Abstract
The gonads (ovary, testis) produce gametes (oocytes and spermatozoa). The pituitary glycoprotein hormones luteinizing hormone (LH, lutropin) and follicle-stimulating hormone (FSH, follitropin) are gonadotropins because their targets are the gonads. Another hormone produced by the syncytiotrophoblast of the human placenta is human chorionic gonadotropin (hCG, choriogonadotropin), and its activity is like LH. This review describes the protein structure, gene structure, and regulation of biosynthesis of both the gonadotropins and their membrane bound receptors. The functions of both hormones and receptors are covered extensively, as well as naturally occurring mutations that have been identified in disease states and therapies derived from their study.
Keywords
Gonadotropin, gametogenesis, reproduction, structure and function, mutations, G protein-coupled receptors, gonadotropin receptors, glycoprotein hormones, protein engineering, signal transduction.
Derived from the Greek term gonos , for “that which generates,” the gonads are the female and male organs that produce eggs and sperm, respectively. Pituitary glycoprotein hormones, which bind to target receptors on granulosa and thecal cells in the ovary or Sertoli and Leydig cells in the testis, have therefore been named gonadotropin hormones. The International Union of Pure and Applied Chemistry nomenclature for human pituitary gonadotropin hormones are follitropin for follicle-stimulating hormone (FSH), lutropin for luteinizing hormone (LH), and for the placental chorionic hormone, choriogonadotropin (CG). Here we will abbreviate them as FSH, LH, and CG. As implied by their names, the role of the gonadotropins is to activate gonadal cells to produce oocytes and sperm necessary for procreation. In addition to their role in stimulation of proliferation and differentiation of gonadal cells that support gametogenesis, they are essential for the production of steroid hormones by their target cells in gonads. Some of these steroids are essential for producing high-quality gametes and secondary sexual characteristics associated with sexual maturity, while still others are essential for the receptivity of the uterus for implantation and maintenance of pregnancy.
A unique feature of gonadotropins regardless of origin is that, at the level of primary sequence, they share one identical or common subunit encoded by a unique gene, and each has a second unique subunit that combines with the identical/common subunit to form active trophic hormones. The common subunit has been named the α-subunit, and the unique subunit has been named the β-subunit. They are therefore heterodimeric, and incidentally, the three gonadotropins share this common α-subunit with another pituitary hormone, thyroid stimulating hormone, or thyrotropin (TSH).
The three gonadotropins act via two G-protein-coupled receptors (GPCRs). The LH receptor recognizes both LH and hCG; thus it is referred to as luteinizing hormone/choriogonadotropin receptor [LHCGR] in primates, since CG is only expressed in primates (and equids, which express equine CG), and the FSH receptor (FSHR), which is specific for FSH. Both receptors are expressed in the male and female gonads; the LHCGR is expressed in Leydig, theca, granulosa, and luteal cells, whereas the FSHR is expressed in granulosa and Sertoli cells. Although the presence of the gonadotropin receptors in extragonadal tissues has been reported, their physiological significance is still under debate.
Gonadotropin Proteins (LH, hCG, and FSH)
Protein Structural Attributes
The three gonadotropins and TSH comprise the better characterized members of a family of complex proteins known as the glycoprotein hormones (GPHs). They are noncovalently bound heterodimers composed of a common α-subunit and distinct β-subunits. The common α gonadotropin subunit (α-subunit), encoded by the CGA gene, contains 92 amino acid residues, and LHβ, FSHβ, and hCGβ subunits are, respectively, 121, 110, and 145 amino acid residues in length. The additional length of the hCGβ subunit is due to a carboxy-terminal extension arising from a frameshift mutation in an ancestral LH β-subunit gene, resulting in a read-through into an untranslated region of LHβ subunit and an extension of the open reading frame. This extension is known as the carboxy-terminal peptide (CTP). The amino acid sequences of the human (h) subunits are shown in Fig. 2.1 , and it can be seen that the α- and β-subunits are relatively rich in Cys residues and that considerable homology exists in the β-subunits.

Crystal structures have been determined for partially active deglycosylated hCG, glycosylated, antibody-bound hCG, a partially deglycosylated fully active hFSH, and a partially deglycosylated complex of a single chain hFSH bound to a large N-terminal fragment (residues 1 to 268) of the hFSHR ectodomain (ECD) and FSH-FSHR complex containing the entire ECD including the hinge region that is required for signal specificity. Fig. 2.2 shows the crystal structures of hCG and hFSH.

The conformations of hCG and hFSH are quite similar, each being highly elongated molecules with the two subunits intertwined one with another in a slightly twisted manner. Despite the absence of any striking sequence homology, the two subunits in both heterodimers have similar folds, characterized by three major loops, and each subunit contains a cystine knot motif, consisting of three disulfides located in the core of each subunit. The α- and β-subunits contain, in addition to the three disulfides in the cystine knot, two and three disulfides, respectively. A 20-amino acid residue region of the β-subunit, denoted as the seatbelt , wraps around a portion of the α-subunit like a molecular seatbelt held in place with disulfide bonds. A major difference in the structures of hCG and hFSH is in the C-terminal portions of the seatbelts, which exhibit distinct conformations. In both hCG and hFSH, the two subunits are associated in a head-to-tail arrangement (see Fig. 2.2 ). Although there are no structures available for LH and TSH, the similarities of the hCG and FSH structures engender confidence that the overall conformations of LH and TSH will be closely related to these known structures. Solution structures have also been obtained for deglycosylated human α-subunit using nuclear magnetic resonance spectroscopy. The overall ensemble of structures determined for α-subunit is similar to that obtained in the crystal structures of hCG and hFSH.
With crystal structures available for FSH and an FSH-FSHR ECD complex (discussed elsewhere in this chapter), it is possible to delineate the conformational changes of the free heterodimer that occurred following binding to receptor. The unbound form of the hormone is more flexible than that of the bound form. It is the C-terminal region of the α-subunit, however, that undergoes the greatest change in conformation. In addition, the two C-terminal residues in the α-subunit are unordered in the crystal structure of FSH, but in the complex with receptor they are fully ordered.
Glycosylation
The gonadotropins are trafficked from the endoplasmic reticulum (ER) to the cis-Golgi and undergo glycosylation as they traverse the Golgi reaching the trans-Golgi, to yield the mature hormones. The human subunit primary sequences contain N-linked glycosylation sites (consensus sequence Asn-X-Ser/Thr, where X is any amino acid except proline): two on the α-subunit at Asn52 and Asn78, two on the hCG β-subunit (Asn13 and Asn30), two on FSHβ (Asn7 and Asn24), and one on the LH β-subunit at Asn30. In addition, hCG β-subunit contains four mucin-type O-linked glycans at serine 121, 127, 132, and 138, located on the CTP ( Fig. 2.3 ), resulting in a longer half-life of hCG compared with LH. The carbohydrate moieties appear to be important in subunit assembly and stabilization, secretion and circulatory half-life. Although earlier studies suggested a role of the N-linked glycan at Asn52 on α-subunit in receptor activation, more recent evidence indicates that the glycan acts more as a conformational or stabilizing determinant of the protein. Moreover, there is growing evidence that the particular type of glycosylation may influence biological activity. Of note is that the oligosaccharides on the α-subunit differ in a hormone-specific manner, apparently influenced by its cognate partner since characterization of the oligosaccharides, released from the α-subunit, can identify the β-subunit with which it was associated.

The biantennary N-linked glycans on hFSH and hCG terminate in sialic acid (and to a lesser extent sulfate), and the number of such moieties varies from 0 to 2, accounting in large part for the microheterogeneity of these GPHs. In LH the biantennary N-linked structures tend to terminate mainly in sulfates, resulting in a decrease in its circulatory half-life compared with the sialic acid-containing hormones. This arises from a hepatic receptor that recognizes the terminal N-acetyl galactosamine-sulfate, rapidly removing it from circulation. Indeed, ablation of the gene in mice that encodes GalNAc-4-sulfotransferase, the enzyme responsible for modifying the terminal GalNAc in LH with sulfate, resulted in increased half-lives and circulating levels of LH.
These are but generalizations since, for example, hFSH also contains triantennary and tetraantennary N-linked glycans, and some hFSH β-subunits lack N-linked structures completely. A variety of glycosyltransferases are responsible for N- and O-glycan biosynthesis—notably, sulfation in the pituitary requires N-acetylgalactosamine transferase and sulfotransferase, both of which are missing in the placenta. Also, fucose is often found in the GPHs. As summarized, nearly 50 different N-linked and O-linked glycans have been reported in hCG α and β preparations. The major gonadotropin N-linked and C-linked glycans are shown in Fig. 2.3 .
Folding and Assembly
Investigations into the kinetic folding pathways of the hCG subunits led to the interesting suggestion that disulfide exchange occurs during the maturation process and that subunit association occurred before completion of protein folding and disulfide formation. Moreover, it was posited that subunit association occurred before the seatbelt was latched by closure of Cys26 and Cys110 (wraparound model). In contrast to these reports, a different mechanism in which subunit assembly involved closure of the seatbelt latch, followed by threading of α loop 2, has been proposed (threading model). Others have also studied the folding patterns of hCG and reported that subunit association occurred between an almost completely folded α-subunit and an immature β-subunit. A recent study suggests that the LH β-subunit is not completely folded prior to assembly with the α-subunit, and that the α-subunit serves as a chaperone to facilitate formation of the cysteine knot and the seatbelt latch.
