyzing direct hydride transfer from NADPH to the carbon 5 position of the steroid substrate. They produce either 5α or 5β-dihydrosteroids.
5α-Reductases
Two different human 5α-reductases sharing 50% similarity in amino acid sequence and a molecular weight of approximately 29 kDa have been identified. The genes for type 1 (SRD5A1) and 2 (SRD5A2) 5α-reductases each have five exons. The substrate-binding domain of the 5α-reductases is encoded by exon 1, and the cofactor-binding domain is encoded by exons 4 and 5. SRD5A2 is located on chromosome 2p23, whereas SRA5A1 is located on chromosome 5p15 with a pseudogene on Xq24-qter. A third enzyme containing a 5α-reductase domain, SRD5A3, has been identified, but its role in steroid metabolism has not been established.
SRD5A2 is predominantly expressed in male genital structures, including genital skin and prostate, where it reduces testosterone to yield the more potent androgen, 5α-dihydrotestosterone. SRD5A1, which catalyzes a similar reaction on both C21 and C19 steroid hormones, is expressed in the liver, kidneys, skin, and brain. Although this enzyme can also make 5α-dihydrotestosterone, its tissue distribution suggests that its predominant function is to inactivate steroid hormones.
SRD5A2 has an acidic pH optimum and a K m for testosterone in the nanomolar range, whereas SRD5A1 has a broad alkaline pH optimum and a lower affinity for its substrates in the micromolar range. SRD5A2 can also be distinguished from the type 1 enzyme by its selective inhibition by finasteride with an inhibition constant (K i ) of 3 nM.
Inactivating mutations in SRD5A2 cause male pseudohermaphrodism (46,XY DSD). The enzyme defect is characterized by abnormal testosterone-to-5α-dihydrotestosterone ratios. Affected males have varying degrees of abnormal development of the external genitalia, ranging from mild hypospadias to severe defects in which the external genitalia are essentially female. Wolffian ducts develop normally in response to adequate levels of testosterone.
Females carrying mutations in the SRD5A2 gene have a normal phenotype and normal menstrual cycles. They have a low incidence of hirsutism and acne, and similar to males with the disease, they have low ratios of 5α- to 5β-dihydrosteroid metabolites in the urine. The infrequency of acne in both affected males and affected females, the rarity of hirsutism in affected females, the absence of male pattern baldness, and the finding of an atrophic prostate in affected males indicates that SRD5A2 plays an important role in androgen metabolism in skin and in the androgen-dependent growth of the prostate.
Among the mutations reported are deletions that inactivate SRD5A2 and missense mutations that impair enzyme activity by affecting substrate or cofactor binding. A SRD5A2 variant (Ala49Thr) that has increased catalytic activity has been associated with increased risk of prostate cancer.
Mutations have not been described in the human SRD5A1 gene. However, targeted deletion of the murine counterpart results in a female phenotype of reduced fecundity and a parturition defect caused by failed cervical ripening. This defect can be reversed by administration of 5α-androstanediol.
The only known human 5β-reductase (SRD5B1 or AKR1D1) is a member of the AKR superfamily. This enzyme is involved in steroid hormone inactivation in the liver. Its reaction mechanism is similar to that described for 5α-reductase, except that an A/B cis -fused ring product is formed. The enzyme efficiently catalyzes the NADPH-dependent reduction of the Δ 5-4 double bond in C27, C21, and C19 steroids to yield the 5β-dihydrosteroids with a distinct preference for C27 steroids. In keeping with this observation, mutations in the SRD5B1 (AKR1D1) gene that encodes this enzyme result in abnormal bile acid synthesis along with a marked reduction in the primary bile acids and 5β-reduced steroid metabolites.
Sulfotransferases
A family of enzymes introduce the sulfonate ( ![]() ) anion from an activated donor, 3 ′ -phosphoadenosine-5 ′ -phosphosulfate (PAPS), to a steroid hydroxyl acceptor, inactivating the hormone. The major enzymes performing this reaction include estrogen sulfotransferase (SULT1E1), an enzyme that sulfonates the 3-hydroxyl function of phenolic steroids, and the hydroxysteroid sulfotransferases encoded by the closely linked SULT2A1 and SULT2B1 genes on chromosome 19q13.4.