In addition to the heterodimeric nature of the hormones, homodimers have also been found for LH β-subunit, and for the α-subunit. Whether these homodimeric forms of the GPHs have any associated bioactivity remains to be shown, although it has been reported that the free α-subunit potentiates progesterone-mediated decidualization.
Homologous Proteins
A search of the human genome revealed two glycoproteins GPA2 and GPB5 similar to the GPH α and β subunits, respectively. Heterodimers can be formed between GPA2 and GPB5, and the complex is capable of stimulating the TSH receptor. This heterodimer, named thyrostimulin, is postulated to act in a paracrine manner to activate the pituitary thyroid-stimulating hormone receptor (TSHR). Comparative structure analysis of the GPH subunits and GPA2 and GPB5 suggests that GPA2 and GPB5 cannot form stable heterodimers at low concentration in circulation, as they lack the cystine residues potentially involved in the cystine knot structure and GPB5 does not possess a sequence homologous to the seat belt. This suggests a paracrine, rather than endocrine, role for thyrostimulin. Phylogenetic studies suggest that the thyrostimulin may in fact be an “old” hormone with the subunits appearing with the emergence of bilateral metazoa more than 500 million years ago. In the basal chordate, amphioxus, thyrostimulin appears to play a role in embryonic development, and heterodimerization may not be required for its activity. Recent studies suggest that thyrostimulin may function in other mammalian tissues. In the rat ovary, it is expressed in the oocyte where it functions as a paracrine regulator of TSHR that is present in the granulosa cells and is capable of stimulating epithelial ovarian cancer cell proliferation via transregulation of the epidermal growth factor (EGF) receptor (EGFR). Another study suggests that it may negatively regulate osteoblastic bone formation during skeletal development.
Structure-Function Studies
- ◆
Prior to structural determination, extensive structure-function relationships were established through mutagenesis, which ultimately confirmed the authenticity of the gonadotropin-receptor crystal structures.
- ◆
These mutants established the requirement of posttranslational modifications for the assembly, secretion and biological activity of the gonadotropins.
- ◆
Protein engineering efforts have been directed at producing longer acting and superagonist forms of gonadotropins.
Site-Directed Mutagenesis
As discussed elsewhere in this chapter, only a limited number of naturally occurring mutations have been identified in α- and β-subunits of the GPHs. In contrast, there is a wealth of information available from site-directed mutagenesis, followed by biological characterization of the mutant hormones. Few mutants have been described in which there was a significant increase in bioactivity; most mutations either have no effect or induce a loss of function, disrupting folding, subunit assembly, or receptor binding/activation. Gain-of-function mutations in hCG were obtained by replacing single or multiple amino acid residues at the N-terminal region of the α-subunit with Lys. A twofold increase in potency of hCG was obtained with a single replacement of Phe with Thr at position 18 of α-subunit. Mutant forms of α-subunit missing the N-linked oligosaccharide at Asn52 are capable of associating with the hCG β-subunit or FSHβ-subunit, giving a heterodimer that binds to the cognate receptor but has diminished signaling efficacy. Although still controversial, it appears that the role of N-linked glycosylation at Asn52 is to stabilize the active conformation of the heterodimer by formation of a hydrogen bond with a Tyr on the β-subunit. Mutations in the central region and at the C-terminus of the α-subunit yielded mutants that associated with the β-subunits of hCG and hFSH but displayed compromised functionality in receptor binding.
A large number of β-subunit mutations have been prepared and characterized. As with mutations in α, many interfere with folding, subunit assembly, or receptor binding, with the data, overall, being consistent with the crystal structures of hCG and FSH. Unlike α-subunit mutants, however, there have been no reports of β-subunit mutants that retain the ability to form heterodimers and bind to receptor, yet do not signal. This suggests that the α-subunit may play a predominant role in activation of gonadotropin receptors following the initial binding event.
Protein Engineering
Protein engineering has been used to produce a variety of subunit deletion derivatives, single chain hormones and chimeric hormones, yielding quite interesting results. C-terminal deletion mutants of the α-subunit result in N-terminal fragments that retain the ability to bind to the β-subunit, but the resulting heterodimers exhibit minimal, if any, receptor binding capability. Deletion mutants at the N- and C-termini of the hCG β-subunit have been reported by several groups, and the shortest form that retains minimal functionality in subunit assembly and subsequent receptor binding and activation is a fragment consisting of residues 8 to 100.
A number of GPH chimeras have been designed and characterized, providing useful information on specific amino acid residues involved in receptor binding and activation. In general, these results emphasize the role of the β-subunit seatbelt region in receptor binding, although different portions are important in receptor specificity.
A novel approach to study gonadotropin structure-function relationships involved the design of single chain hormones, derived by fusion of α- and β-subunits using an intervening peptide sequence, or by simply aligning the cDNAs for α- and β-subunits and removing the stop codon of the N-terminal protein, thereby creating a single contiguous protein composed of both subunits. The first reports of a yoked or tethered hCG demonstrated that the single chain gonadotropin, in the configuration N-hCG β-subunit-α-C, was bioactive. Subsequently, similar fusion proteins of LH and FSH were also found to be bioactive. Later, single chain proteins of hCG in the N-α-hCG β-subunit-C configuration and proteins with different linkers were expressed and characterized, in many cases with interesting mutations in one or both subunits. From these studies, it was concluded that the N-α-hCG β-subunit-C configuration was also bioactive and, quite surprisingly, that each disulfide of the subunits could be eliminated without a loss of activity. The single chain hormones also displayed increased stability and heat resistance in vitro, compared with their heterodimeric counterparts. Extending the approach of covalently linking the two subunits, disulfide-linked heterodimers were designed and expressed. These gonadotropin analogues substantiated the notion that α-Asn52 contributed to heterodimer stability and was not involved directly in signal transduction, and also led to suggestions that the C-terminus of the α-subunit is not required for LHR binding. This latter finding is consistent with the observation that the five C-terminal residues can be removed from the α-subunit without a loss of LHCGR binding. This is in contrast to the observation that mutations of hFSH αS85A, αT86A, αK91A, or α S92A in the context of heterodimeric hFSH only retained 10% or less of hFSH receptor-binding activity. The use of single chain gonadotropins, particularly in the N-α-β-C configuration, also raises interesting questions about the role of the C-terminal region of α-subunit in FSH, where the structure of the hFSH-hFSHR ECD complex shows a large movement of α-subunit in the receptor complex compared with the heterodimer. This riddle is unlikely to be solved until the crystal structure of single chain gonadotropin in complex with receptor is determined. Of great interest was the report that single chain hCG β-β homodimers bind to LHCGR, albeit with an affinity about three times lower than wild type hCG; this engineered homodimer does not elicit a biological response, and blocks hCG binding to LHCGR. A second single chain hCG antagonist was designed by mutating three of the four N-linked glycosylation sites that are associated with LHCGR activation (Asn13 and Asn30 in the β-subunit and Asn52 in the α-subunit). Asn78 in the α-subunit, associated with receptor binding, was left intact. This analogue behaved as a competitive antagonist and suppressed ovarian hyperstimulation syndrome (OHSS) in rats.
The approach of converting GPHs to single chains has been extended to produce fusion proteins with dual and triple activities. For example, a three-domain fusion protein of the form N-FSHβ-hCG β-subunit-α-C exhibited both LH and FSH activities. Interestingly, the bifunctional gonadotropin is secreted from cells as two species, one with LHCGR and the other with FSHR activities. This, of course, raises the simple explanation that heterodimers are forming between two fusion proteins. Here again, until a crystal structure can be obtained for these analogues, their mechanism of action remains unclear. A four-domain fusion protein, N-TSHβ-FSHβ-hCG β-subunit-α-C, although secreted inefficiently, was found to exhibit three distinct bioactivities both in cellular and whole animal studies. These results raise intriguing questions regarding subunit association and conformation, as manifested in receptor binding and activation. A most provocative finding was based on a bifunctional, triple domain fusion protein of the form N-FSHβ-hCG β-subunit-α-C. These investigators showed that disruption of heterodimer formation by mutation of either Cys10-Cys60 or Cys32-Cys84 did not eliminate bioactivity, and thus concluded that αβ contacts are not required for receptor binding and activation. These selected results, along with others not covered here, indicate that single chain GPHs exhibit some properties distinct from those of heterodimers. The increased stability of the single chain proteins, the single chain hCG antagonists and the dual activities, bioactive LH/hCG-like and FSH-like, make them interesting formulations as candidates for clinical utility.
The approach of single chain gonadotropins was extended to produce fusion proteins of single chain hCG with LHCGR, which when expressed, led to constitutive receptor activation in transfected cells and transgenic mice. This model was also used to demonstrate that protein fusions of the individual subunits with LHCGR were devoid of bioactivity.
Genes and Transcripts
- ◆
With the exception of the hCG β-subunit, gonadotropin subunits are encoded by one gene.
- ◆
Mutations in the gonadotropin genes discovered in symptomatic patients often affect assembly, conformation, and hence biological activity.
- ◆
Polymorphisms in the protein coding regions of the gonadotropin genes can have either detrimental or no effect, depending on whether the polymorphism results in changes in the amino acid sequence and whether the change in primary structure affects function.
- ◆
Polymorphisms in noncoding regions can affect transcription or mRNA processing which may or may not be associated with clinical conditions.
Gonadotropin Subunit Genes
The common α-subunit and the β-subunits of LH and FSH are each encoded by unique genes; in contrast, the CG β-subunit, expressed in primates, is encoded by six genes. However, in equids the CG β-subunit and LH β-subunit are products of the same gene. It has been suggested that the GPH α- and β-subunits diverged from a common ancestral gene more than 900 million years ago, with the β-subunit gene undergoing duplications and mutations to yield the current family. In humans, the gene encoding the common α-subunit is on chromosome 6, that for FSHβ is on chromosome 11, and those for LHβ and CGβ are on chromosome 19 ( http://www.ensembl.org ). The gene for human α-subunit is 9.4 kb and contains four exons and three introns; that for FSH β-subunit is 4.2 kb with three exons and two introns; for LH β-subunit the gene is 1.1 kb with three exons and two introns; and those for CG β-subunit are variable in length.
The one LH β-subunit and six CG β-subunit genes exist in a large cluster spanning about 52 kbp. The six genes encoding the β-subunit of CG (i.e., CGB, CGB1, CGB2, CGB5, CGB7, and CGB8 ) exist as tandem and inverted repeats. The genes for CG β-subunit have been analyzed in detail. Four of the CG β-subunit genes, CGB, CGB5, CGB7 , and CGB8 , exhibit 97% to 99% sequence identity, while their identity with the LHB gene is 92% to 93%. These gene similarities lead to protein amino acid sequences that are 98% to 100% identical for the four CG β-subunit gene products and 85% identical with the LH β-subunit. CGB, CGB5, CGB7, and CGB8 encode proteins with a carboxy-terminal extension, the CTP, that appears to have arisen from the LHB gene by frameshift, leading to a read-through into a previously untranslated region.
Gonadotropin Subunit Transcripts
The available evidence indicates single transcripts for the gonadotropin genes, with the exception of the human FSHB gene for which four mRNA species have been described, arising from alternate splicing and the utilization of two polyadenylation sites. The CGB family is interesting in that all six genes appear to express transcripts of varying lengths, albeit sometimes without detectable protein production. CGB5 and CGB8 are highly expressed in the placenta. Although expressed in placenta, pituitary, testis, and breast cancer, no proteins have yet been identified for the CGB1 and CGB2 genes. The predicted sizes of CGβ1 and CGβ2 proteins are smaller than that of the hCG β-subunit; this observation, coupled with the distinct amino acid sequences predicted, suggests that these proteins, if biosynthesized, may have quite different functions than those of hCG. Using transgenic mice expressing a 36 kb cosmid insert that contained the six CGB genes, transcripts of CGB1 and CGB2 genes were found to be present in the brain at levels comparable to those of the other four CGB genes. The human LHB mRNA is 700 nucleotides in length, and depending upon the species, the CGA gene encodes an mRNA for α-subunit of 730 to 800 nucleotides.
Naturally Occurring Mutations
Mutations in the gonadotropin hormone genes, although rare, help in elucidating their physiological roles and defining the important structural domains of the hormones. The only mutation reported in the CGA gene is that from a human carcinoma, Glu56Ala, resulting in a mutant form of α-subunit that does not associate with LH β-subunit. In contrast, there are several reports of mutations in the genes encoding the three gonadotropin β-subunits, resulting in loss of function and thus hypogonadism.
The first report of a mutation in the LHB gene was that of a missense mutation in a male presenting with delayed puberty and hypogonadism. This mutant led to a replacement of Gln54 with Arg; while subunit assembly could occur, the heterodimer was unable to bind to LHCGR. Other studies showed that LH β-subunit and hCG β-subunit with Gln54 replacements formed heterodimers with α-subunit, but these heterodimers exhibited reduced binding to LHCGR. Another missense mutation in the LH β-subunit was that of Gly36 to Asp, reported in a male with delayed puberty and infertility. Gly36 is part of the CAGYC sequence in the LH β-subunit that is critical to the formation of the cystine knot; presumably, an Asp at this position prevents at least one of the disulfides from forming.
The third identified mutation was a G-C substitution at the +1 position of intron 2 (a 5′ splice-donor site) that leads to a hypothetical aberrant protein with a 79-amino acid residue insert beginning after Met41 and a frameshift in exon 3, thus removing the essential seat belt loop of β and important cysteines. The offspring of consanguineous parents who were heterozygous for the mutation were studied. Three homozygous siblings presented with hypogonadism and infertility, undetectable levels of LH, and high levels of free α-subunit, while their heterozygous siblings were fertile. The two males had elevated FSH and low testosterone, while the female had FSH, estradiol, and progesterone values in the normal range and underwent normal pubertal development and menarche at age 13 years.
A 9-bp deletion in exon 2 resulting in deletion of amino acid residues 10 to 12 of LHβ was reported in a man and his sister. Both were homozygous for the deletion, while two additional unaffected siblings were heterozygous. In spite of undetectable levels of LH, low serum, and intratesticular testosterone concentrations, the man had complete spermatogenesis and a normal sperm count. Presumably, the low activity of the mutant LH detected in vitro was sufficient for normal spermatogenesis. The sister underwent normal puberty and menarche, but subsequently had amenorrhea, infertility, ovarian cysts, and low estradiol levels.
A recent report described a compound heterozygous mutation in a 31-year-old male with delayed puberty, azoospermia, and hypogonadism due to lack of LH. The first mutation was identified as a 12-bp deletion in exon 2 of the LHB gene, causing a deletion of four leucine residues in the signal peptide, and the second mutation was a G to T mutation at the 5′ splice site of intron 2, resulting in aberrant RNA splicing. The patient’s 16-year-old sister harboring the same mutations had normal pubertal development but developed oligomenorrhea. A homozygous 3-bp deletion in the LHB gene resulting in the deletion of Lys40 in LHβ was identified in two brothers with LH deficiency and hypogonadism. The mutated LH β-subunit was able to heterodimerize with α-subunit, but was not secreted.
Rare missense heterozygous mutations in the hCGβ subunit have been identified in a Northern European population. A Val56Leu missense variant in the CGB5 gene was identified in a patient with recurrent miscarriage (RM), which impaired subunit assembly but elicited a strong signaling response. A Pro73Arg variant found in five individuals (three RM and two controls) resulted in altered conformation, but did not affect biological activity. Individuals homozygous for this variant have not been identified, perhaps because such genotypes would result in pregnancy failure.
To date, 10 patients with mutations in exon 3 of FSHB have been identified, and they present with an absence of pubertal development, amenorrhea, and infertility in females and delayed or normal puberty with azoospermia and infertility in males. The first reported mutation was a homozygous 2-bp deletion at codon 61 (Val61X) causing a frameshift and premature termination of the β subunit in a 27-year-old woman, and later also identified in an 18-year-old male with delayed puberty. These observations were followed by a report of a compound heterozygous mutation, with one being the Val61X mutation and the other a missense mutation resulting in a Cys51Gly replacement. Other identified mutations include a nonsense mutation (Tyr76X), a frameshift mutation at codon 79 (Ala79X) caused by a 1-bp deletion resulting in premature termination, and two missense mutations resulting in Cys82Arg and Cys122Arg replacements. These mutations result in loss of bioactivity due to production of a truncated protein or due to aberrant tertiary structure as a result of mutations in the cysteine residues involved in the cystine knot structure and inability to associate with the α-subunit. One case of hypoglycosylation, likely caused by altered conformation, was reported for FSH, resulting in a hormone with diminished activity.
Overall, the observed phenotypes associated with the naturally occurring mutations in LHB, FSHB , and CGB are consistent with the known structure activity relationships of the gonadotropins, although fertility in men does not always appear as sensitive to some FSH mutations.
Polymorphisms
A fairly well characterized variant in the LHB gene (V-LHβ) appears in variable frequencies in ethnic groups throughout the world and results from two single nucleotide polymorphisms (SNPs) that are found together on one allele. The replacement of Trp8 with Arg caused altered immunoreactivity of the hormone, and substitution of Ile15 with Thr introduces an extra glycosylation site in the LHβ subunit. V-LH demonstrates increased biopotency in vitro with altered half-life in the circulation. The association of the V-LH with various clinical conditions has been assessed. A number of studies have addressed the association between V-LH and various clinical conditions such as infertility, polycystic ovarian syndrome, and menstrual disorders. No clear association was found with PCOS. Studies have found an association with female but not male infertility.
Another LH β-subunit variant is a replacement of Gly102 with Ser, resulting in reduced LH biopotency in vitro, and has been associated with reproductive disorders in some populations. The frequency of this polymorphism was recently reported to be higher in a population of Chinese Han women with PCOS, and PCOS carriers had lower LH and higher fasting glucose levels, but not in a population of Korean women.