) anion from an activated donor, 3 ′ -phosphoadenosine-5 ′ -phosphosulfate (PAPS), to a steroid hydroxyl acceptor, inactivating the hormone. The major enzymes performing this reaction include estrogen sulfotransferase (SULT1E1), an enzyme that sulfonates the 3-hydroxyl function of phenolic steroids, and the hydroxysteroid sulfotransferases encoded by the closely linked SULT2A1 and SULT2B1 genes on chromosome 19q13.4.
The SULT2A1 enzyme, also known as DHEA sulfotransferase, has a broad substrate range including the 3α-, 3β-, and 17β-hydroxy functions of steroid hormones. SULT2A1 enzyme is expressed at high levels in the fetal zone of the adrenal cortex, as well as in the zona reticularis after adrenarche, and in the liver, gut, and testes. This enzyme is developmentally regulated in the adrenal cortex, with expression increasing between ages 5 and 13 years in association with adrenarche. The GATA-binding factor 6 (GATA6) transcription factor, which is known to increase transcription of other genes involved in androgen biosynthesis, also activates expression of the SULT2A1 gene.
The SULT2B1 gene gives rise to two protein isoforms through alternative splicing. The SULT2B1a isoform sulfonates pregnenolone, whereas the SULT2B1b isoform preferentially sulfonates cholesterol. They both sulfonate dihydrotestosterone. Unlike the SULT2A1 gene that is expressed in a limited number of tissues, SULTB1 isoforms are present in a variety of hormone-producing and hormone-responsive tissues, including the placenta, ovary, uterus, and prostate.
SULT1E1 is expressed in many tissues, including the adrenal gland, liver, kidneys, muscle, fat, and uterus. In the endometrium, progesterone increases its activity, contributing to the inactivation of estradiol in the secretory phase. The importance of estrogen sulfotransferase in modulating local levels of bioactive estrogens has been shown in mice with targeted deletions of the gene. Male mice with estrogen sulfotransferase deficiency have Leydig cell hyperplasia and become infertile with age due to elevated testicular levels of bioactive estrogen. Adipose tissue mass also increases. Hydroxylated metabolites of polychlorinated biphenyls are potent inhibitors of estrogen sulfotransferase with IC50 values in the picomolar range. The blockade of estradiol inactivation by these compounds may account for the reported estrogenic activity of polychlorinated biphenyls.
Mutations in the gene that encodes one of the enzymes that synthesizes the sulfate donor, PAPS (PAPS synthase type 2 [PAPSS2]), results in increased free DHEA levels, with the subsequent conversion of DHEA into biologically active androgens.
Steroid Sulfatase
The sulfonate function on steroids is cleaved by steroid sulfatase, an enzyme encoded by the STS gene on chromosome Xp22.3. This enzyme plays a key role in controlling the generation of biologically active steroids from the inactive sulfated molecules (e.g., estrone sulfate and DHEA sulfate). The sulfated steroid substrates, which are hydrophilic and not membrane-permeable, are taken up by cells via specific transport proteins, including a sodium-dependent anion transporter (SLC10A6) .
The syncytiotrophoblast is enriched in steroid sulfatase, which plays a key role in placental estrogen synthesis by liberating sulfonated androgen precursors produced in the fetal compartment prior to aromatization. The enzyme is also expressed in skin where it metabolizes cholesterol sulfate and sulfated estrogens.
Sulfatase deficiency is associated with marked impairment of placental estrogen synthesis during pregnancy and ichthyosis developing after birth. It occurs mostly in males because of the X chromosome location of the sulfatase gene at a frequency of 1 : 2000 to 1 : 6000 live born males. The majority of subjects with steroid sulfatase deficiency have a deletion of the entire gene that results from recombination of repetitive elements that flank the locus. The large deletions of the STS gene occur in association with mutations in the adjacent Kallmann syndrome gene (KAL1) . Partial deletions in the STS gene causing enzyme deficiency have also been described.