An unusual polymorphic variant of LH β-subunit involves an Ala to Thr replacement of three residues before the signal peptide cleavage site. Using in vitro assays, it was found, rather surprisingly, that the mature protein from the variant appears less potent than “wild type” (WT) LH in cAMP production, but more potent in inositol phosphate production. The SNP-related alteration may interfere with proper processing of the β-subunit, although studies have not addressed this possibility.
A polymorphism has been reported in exon 3 of CGB5 , resulting in a Val79 replacement with Met. This SNP results in a β-subunit deficient in folding and interaction with the α-subunit. The frequency and physiological consequences of this variant are unknown; one sampling of just under 600 samples from four European groups failed to detect a single case. Other variants have been detected, but these were silent or located in intron regions. In a case study in which the CGB5 and CGB8 genes were analyzed in RM and control fertile patients from Estonia and Finland, 71 polymorphisms were identified, of which 48 were novel. A protective effect against RM was associated with two SNPs located at identical positions in CGB5 and CGB8 , and with four CGB5 promoter variants. A follow-up study that included a Danish cohort with RM in addition to the Estonian and Finnish subjects confirmed that two SNPs in the CGB5 promoter region seemed to offer protection against RM, but variants in the CGB8 promoter region had no effect. These polymorphisms can be found in the dbSNP database ( http://www-ncbi-nlm-nih-gov.easyaccess1.lib.cuhk.edu.hk/SNP/ ).
A limited number of silent variants of FSHB have been reported. Recently, an FSHB promoter polymorphism (–211G/T) identified in a cohort of European men has been associated with reduced serum FSH levels in GT heterozygotes and TT homozygotes. This polymorphism was found to be more prevalent among infertile men. The low serum FSH levels associated with this SNP have been demonstrated to be due to reduced binding of the LHX3 transcription factor and reduced FSHB transcription. The LHB and FSHB polymorphisms can be found in the SNP database ( http://www-ncbi-nlm-nih-gov.easyaccess1.lib.cuhk.edu.hk/projects/SNP/snp_ref.cgi?geneId=3972 and http://www-ncbi-nlm-nih-gov.easyaccess1.lib.cuhk.edu.hk/projects/SNP/snp_ref.cgi?geneId=2488 ).
Expression and Secretion of the Gonadotropins
- ◆
Production of biologically active gonadotropin is regulated in the hypothalamic-pituitary axis at the level of transcription, glycosylation, and secretion.
- ◆
Trophic factor initiated intracellular signaling pathways regulate transcription of the gonadotropins.
- ◆
Glycosylation and secretion, which ultimately determine production of cognate protein, are dependent on and guided by appropriate nascent protein primary structure.
- ◆
Variation in normal or presence of aberrant proteins provide for the design of diagnostic markers or useful therapeutics.
Transcriptional Regulation
The neuroendocrine reproductive axis, composed of the hypothalamus, anterior pituitary, and gonads, is now known to be regulated, apparently in large part, by kisspeptin, a product of KISS1 , acting via the GPCR, GPR54, located on GnRH neurons. The three gonadotropin subunit genes in the pituitary are differentially responsive to GnRH pulse frequency and magnitude: LHB is preferentially transcribed at high GnRH pulse frequencies and FSHB at lower frequencies. Although CGA is preferentially transcribed at high pulse frequencies, its regulation is less important, because α-subunit is produced in excess of LHβ and FSHβ at both high and low GnRH pulse frequencies. Sex steroid-mediated regulation occurs mostly at the hypothalamic level, although there are also some direct actions at the pituitary, and recent evidence suggests a critical role of the kisspeptin-GPR54 system in sex steroid action.
Considerable research has been devoted to understanding the mechanism by which GnRH regulates the synthesis and secretion of LH and FSH. Binding of GnRH to its receptor in the gonadotropes primarily activates Gα q/11 , resulting in the activation of phospholipase Cβ, calcium influx, activation of protein kinase C (PKC), and calcium-calmodulin kinase II. PKC mediates the downstream activation of the mitogen activated protein kinase (MAPK) cascades: extracellular signal regulated kinase (ERK1/2), jun N-terminal kinase (JNK), and P38. The MAPK pathway can also be activated by GnRH stimulation of Gα s The signaling mechanisms that differentially regulate LHB and FSHB transcription in response to differential pulse frequencies of GnRH are not well understood. Current evidence suggests that GnRHR couples to different G proteins to differentially activate distinct signaling pathways in response to varying GnRH pulse frequencies.
Our understanding of the mechanisms that mediate transcription of the pituitary gonadotropin genes has primarily come from studies on the rodent genes in two murine immortalized gonadotrope cell lines, αT3-1 and LβT2, and more recently from in vivo studies in mouse models. Several excellent recent reviews on the GnRH and steroid mediated transcriptional regulation of these genes are available. Briefly, GnRH activation of MAPK signaling cascades increases transcription of the immediate early genes, including Egr1 , Jun , and Atf3 , which encode the DNA proteins early response protein 1 (EGR1), JUN, and activating transcription factor 3 (ATF3), respectively. EGR1, SF-1, and PITX1 form a tripartite complex that binds to a highly conserved proximal promoter sequence to synergistically activate the LHB/Lhb gene. Activator protein 1 (AP1), NR5A1, nuclear factor Y, PITX1, and LHX3 are some of the factors implicated in Fshb expression. β-catenin regulates both Lhb and Fshb in response to GnRH, while the forkhead box transcription factor, FOXO1, inhibits Lhb and Fshb transcription in LβT2 gonadotrope cells. Interestingly, a recent study demonstrated that β-catenin is not essential for gonadotropin synthesis in vivo.
Activin, a member of the transforming growth factor β (TGFβ) superfamily, is an important regulator of Fshb gene expression, and the transcription factor FOXL2 is required for activin responsiveness. Activin signaling is mediated by SMAD proteins. Cell culture and in vivo studies using knock-out mice have demonstrated that SMAD 4 and FOXL2 function synergistically to regulate Fshb transcription.
It has been proposed that in the trophoblast layer of the placenta, association of the cAMP response binding protein (CREB) and ETS2, augmented by protein kinase A, regulates CGA expression. An upstream regulatory element on CGA contains binding sites for several transcription factors, and a second control element, α-ACT, binds a GATA factor and AP2γ. Expression of hCGβ is regulated by transcription factors such as AP2, SP1, and SP3.
Posttranslational Regulation (Glycosylation)
As discussed earlier, the carbohydrate structure among the gonadotropins is highly variable and may be the result of microheterogeneity, due to structural heterogeneity of carbohydrates at the same site, or macroheterogeneity due to absence of one or more glycan chains at known glycosylation sites. It is now well established that carbohydrate microheterogeneity of GPHs can vary with physiological states. Examples include a shift in the structures of hCG N-linked oligosaccharides during pregnancy in the differentiation of cytotrophoblasts to syncytiotrophoblasts, changes in LH and FSH N-linked glycosylation during the menstrual cycle and with increasing age, and alterations in FSH N-linked glycans during adolescence in boys. Oligosaccharide complexity of recombinant hFSH has been shown to differentially affect steroidogenesis and gene expression in human granulosa cells.
Various laboratories have shown that a hyperglycosylated variant of hCG is produced in early pregnancy (i.e., from cytotrophoblasts), and in gestational trophoblastic diseases, where hCG is secreted by syncytiotrophoblast. Hyperglycosylated hCG molecules have enhanced branching into triantennary and unusual biantennary N-linked oligosaccharides and a prevalence of the more complex Core 2, as opposed to Core 1, O-linked sugars. In clinical studies hyperglycosylated hCG is identified by the binding of a monoclonal antibody B152 that specifically recognizes the Core 2 O-linked sugars at Ser 132. It has been suggested that in addition to the canonical physiological function of “rescuing” the corpus luteum, hyperglycosylated hCG acts in an autocrine/paracrine manner to promote trophoblast invasion. There are also data suggesting that the hCG produced by cytotrophoblasts and choriocarcinoma have distinct carbohydrate moieties. The N-linked oligosaccharides on hCG from invasive mole and testicular cancer are characterized by both biantennary and triantennary structures, and often more heavily fucosylated glycans, while the four O-linked units tend to have more Core-2 type structures.
Samples from patients with choriocarcinoma, testicular cancer, and invasive moles showed interesting differences in their glycan complexity. Triantennary N-linked glycans increase in choriocarcinoma at Asn30 but not at Asn13, while monoantennary N-linked glycans were observed at both Asn13 and Asn30. The status of hCG fucosylation in pregnancy and cancer patients has been investigated by several groups, with some conflicting results. In malignancies, fucosylation was reported to increase at Asn13, but not at Asn30.
More recently, four naturally occurring human pituitary FSH glycosylation variants have been identified based on the loss of one or more N-glycans in FSHβ. , These glycoforms are named for their predicted molecular mass of the FSHβ subunit in western blot analysis. A depiction of what these forms may look like is represented in Fig. 2.4 . The fully glycosylated FSH possessing N-glycans at both Asn7 and Asn24 migrates as a 24-kDa band and is designated as hFSH24, while absence of both glycans produces a 15-kDa band (FSH15). Single hypoglycosylated variants are a mixture of hFSH18 and hFSH21 and represent loss of glycosylation at Asn7 and Asn24, respectively. Three variants, hFSH18, hFSH21, and hFSH24, are secreted, and most pituitary and urinary hFSH preparations consist of a mixture of the more abundant hFSH24 and hFSH21 in an 80 : 20 ratio. Deglycosylated hFSH15 is not secreted and in vivo studies utilizing Fshb null mice have shown the N-linked glycans on the β-subunit are required for efficient assembly with the α-subunit in the pituitary and subsequent secretion.

A progressive decrease of hFSH21 has been detected in women between the ages of 24 and 55, resulting in a decrease in the hFSH21 to hFSH24 ratio. This decrease in hFSH21 suggests a loss in biological activity of circulating hFSH, as hypoglycosylated hFSH containing a mixture of hFSH18 and hFSH21 was more active in vitro in receptor binding assays and in stimulating the cAMP pathway and steroidogenesis in human granulosa cells compared with hFSH24. A recent study suggests that hFSH21/18 and hFSH24 may activate distinct biological pathways. It has been proposed that the loss of the hypoglycosylated FSH may contribute to the loss of ovarian function associated with aging.
Regulation of Secretion
Pathways of secretion and polarity of hormone release are different for the various gonadotropins. Although LH and FSH are synthesized by the same cell, LH is packaged in dense storage granules, with regulated secretion occurring from the basolateral surface under the control of the pulsatile secretogogue, GnRH. In contrast, FSH secretion is constitutive, linked to its synthesis, and exhibits no apparent polarity of secretion. The sorting determinant for the regulated secretion of LH lies in the hydrophobic heptapeptide at the carboxy terminus of LHβ, and a single leucine residue at position 118 contributes to the sorting of LH via the regulated pathway. The heptapeptide directs the LHβ subunit to a perinuclear sub-domain of the ER, which suggests that entrance into the regulated secretory pathway is a pre-Golgi event. In vivo studies have demonstrated that FSH containing the heptapeptide is released via the regulated pathway similar to LH and can enhance ovulation efficiency and prolong ovarian follicle survival. In addition, sulfation of LH does not play a role in its regulated secretion but may be important for its extracellular bioactivity.
Although LHβ and CGβ share 85% sequence identity and are functionally interchangeable, hCG is not stored in granules, but rather is secreted constitutively into the maternal circulation at the apical side of trophoblasts. The CTP sequence, unique to hCG, is the important determinant in the constitutive secretion, and the O-linked oligosaccharides in the CTP are essential for the apical release of hCG.
Physiological and Pathophysiological Conditions
The accepted physiological action of hCG is in the initiation and maintenance of pregnancy. In the gonads, it maintains functionality of the corpus luteum and progesterone production, particularly during the first trimester of pregnancy. It also mediates multiple placental, uterine, and fetal functions including trophoblast invasion, development of syncytiotrophoblast cells, angiogenesis in the uterine endometrium, uterine growth and differentiation, placental development, and localized suppression of the immune system. In addition, hCG is located in the pituitary, but its physiological significance there is unknown. As with pituitary hCG, there are reports of small amounts of GPH synthesis in various nonpituitary and nonplacental tissues, but specific functions have not been ascribed to these ectopically produced hormones. The major forms of circulating LH and hCG have been delineated, along with their patterns in normal physiological conditions and various disorders.
There is ample evidence supporting ectopic production in a variety of disorders. It is well known that hCG is expressed in malignant forms of gestational trophoblastic disease (e.g., invasive mole and choriocarcinoma). In men and women, hCGβ, hyperglycosylated hCGβ, and only occasionally intact hCG, is expressed in a variety of other malignancies, including breast, bladder, colorectal, gynecological, head and neck, hematological, lung, neuroendocrine, oral/facial, pancreatic, prostate, and testicular cancer. The detection of free CGβ subunit in these malignancies is generally associated with poor prognosis.
In an analysis of human GCA , LHB , and CGB gene expression in breast cancer, studies showed that most normal tissues expressed only CGB7 , while CGB3 , CGB5 , and CGB8 were expressed in trophoblastic tissues and correlated with the malignant transformation of breast cancer and other nontrophoblastic malignancies. Human CG A, LHB, CGB1, CGB2, and CGB7 were not, however, upregulated in breast cancer.
A possible role of LH in the etiology and progression of Alzheimer disease has been postulated. Reduction in serum testosterone and increase in LH are associated with decline in cognitive function and both LH and LHCGR are reported to be expressed in the brain. Of interest is the observation that LH and hCG modulates the processing of the amyloid-β precursor protein, yielding deposition of amyloid-β peptide. Genetic ablation of Lhcgr in amyloid precursor protein transgenic mice improved the amyloid pathology, suggesting that chronic elevation of LH may promote amyloid-β plaque formation.
Diagnostic and Therapeutic Applications of the Gonadotropins
Immunoassay-based measurements of the serum concentrations of pituitary-derived gonadotropins have been the mainstay of monitoring functionality of the hypothalamic-pituitary-gonadal axis. Detection of hCG is the gold standard for pregnancy diagnosis, as well as monitoring trophoblastic malignancies. Specialized biomarkers include hyperglycosylated hCG for the early detection of pregnancy or its complications, and as a component of serum screening to detect Down syndrome, coupled with other markers. hCGβ and hyperglycosylated hCGβ are used as tumor markers for the detection of malignancies. Immunocytochemistry is also commonly used in evaluating the expression of hCG β-subunit in suspected tumor tissue sections. It has been recognized for decades that multiple variants of hCG are present in normal pregnancy, giving rise to micro- and macroheterogeneity. These include intact or heterodimeric hormone, nicked hCG, heterodimeric hormone with bond cleavages in the hCG β-subunit 43 to 48 region, free α- and β-subunits, nicked hCG β-subunit core fragment (i.e., free β-subunit with bond cleavages in the 43 to 48 region), and hCG β-subunit core fragment, consisting of two disulfide-linked fragments, 6 to 40 connected to 55 to 92. Hyperglycosylated hCG can present with a similar number of derivatives. Some of these variants may be useful diagnostic tools for germ cell and gynecological malignancies. Therefore hCG assays must be capable of distinguishing the variants. Several reviews, workshop proceedings, and reports have addressed this issue and the challenges of obtaining and using appropriate standards. The preparation and adoption of universal standards, coupled with complete characterization and disclosure of antibody specificities, will greatly facilitate standardization of GPH immunoassays.
While immunoreactivity is the primary technique for determining hormone concentrations in body fluids, it is often necessary to measure bioactivity. The earlier cumbersome in vivo assays for the GPHs have mostly been replaced with radioreceptor and signaling assays in transfected cells. Such measurements provide quantitative data on hormone-receptor binding and efficacy of signal transduction, but they give no information on circulatory half-life and thus in vivo potency. For this, animal and human studies are obviously required.
The dawn of molecular biology allowed for the preparation of recombinant gonadotropins expressed in Chinese hamster ovary (CHO) cells that are used in the treatment of infertility. The longer circulatory half-life of hCG, attributed to the β-subunit CTP, has been utilized most effectively in producing long-acting analogues of FSH and TSH, with a CTP engineered to the β-subunit C-terminus. Another approach that has proven successful in extending the half-life of FSH is an engineered extension at the α-subunit N-terminus with two sites of N-glycosylation. The resulting analogue was glycosylated as judged by mobility on SDS-PAGE and exhibited increased circulatory half-life and in vivo potency.
Recently, a number of reports have appeared, suggesting that hCG, in particular, could be used therapeutically in the treatment of cancer. A recent phase I clinical trial has shown that administration of hCG to postmenopausal breast cancer patients led to a reduction in the proliferative index (Ki67) and the levels of estrogen and progesterone receptors. In contrast, female transgenic mice overexpressing hCG or LH exhibit multiple sites of tumorigenesis. A conjugate of the lytic peptide, Hecate (a 23-amino acid residue peptide similar to bee venom melittin that disrupts cell membranes), to a 15-amino acid residue peptide from hCG β-subunit (residues 81 to 95) was found to kill cultured breast, prostate, and ovarian cancer cells and reduce the tumor xenografts in nude mice. In a transgenic mouse model, it was found that the hecate-hCG β-subunit 15-amino acid residue peptide reduced malignant Leydig and granulosa cell tumors via necrosis or necrosis-like cell death. While it is surprising that such a short hCG β-subunit peptide is capable of LHCGR binding, the results are quite dramatic and may lead to specific therapies of LHCGR-positive tumors.
Gonadotropin Receptors
- ◆
Gonadotropin receptors belong to the Rhodopsin family of G-protein coupled Receptors (GPCR).
- ◆
They are composed minimally of a single polypeptide with an extracellular N-terminus domain, an intracellular C-terminus domain, and a seven-membered alpha helical transmembrane domain.
- ◆
Upon hormone activation the receptor undergoes a conformational change that dissociates G-proteins complexes into substituents, which activate downstream effectors.