Characteristically, in pregnancies hosting an affected fetus, maternal plasma estriol and urinary estriol excretion are quite low at approximately 5% of the levels found in normal pregnancies. Excretion of estrone and estradiol are also reduced at approximately 15% of normal. Maternal serum 16α-hydroxydehydroepiandrosterone levels are elevated, and intravenous administration of DHEA sulfate to the mother does not lead to an increase in estrogen excretion, whereas administration of DHEA does lead to increased estrogen excretion.
Steroid sulfatase is expressed in estrogen target tissues, including endometrium, bone, and breast. Increased sulfatase expression may contribute to greater bioavailability of estradiol in certain tumors, including breast and endometrial cancers. Inhibitors of sulfatase have been developed and tested in clinical trials (Irosustat).
UDP-Glucuronosyl Transferases
Glucuronidation, catalyzed by a family of uridine 5′-diphospho UDP-glucuronosyl transferases, is part of the metabolic clearance mechanism for steroid hormones by the liver and extrahepatic tissues. There are 18 known UDP-glucuronosyl transferases that have been divided into three subfamilies: UGT1A, UGT2A, and UGT2B. UGT1A enzymes are encoded by a single gene that gives rise to alternatively spliced products capable of acting on estrogens. The UGT2 enzymes are products of separate genes UGT2A and UGT2B . UGT2A is expressed in olfactory epithelium, and UGT2B is expressed in the liver, kidney, breast, lung, and prostate. At least seven members of the UGT2B family have been identified with different steroid substrate specificities. Three UGT2B enzymes, UGT2B7, UGT2B15, and UGT2B17, appear to be responsible for glucuronidation of 5α-dihydrotestosterone, androsterone, and androstane-3α-17β-diol. Of these, UGT2B15 and UGT2B17 are expressed in the prostate where they are proposed to modulate steroid action. Polymorphisms in genes encoding UDP-glucuronosyl transferases have been associated with estrogen levels, suggesting a role for these phase II biotransformation enzymes in regulating the bioactive steroid economy.
Other Steroid Hormone Metabolic Pathways
- ◆
Alternative pathways can produce bioactive androgens in the fetal testes and polycystic ovary syndrome.
- ◆
Some pathways modify molecules that, although steroidal in structure, act via mechanisms that do not involve classical steroid hormone receptors.
There are several metabolic fates of steroid molecules that have a significant role in the synthesis of bioactive hormones. A pathway for 5α-dihydrotestosterone production, referred to as a backdoor pathway, is one example. Among the other fates of steroid hormones are esterification to long-chain fatty acids, formation of catechol estrogens, and the 7α-hydroxylation of androgens. Equine steroidogenic tissues also have a novel pathway for biosynthesis of estrogens, resulting in a phenolic A ring and an unsaturated B ring.
An Alternative Pathway for Androgen Synthesis
Fetal androgen synthesis in marsupials involves a novel pathway that leads from 17α-hydroxyprogesterone to 5α-dihydrotestosterone without going through androstenedione or testosterone as intermediates ( Fig. 4.10 ). This pathway, recently identified in humans, is initiated when either progesterone or 17α-hydroxyprogesterone is 5α-reduced. The resulting 5α-C 21 steroids are reduced by 3α-HSDs (AKR1C2/4) yielding substrates for P450c17. Androsterone produced by P450c17 can ultimately be converted to 5α-dihydrotestosterone by HSD17B6. This pathway (the so-called backdoor pathway) is important for fetal testicular production of bioactive androgens and contributes to androgen excess in the context of 21-hydroxylase deficiency. It has been reported to be active in the polycystic ovary syndrome theca cells.