Gonadotropin Receptor Proteins
Protein Structural Attributes
The gonadotropin receptors and the TSHR belong to the highly conserved subfamily of GPCRs, the so-called Rhodopsin family, and more specifically to the δ-group of this large class of GPCRs, according to the phylogenetic classification proposed by Fredriksson. GPCRs are membrane receptors that vary considerably in molecular size but share a common molecular topology consisting of a single polypeptide chain of variable length that traverses the lipid bilayer, forming seven characteristic transmembrane hydrophobic α-helices (transmembrane domains [TMDs]) connected by alternating extracellular and intracellular sequences or loops (EL and IL, respectively), with an extracellular NH 2 -terminus and an intracellular carboxyl-terminal domain (Ctail). These receptors characteristically bind one or several heterotrimeric G proteins that become activated upon agonist binding, which in turn act as mediators of effector (enzymes and/or ion channels) activation and intracellular signaling. In particular, the GPH receptors (GPHRs) are characterized by the presence of a large extracellular domain (ECD) or ectodomain containing several leucine-rich repeats (LRRs), where recognition and high-affinity binding of the corresponding GPHs occur.
The human (h) LHCGR and hFSHR ( http://www-ncbi-nlm-nih-gov.easyaccess1.lib.cuhk.edu.hk/gene/3973 and http://www-ncbi-nlm-nih-gov.easyaccess1.lib.cuhk.edu.hk/gene/2492 ) are 699 and 695 amino acid residues long, respectively ( Figs. 2.5 and 2.6 ). By convention, the primary sequences of the hLHCGR and hFSHR are numbered from the initiator methionine of their precursor sequences obtained by virtual translation of the open reading frames of the cognate cDNA, which includes the signal peptide. The most likely sites of cleavage of signal peptides are predicted to be between residues 24 and 25 of the hLHCGR and residues 17 and 18 of the hFSHR, which yields fully processed, mature proteins of 675 and 678 amino acid residues long with apparent molecular weights of 68 to 75 kDa and approximately 75 kDa for the immature forms and 85 to 95 kDa and approximately 80 kDa for the mature, fully glycosylated membrane expressed forms, respectively. Thus the predicted NH 2 -terminus start sites for the hLHCGR and the hFSHR are Leu and Cys ( Figs. 2.5 , 2.6 , and 2.7 ).
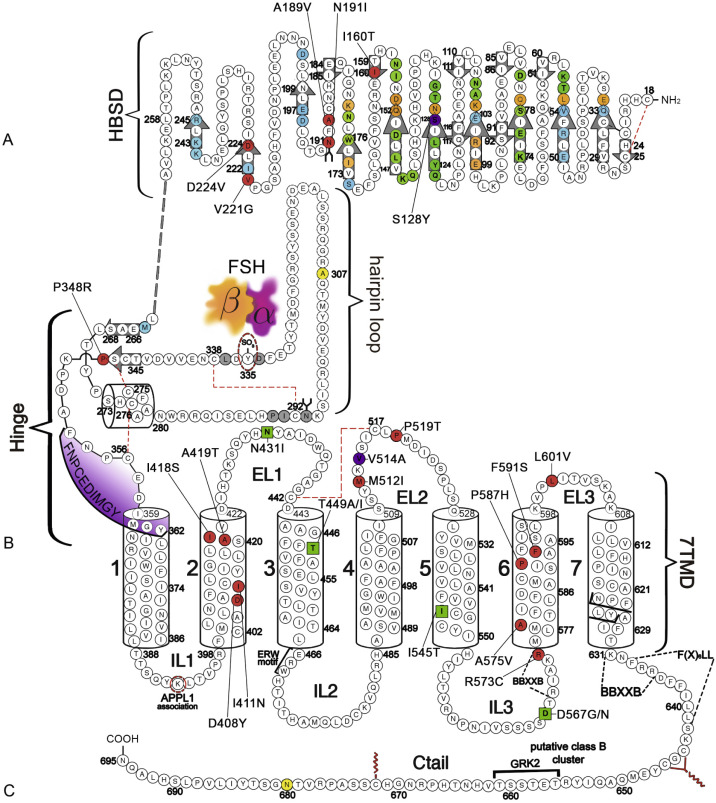


Both the FSHR and the LHCGR exhibit a high degree of amino acid sequence homology. Whereas the ECD amino acid sequences of the gonadotropin receptors are approximately 46% identical, the seven transmembrane domain (7TMD) sequence portions of the receptors share nearly 72% homology. Among the three domains of the gonadotropin receptors, the intracellular regions exhibit the lowest amino acid sequence homology (~27% identity), with the exception of the amino-terminal portions of their carboxy-terminal tail. The ECD of the receptors is essential for hormone recognition and binding, as well as for initiating receptor activation, whereas the 7TMD propagates the conformational changes induced by orthosteric agonist binding to the ECD. In turn, these conformational changes, including cytosolic regions of the receptor, promote binding and activation of downstream signaling effectors. The intracellular domains are closely related with coupling and activation of effectors, as well as with receptor trafficking, agonist-stimulated uncoupling, and desensitization.
Extracellular Domain
As mentioned previously, GPHRs are characterized by a large ECD where recognition and binding of their cognate ligands occur. This domain can be divided into three subregions: (1) a NH 2 -terminal cysteine-rich region; (2) a region composed of several copies of a structural motif rich in leucine residues (the LRR; 12 in the hFSHR and 9 in the hLHCGR and hTSHR), which is shared with a number of other membrane receptors involved in ligand selectivity and specific protein-protein interactions (see Figs. 2.5 and 2.6 ); and (3) a carboxyl-terminal cysteine-rich domain. This latter domain displays the so-called hinge region, which structurally links the leucine-rich ECD with the serpentine, 7TMD of GPHRs that is involved in high-affinity hormone binding, receptor activation, intramolecular signal transduction, and/or silencing the basal activity of the receptor in the absence of ligand. In the carboxyl-terminal end of this region, there is a particularly important amino acid sequence (FNPCEDIMGY) that behaves as an internal agonistic unit upon structural changes in the ECD provoked by hormone binding. The ECD of gonadotropin receptors contains several putative glycosylation sites—six in the hLHCGR and three in the hFSHR. The only direct biochemical evidence that exists as to which sites are glycosylated in either comes from the crystal structures of the FSHR ECD residues 25 to 250. The structures show that carbohydrate is attached at residue N191, which protrudes into solvent, whereas no carbohydrate is attached at residue N199, which protrudes from the flat β-sheet into the hormone-receptor binding interface. No structural information is available for residues 293 and 318 at this time. Nevertheless, studies with the rat FSHR have suggested that the human receptor might be glycosylated at two of three glycosylation consensus sequences (191, 199, and 293), whereas in the rat LHCGR, at least five or perhaps all six of the glycosylation sites have been reported to be glycosylated. Although carbohydrate does not play any role in hormone binding, its role in folding and trafficking of the receptor to the cell surface will be discussed later in this chapter. The ECD of gonadotropin receptors also has several disulfide bonds (five in each receptor) that stabilize the three-dimensional structure of the ECD, including the hinge region (see Figs. 2.5 and 2.6 ). It is questionable, however, if the inner Cys-bridge in the hinge region of the LHCGR is essential because in new world monkeys, LHCGR (“LHCGR type 2”) with constitutive skipping of exon 10 (which encodes 27 amino acids in the hinge region) still functions normally when stimulated by CG.
The first crystal structure of a large portion of the ECD of the hFSHR (residues 18–268; Figs. 2.7 and 2.8 ) complexed with a single chain hFSH analogue shed important light on the three-dimensional structure of this receptor region and its involvement in ligand binding and receptor activation. Although previous biochemical and in silico studies had predicted the relationship between the GPHR ectodomain and GPH agonist binding and recognition, it was not until the first structure of the FSH in complex with the extracellular-hormone binding domain of the FSHR (FSHR HB ) was established that this relationship was fully documented; this structure, however, did not include the hinge region, which had been considered as a separate structure essential for FSHR activation, as had been suggested by biochemical studies. The Fan-Hendrickson structure showed that hFSH binds to hFSHR HB like a “handclasp,” as previously predicted by in silico studies; that most β-strands in the inner surface participate in FSH binding; that both hormone subunits are involved in the specificity of binding to the receptor with charge and stereochemical dominance dictating specificity; and more importantly, that carbohydrates do not participate in the binding interface of the FSH–FSHR HB structure, but rather are sequestered to the periphery of the complex. A comparison of the crystal structure of free hFSH with that of the receptor-bound hormone also revealed that the structures of the free and bound hFSH are quite similar but the hormone is more rigid when bound to the receptor, perhaps because the β-sheet structure of the receptor extracellular domain is rigid. The most obvious change in the hormone is on the carboxyl-terminus of FSHα-subunit, which becomes buried at the receptor interface where it forms contacts with receptor residues that are highly conserved among the three GPHR.

More recently, the crystal structure of FSH bound with the entire FSHR ectodomain (FSHR ED ) was determined ( Fig. 2.9 ). The structure described more clearly the role of the hFSHR ECD (and other GPHR as well) in ligand binding and, more importantly, receptor activation. The new structure suggests that FSH is initially recruited by the previously described FSHR HB through high-affinity interactions between the gonadotropin and the concave surface of LRRs 1 to 8. However, the interface between the FSH and the FSHR ED is broader than that previously identified on the FSH-FSHR HB structure due to the presence of secondary interaction sites. Accordingly, binding of FSH to the FSHR into the hormone binding domain provokes conformational alterations in the L2β loop (V38β to Q48β) of FSH that lead to interactions between residues in this loop and LRRs 8 and 9, as well as to interactions of FSHR residues located in the hinge region (discussed later) with residues on both FSH subunits. Specificity of the FSHR for its ligand is determined by several residues, including L55, E76, R101, K179, and I222, among which L55 and K179 are important to distinguish between LH, hCG, and FSH, due to their interaction with the “seatbelt” of FSHβ, whereas the other residues dictate specificity for TSH. A detailed map of interaction between residues from FSH and the FSH ED is shown in Fig. 2.10 .


The FSHR ECD structure identified the hinge region (or signal specificity subdomain [SSSD]) as an integral part of the ECD (see Fig. 2.5 ); the structure determined also confirmed that this particular region plays an essential role in mutation-provoked receptor activation, as earlier biochemical studies on the FSHR and TSHR had indicated. In fact, these and more recent biochemical studies strongly suggest that the ECD of the GPHRs acts as a tethered inverse agonist, which switches to an agonist upon ligand binding (i.e., the unoccupied hinge region is inhibitory of receptor activation) and activation of the internal FNPCEDIMGY sequence unit located in the carboxy-terminal of the hinge region (see Figs. 2.5 and 2.6 ). The hinge region of the FSHR bears a sulfated Tyr residue (Y335; Y331 in the hLHCGR) that interacts with a binding pocket located in the interface of the α- and β-subunits of FSH and formed via conformational changes in the ligand occurring after binding to the hormone-binding subdomain (see Fig. 2.5 ). The sulfated Y335 is located right after a rigid hairpin loop, which is thought to lift when Y335 binds to the α-β pocket; this shift of the loop is thought to unlock the inhibitory effects of the loop on the 7TMD, leading to conformational changes in the latter domain and eventually to activation of the receptor. This tethered inverse agonist region in the ECD had been previously mapped to the hairpin loop segments 296 to 331 of the receptor. As part of this process, a fixed short helix formed by residues S273 to A279 (S277 and R283 in the hLHCGR) rotates, functioning as a pivot, additionally contributing to the conformational change of the FSHR SSSD. The importance of this helix movement in FSHR activation is emphasized by the finding that substitution of the S273 and S277 residues in the hFSHR and hLHCGR, respectively, with nonpolar hydrophobic residues (e.g., S273I and S277I mutants) leads to constitutive activation of the receptors. In addition, the disulfide bond C275-C346 (see Fig. 2.5 ; C279-C343 in the hLHCGR) fastens the last β-strand (LRR12) to the short helix forming a rigid body. Meanwhile, the disulfide bridge C276-C356 (C280-C353 in the hLHCGR) ties this helix to the last few residues before the first TMD. Due to these constraints, the movement of the hairpin loop that occurs upon ligand binding could directly influence the conformation of the TMD helix 1, thereby promoting rearrangement within the remaining TMDs, ultimately leading to receptor activation. Given the similarity among the structures of GPH and GPHR, it is highly possible that all GPHRs share this two-step recognition process (ligand recruitment by the HBSD followed by SSSD sulfated tyrosine docking—Y335 in the FSHR, Y331 in the hLHCGR, and Y385 in the TSHR)—albeit with some differences in the spatial arrangements when hLH and hCG interact with the sulfated Y331 residue in the hLHCGR compared with hFSH interaction with Y335. In fact, mutation of GPHR in this critical sulfated tyrosine residue led to loss of sensitivity of their corresponding ligands. Further, substitutions in amino acid residues located below the sulfated tyrosine binding pocket (αPhe74Glu) or at the potential exosite (βLeu73Phe) increased signaling of the hFSH mutants, perhaps by forcing the hairpin loop upward at the top of the pocket. Although both LH and hCG bind the same receptor, differences in hLHCGR binding, activation, and signaling have been suggested on the basis of homology modeling and site-directed mutagenesis on the hinge region, specifically in the primate-specific exon 10, which is essential for full LHCGR activation by hLH but not by hCG. In this vein, it is important to note that the carboxyl-terminal flanking sequence of the L2β loop (which participates in hLH and hCG binding to the hinge region of the hLHCGR) differs between hLH and hCG with respect to proline residues, leading to differences in the interaction between hLH and hCG with the sulfated Y331.
The crystal structure of a large region of the ECD of the TSHR in complex with a TSHR autoantibody has also been solved. Although the number of LRRs is different between the TSHR and the FSHR, the overall structure of their corresponding ECDs is very similar. Interestingly, the TSHR surface that binds the autoantibody is remarkably similar to the surface of the FSHR that binds FSH.
An interesting nuance is hFSHR (and hTSHR as well, but not hLHCGR) promiscuity for ligand specificity, occurring upon structural modifications in the ECD caused by particular mutations (see Figs. 2.5 and 2.11B ), a phenomenon that may have important consequences in the clinical arena. GPH-GPHR pairs have evolved in such a manner that a limited number of residues in both the “seatbelt” domain of the ligand and the LRRs of the receptor at the hormone binding domain participate in electrostatic interactions at the receptor-hormone interface to define binding and specificity. Due to the structural similarities between the GPH and the GPHR, it is conceivable that “cross-activation” of a given GPHR by other than its cognate ligand may occur, albeit with a low binding affinity, without triggering basal detectable receptor activation in physiological conditions. Therefore it is not surprising that substitutions in key residues that directly or indirectly participate in the interaction of the receptor with its cognate ligand may decrease structurally related ligand discrimination, resulting in the interaction of the altered receptor with other than its own cognate GPH. This is the case, for example, of the Ser128Tyr mutation at the hFSHR (see Fig. 2.5 ), which leads to pregnancy-associated OHSS. In its severe form, OHSS may be life-threatening due to increased responsiveness of the FSHR to hCG, which circulates at very high levels during the first trimester of pregnancy. In this particular mutation, the Ser→Tyr replacement allows the hFSHR to hydrogen bond αArg95 of hCG, leading to receptor activation. A different panorama is observed in the case of mutations in the 7TMD, leading to constitutive activation of the FSHR and concomitantly promiscuous binding of hCG and/or TSH, as will be discussed later.

Extracellular Loops
The ELs, or exoloops, of GPHRs transduce the signal generated by the ligand-ECD interactions to the transmembrane helices, either through direct hormonal contact and/or by modulating the interdomain interactions between the hinge region and the TMD. In fact, EL1 and 3 are solvent exposed and accessible to gonadotropin hormone, representing potential secondary binding sites for the gonadotropin, specifically at its α-subunit tips in the bound state, as supported by structural studies on the FSHR. Moreover, according to mutagenic and functional studies and the 7TMD theoretical model of the hFSHR proposed by Jiang et al., it seems that the ELs interact with the hairpin loop of the hinge region to trigger FSHR activation; apparently, lifting of the sulfated Y335 to the FSHα/β binding pocket ( Fig. 2.5B ) frees the hinge-tethered exoloops, releasing the inhibitory influence of the ECD on receptor activation. Accordingly, mutations in residues located at the hFSHR and hLHCGR ELs 1 to 3, besides altering the intracellular traffic of the receptor, may also attenuate agonist binding, alter hormone-stimulated signal transduction, or provoke constitutive activation of the receptor. The relationship between the exoloops of the gonadotropin receptors and the hinge region is further supported by studies on the TSHR, in which particular residues (e.g., Tyr563 and Lys565 at the exoloop 2 [Tyr511 and Lys513 in the FSHR, and Tyr508 and Lys510 in the LHCGR]) are crucial for ligand-stimulated receptor activation.
Transmembrane Domains
As described previously, the α-helices forming the 7TMD of the gonadotropin receptors are joined by three alternating IL and ELs (see Figs. 2.5 and 2.6 ). Although a three-dimensional structure of the 7TMD of the GPHRs is lacking, the three-dimensional structure of a number of other GPCRs with short extracellular domains have now been solved (also see http://gpcr.usc.edu ), and the TMD of the gonadotropin receptors is likely to be very similar, particularly among the rhodopsin/β 2 -adrenergic receptor (AR)–like subfamily of GPCRs. TMD residues that are highly conserved among this subfamily of GPCRs are highlighted in Fig. 2.7 . Both gonadotropin receptors exhibit the general ERW and NPXXY motifs (at the TMD3-IL2 junction and within TMD7, respectively), which are common features of the rhodopsin-like GPCR subfamily and play a crucial role in receptor activation. Further, the importance of the conformational changes on the 7TMD in receptor activation is emphasized by the fact that the majority of naturally mutations leading to constitutive activation of the GPHRs are located in this receptor domain (see Figs. 2.5 , 2.6 , and 2.11 ). In addition, several amino acid residues in the cytoplasmic face of the 7TMD of gonadotropin receptors (i.e., at the ILs) also play important roles as interactors with effectors and adapters involved in intracellular signaling and postendocytic processing (discussed later).