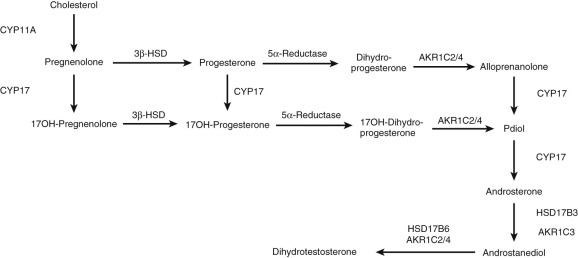
Catechol Estrogens
Catechol estrogens are generated by the actions of genes encoded by CYP1A1 and CYP1A2 (which catalyze 2-hydroxylation of estrogens), and CYP1B1 , which is an estrogen 4-hydroxylase. Although the catechol estrogens are short-lived in vivo and are postulated to have physiological functions as locally generated signaling molecules, they also yield potent genotoxic molecules implicated in carcinogenesis. The 4-hydroxyestrogens can be oxidized to quinone intermediates that react with purine bases of DNA, resulting in depurinating adducts that generate highly mutagenic apurinic sites ( Fig. 4.11 ). Quinones derived from the 2-hydroxyestrogens produce stable DNA adducts and are presumed to be less genotoxic. Metabolism of catechol estrogens may also generate oxygen-free radicals.
Catechol estrogens are methylated by catechol- O -methyltransferase, resulting in a catecholamine-like substance. The methylated catechol estrogens have antiangiogenic and antitumor activity through inhibition of hypoxia-inducible factor-1α (HIF-1α), a proangiogenic transcription factor. There is evidence that altered production of 2-methoxyestradiol may contribute to the pathogenesis of preeclampsia through its modulation of antiangiogenic factor production by the placenta. Levels of antiangiogenic (catechol estrogens) as well as proangiogenic estrogen metabolites are correlated with luteal function.
Steroid Fatty Acid Esters
Steroids esterified to long-chain fatty acids are present in blood bound to lipoproteins, or are found in tissue, particularly steroidogenic glands and fat. These hydrophobic molecules may serve as a depot form of steroid, but they also have unique biochemical attributes. Estradiol 17-esters are produced in blood by the action of lecithin-cholesterol acyl transferase and in tissues by SOATs. The fatty acid esters of estradiol have pronounced antioxidant activity. Fatty acid esters of other steroids, including pregnenolone, testosterone, DHEA, and glucocorticoids, have also been described.
Steroid 7α-Hydroxylation
Substituents on carbon 7 of the steroid nucleus can have a significant effect on activity. A cytochrome P450 encoded by the CYP7B gene catalyzes 7α-hydroxylation of steroid hormones and oxysterols. The 7α-hydroxylation of DHEA produces a molecule with enhanced immunostimulatory activity, a property demonstrable in animal bioassays.
B-Ring Unsaturated Steroids
The pregnant mare produces estradiol and estrone, but also B-ring unsaturated estrogens (equilin [with an 8,9 olefinic bond], equilenin [with a phenolic B ring], 17α-dihydroequilin, 17α-dihydroequilenin, 17β-dihydroequilin, and 17β-dihydroiequilenin; Fig. 4.12 ).
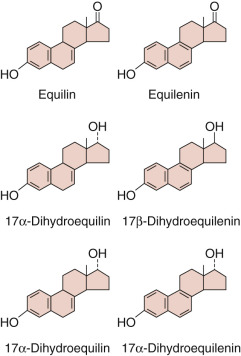
These B-ring unsaturated compounds are potent estrogens in vivo . Although 7-dehydrocholesterol can be converted into B-ring unsaturated estrogens (as occurs in Smith-Lemli-Opitz syndrome), the biosynthesis of these compounds in the pregnant mare occurs by a pathway not requiring the synthesis of squalene or cholesterol, and thus does not involve 7-dehydrocholesterol. Evidently, a C25 sesterterpene pathway coexists with the normal biosynthetic route of “standard” estrogens from a cholesterol precursor.
Vitamin D Synthesis and Metabolism
- ◆
Vitamin D 3 is produced endogenously in the skin from 7-dehydrocholesterol.
- ◆
Vitamin D 2 is obtained from the diet from plant sources.
- ◆
Vitamin D 2 and D 3 are metabolized by the same enzymes to produce biologically active compounds: 1α,25-dihydroxyvitamin D.
Increasing epidemiological evidence indicates that in addition to calcium and bone metabolism the secosteroid, vitamin D, has important roles in immune function and reproduction. Vitamin D deficiency or deficiency of its active metabolites is associated with hypogonadism, reduced fertility, polycystic ovary syndrome, endometriosis, and leiomyomas.
The impact of vitamin D on reproduction is demonstrable in genetically engineered mice. Vitamin D receptor (Vdr) and 1α-hydroxylase (Cyp27b1) knockout female mice display reduced fecundity, impaired folliculogenesis, and anovulation. These reproductive abnormalities are evidently due to dysfunctional calcium homeostasis since high calcium diets partly restore fertility and increase the rate of conception in Vdr null mice.
Vitamin D 3 (cholecalciferol), the endogenous member of the vitamin D family in humans, is produced in the innermost layers of the skin (stratum basale and stratum spinosum) through the action of ultraviolet light on 7-dehydrocholesterol ( Fig. 4.13 ), resulting in the opening of the steroid nucleus B ring. Vitamin D 2 (ergocalciferol) is a plant vitamin D family member, which is metabolized similarly to vitamin D 3 . Vitamin D 3 travels to the liver where it is converted into 25-hydroxyvitamin D, mainly by an endoplasmic reticulum P450, CYP2R1, a highly conserved and substrate-specific 25-hydroxylase. There are other hepatic enzymes that can catalyze 25-hydroxylation, but they have lower affinities for vitamin D molecules. The 25-hydroxylation reaction does not appear to be regulated and the formation of 25-hydroxyvitamin D molecules is determined principally by endogenous formation of vitamin D 3 or dietary intake. Rare inactivating mutations have been described in the CYP2R1 gene leading to hypocalcemia, secondary hypoparathyroidism, and rickets. 25-hydroxyvitamin D 3 in plasma is mainly bound to vitamin D binding protein (also called group specific protein). Megalin and cubilin, endocytic receptors, participate in the uptake of vitamin D binding protein and its 25-hydroxyvitamin D 3 cargo for further metabolism. The 25-hydroxyvitamin D 3 is converted into the biologically active 1α,25-dihydroxyvitamin D 3 by the action of a mitochondrial enzyme, CYP27B1, in the kidney, but also in monocytes, maternal decidua, and placental trophoblast cells. Like mutations in CYP2R1 , inactivating mutations of the CYP27B1 gene are also associated with hypocalcemia, secondary hyperparathyroidism, and the clinical and radiographic features of severe rickets. The CYP27B1 gene and the formation of 1α,25-dihydroxyvitamin D by the kidney are regulated by negative feedback by 1α,25-dihydroxyvitamin D 3 , stimulated by parathyroid hormone (PTH), whose levels reflect calcium status, and inhibited by fibroblast growth factor 23 (FGF23), which reflects phosphate homeostasis.
The cholesterol side-chain cleavage enzyme, CYP11A1, can also act on vitamin D producing 20- hydroxy compounds that have no role in calcium metabolism but have antiinflammatory and antifibrotic effects.
Inactivation of 1α,25-dihydroxyvitamin D molecules is carried out mainly by a mitochondrial 24R-hydroxylase (encoded by the CYP24A1 gene), which catalyzes both 23- and 24-hydroxylation. Mutations in the CYP24A1 gene are associated with severe neonatal hypercalcemia, and a less severe form of hypercalcemia in adults. It should be noted that some believe that 24-hydroxylated vitamin D molecules have biological actions that are distinct from those of 1α,25-dihydroxyvitamin D 3 .
Regulation of Expression of the Steroidogenic Machinery
- ◆
NR5A1 (SF-1) is a key transcription factor regulating expression of genes encoding steroidogenic proteins.
- ◆
Other key transcription factors regulating genes involved in steroidogenesis include NR5A2 (LRH-1), and GATA-4 and 6.
The regulation of expression of genes encoding proteins involved in steroidogenesis in the ovary, testes, and adrenal cortex shares a number of similarities with respect to the involvement of cis elements and transcription factors. SF-1, a nuclear receptor, also known as Ad4BP and by the nuclear receptor family designation NR5A1, is essential for development of steroidogenic glands. Most of the genes encoding key proteins involved in steroidogenesis (e.g., SCARB1, STARD1, CYP11A1, CYP11B2, CYP17A1, CYP19A1, CYP21A2 ) contain one or more NR5A1 response elements in their proximal promoters. These elements are important for basal as well as stimulated expression of these genes, generally by a cAMP-mediated signal transduction pathway.
The importance of cAMP signaling in steroidogenic tissues is reflected in increased steroid production when specific phosphodiesterases are blocked, which elevates intracellular cAMP levels, or when mutations occur in the gene encoding the regulatory subunit of protein kinase A (PRKAR1A) , resulting in unrestrained protein kinase A activity. There appear to be several mechanisms by which NR5A1 action is modified by cAMP, including phosphorylation of the protein by kinases, providing a link between this transcription factor and molecules that transduce signals from plasma membrane receptors.
The importance of NR5A1 to the regulation of steroidogenic tissues was documented by gene targeting. Mice deficient in NR5A1 lacked adrenal glands and gonads, and males were consequently sex-reversed. Haploinsufficiency of Nr5α1 in the mouse resulted in an impaired adrenal steroidogenic response to stress, although basal steroidogenesis was not affected due to compensatory hypertrophy. NR5A1 haploinsufficiency has been reported in humans with primary adrenal failure and 46,XY DSD. It is now evident that variation in the NR5A1 gene is associated with a range of phenotypes such as 46,XY DSD, hypospadias, anorchia, male factor infertility, and primary ovarian insufficiency. Among the NR5A1 mutations reported are missense mutations within the DNA-binding region, a nonsense mutation, and a frameshift mutation predicted to disrupt RNA stability or protein function. Functional studies of the missense mutants (Cys33Ser, Arg84His) and of one nonsense mutants (Tyr138Stop) showed impaired activation of NR5A1–responsive target genes.
In addition to NR5A1, other transcription factors participate in the control of genes encoding the steroidogenic machinery. A related transcription factor, liver receptor homologue-1 (SF-2 or NR5A2) recognizes the same canonical DNA motif to which NR5A1 binds and may share functions with NR5A1 in certain tissues, including the adrenal cortex, testis, and ovary. Both NR5A1 and NR5A2 have been crystallized and found to contain phospholipid-binding pockets, with phosphatidyl inositols being the presumed ligands. These observations suggest that phospholipids may be regulatory molecules controlling expression of genes involved in steroidogenesis.
The tissue-specific regulation of genes expressed in multiple steroidogenic glands (e.g., CYP17A1 ) requires the action of other transcription factors working either independently or in concert with NR5A1 in a combinatorial fashion. In addition, the activity of NR5A1 is regulated by transcription factors that either bind to NR5A1 response elements and prevent activation of transcription (chicken ovalbumin upstream promoter-transcription factor [COUP-TF]) or bind to NR5A1 and block its ability to transactivate promoters (DAX-1, also known as NR0B1). Interestingly, expression of the latter gene is upregulated by NR5A1, so there is a complex control mechanism in place for modulating these antagonistic molecules.
Other transcription factors that are known to be important for the expression of genes involved in steroidogenesis include GATA4 and GATA6, which are members of the GATA family of transcription factors originally identified as being central to hematopoiesis and endoderm development, and LXRalpha.
The human placenta is an outlier in many respects in terms of the regulation of steroidogenesis. First, the placenta does not express certain genes that are pivotal to gonadal and adrenal steroid hormone synthesis, including NR5A1, STARD1, HSD3B2, and CYP17A1. Moreover, HSD3B1 replaces HSD3B2 in the placenta, and a START domain protein, STARD3 (which is not subject to acute regulation), may subserve STARD1’s role in cholesterol movement to the placental side-chain cleavage system.
Unlike the gonads and adrenal cortex, the placenta’s capacity to produce progestins is believed to be largely determined by levels of adrenodoxin reductase (which governs the availability of reducing equivalents) and P450scc. In addition, CYP19A1 transcription in the placenta is driven by a different promoter than that used by the gonads. Thus placental steroidogenesis is controlled in a unique way; the mechanism is more tonic, with steroidogenic capacity determined primarily by differentiation of trophoblast cells and growth of the placenta as opposed to tight regulation by trophic hormones.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