Since no structural data are currently available for gonadotropin receptor 7TMDs, only through homology modeling based on biophysical, structural, and computational modeling data from other GPCRs has it been possible to explore the molecular mechanisms subserving receptor activation and propagation of the activation signal from the 7TMD to the intracellular domains. In particular, three structures proved particularly crucial to understand the activation mechanism: a ligand-free form of opsin co-crystalized with the carboxyl-terminus of the α-subunit of the heterotrimeric visual Gt protein, an agonist-bound β 2 -AR stabilized in the active conformation by a nanobody mimicking a G protein, and an agonist-bound β 2 -AR co-crystallized with heterotrimeric Gs protein (Gα s -β 1 γ 2 ). Based on these structures, agonist-stimulated activation seems to lead to a set of common structural rearrangements. First, the extracellular part of the transmembrane bundle is initially affected by the agonist-induced local structural changes: (1) a small distortion of TMD5; (2) relocation of TMD3 and TMD7; and (3) reorganization of TMD5 and TMD6. Concurrently, a rearrangement of a cluster of conserved hydrophobic and aromatic residues called a “transmission switch” deeper in the receptor core occurs (which involves residues 6.48, 6.44, 5.50, 5.51, and 3.40, †
† The first number corresponds to the TMD, where the residue is located, and the second to the most conserved residue in this TMD, which is arbitrarily assigned to 50, with numbers decreasing toward NH 2 -terminus and increasing toward carboxyl-terminus.
generic amino acid numbering according to the Ballesteros and Weinstein nomenclature ), leading to rearrangement at the TMD3–TMD5 interface, and formation of new noncovalent contacts at the TMD5–TMD6 interface. Many of the residues involved in this transmission switch are highly conserved in the Rhodopsin family of GPCRs, suggesting that they are likely to constitute a common feature of GPCR activation and are now discussed in the context of the FSHR primary structure. Thus local changes would be translated into larger-scale helical conformational changes occurring at the cytosolic side, resulting in rearrangements of TMD5 at its cytoplasmic side associated with a modification of the TMD5–TMD6 interface, yielding the large-scale relocation of the cytoplasmic side of TMD6. As a result, the cleft required for binding of the Gα subunit opens. Residues from the IL2 and the cytoplasmic end of TMD3 (i.e., the arginine residue of the conserved ERW sequence in the gonadotropin receptors; see Fig. 2.5 and 2.6 ) participate in the interaction with the G protein after activation. Specifically, as a consequence of receptor activation, the salt bridge between residue 3.5 † (which corresponds to Arg467 and Arg464 in the hFSHR and hLHCGR, respectively) and residue 6.30 † (Asp567 and Asp564 in the TMD6 of hFSHR and hLHCGR, respectively) present in the inactive state would be broken. These biophysical and structural studies indicate that agonist binding alone may not be sufficient to stabilize fully active states of the receptor, and thus binding of an effector protein on the cytosolic side of the receptor is necessary to reach the fully active state . In addition, there may not be a single active state, and different ligands and allosteric modulators may stabilize distinctly different conformations, thereby giving rise to diverse downstream responses that may differ in magnitude. 271,332,333Carboxyl-Terminus
The Ctail of the gonadotropin receptors are the most divergent of the three domains. These domains host important sequences and motifs, which play crucial roles in receptor function, and include cysteine residues closely associated to the plasma membrane (PM; Cys644 and 646 in the hFSHR and Cys 643 and 644 in the hLHCGR) for palmitoylation, a primary sequence motif [Phe( X ) 6 Leu-Leu] at the NH 2 -end of the Ctail within a helical segment that in other GPCRs is referred to as helix 8 (see http://gpcr.usc.edu ), and that regulates upward trafficking from the ER to the PM, a cluster of five serine and threonine residues (656 and 658 to 661) that are a target of G protein receptor kinases (GRK) that phosphorylate the hFSHR while simultaneously promoting β-arrestin recruitment (which are scaffold proteins that regulate ligand-stimulated internalization and intracellular signaling mediated by extracellular signal-regulated kinases [ERK1/2] ), and residues involved in the postendocytic trafficking of the internalized receptor (Pro688 and Leu689, and Gly687 and Thr688 in the FSHR and hLHCGR, respectively). The importance of these sequences in gonadotropin receptor function is more extensively discussed later.
Gene Expression and Regulation
- ◆
Production of gonadotropin receptors begins with induction of gene expression.
- ◆
Gonadotropin receptor gene structures and transcriptional control are discussed.
- ◆
Physiological correlates pursuant to gene control and production of fully functional receptor are considered.
Genes
The human lutropin and follitropin receptors are encoded by single genes located on the short arm of chromosome 2 ( LHCGR , http://www-ncbi-nlm-nih-gov.easyaccess1.lib.cuhk.edu.hk/gene/3973 and FSHR , http://www-ncbi-nlm-nih-gov.easyaccess1.lib.cuhk.edu.hk/gene/2492 ). The FSHR is about 190 Kb long and is composed of 10 exons, whereas the LHCGR is about 70 Kb in length and is composed of 11 exons. The first 9 exons of the FSHR and the first 10 exons of the LHCGR encode the large extracellular domain of the receptors, including part of the hinge region, while the carboxyl-terminal end of the latter, the 7TMD and the Ctail of the receptors, are encoded by the large exons 10 and 11 of the FSHR and the LHCGR, respectively . The structural, molecular, and genomic similarities among both the gonadotropic hormones and their receptors strongly suggest that the gonadotropin receptor genes evolved from duplication of a common ancestral gene. In fact, the location of the genes is separated by only ~200 Kb. Further, the similarities among the genes of the gonadotropin receptors and other GPCRs also suggest that the precursor for the gonadotropin receptor genes arose from combining a common GPCR ancestor.
Transcriptional Regulation
The 5′ flanking regions of the LHCGR and FSHR genes are rich in GC nucleotides, and the proximal promoters of both genes are devoid of TATA boxes and have multiple transcription initiation sites within a region similar to an initiator element. Despite these similarities, the particular and divergent functional characteristics of key regulatory features present in the LHCGR/Lhcgr promoter and the FSHR / Fshr distal regulatory sites emphasize the differential regulation of each receptor gene. Studies of the transcriptional control of the FSHR gene have been done mostly on rodents. Remarkable features of the control of FSHR gene expression include the following: (1) its regulation by a combination of transcriptional and posttranscriptional mechanisms induced by FSH and activin in the ovary and FSH in the testis; (2) expression of the FSHR/Fshr requires regulatory elements located both within and outside the promoter region (i.e., 97 kb upstream of exon 1 to 57 kb downstream of exon 10), the former including elements that bind NR5A1 (SF1) as well as upstream stimulatory factors (USF), and the latter including DHS-3 (DNase I hypersensitive site), located ~4 kb downstream in intron 1, which binds GATA-binding protein complex and POU2F1 (octamer-binding protein 1) in FSHR -nonexpressing cells, suggesting association with gene silencing ; and (3) in several animal species, including humans, the FSHR promoter shows a common E box element, which contributes significantly to promoter activity, and binds its cognate USF1 and USF2 transcription factors, which regulate differential gene expression in Sertoli and granulosa cells. Studies of Fshr have also shown that USF binding to the E box increases during differentiation and decreases, concomitantly with promoter activity, upon FSH administration through increasing the DNA inhibitor of DNA binding/differentiation-2 protein (ID2), which inhibits E box binding and promoter activity. In some species, including rodents, humans, and/or sheep, the FSHR gene promoter also contains a putative estrogen response element (ERE), as well as a cAMP response element (CRE)–like sequence that may be involved in cAMP mediated transcriptional regulation; some rodent species also contain putative cis-acting elements for transcription factors of the HMG-box SRY/Sox family, which seem important in the positive transcriptional regulation of the FSHR gene. Many of the regulatory factors extending beyond the FSHR gene still remain unknown, as disclosed by studies in transgenic mice carrying a yeast artificial chromosome (YAC) with the entire Fshr plus 50 kbs 5′ and 30 kbs 3′ bordering sequences, which failed to express the receptor in Sertoli cells.
The cis-acting elements and transacting factors that control the transcription of the mouse, rat, and human LHCGR gene have been examined in more detail. The major transcription control sites of the TATA-less LHCGR gene promoter are located within a 173 to 176 bp domain; although in the rat, transcription of the LHCGR gene is inhibited by upstream sequences located from –175 bp to –2056 bp, in the human promoter this inhibition is minimal. Elements regulating transcriptional activation of the LHCGR gene include an imperfect ERE/DR motif, as well as Sp1(II) and Sp1(I) elements located at –79 bp and –119 bp from the codon initiation site (ATG) in the human gene. These elements and their corresponding transcription factors either inhibit or stimulate transcription depending on the regulators bound (EAR2/EAR3 or TR4). The ERE/DR motif binds the nuclear orphan receptors (OR) factors EAR2 and EAR3, which inhibit transcription, and TR4, which stimulates it. EAR2/EAR3 inhibits LHCGR transcription by disrupting the positive interaction between TFIIB-bound to the preinitiation complex and Sp1/Sp3 factors bound to their corresponding elements. Meanwhile, Sp1 and Sp3 bound to the Sp1(I) element directly or indirectly interact with silencing regulatory complex mSim3A/RbAp48 to repress transcription. Active silencing of LHCGR expression proceeds through a crosstalk among COUP-TF1/EARs, Sp1/Sp3, and TFIIB. During gonadotropin stimulation, the concentrations of EAR2/EAR3 change, which correlates with derepression of promoter activity. In the ovarian follicle, the derepression-silencing cycle governed by the orphan factors depends on the stage of follicular maturation, with repression present in small, early follicles and derepression-activation in mid- to preovulatory follicles and luteal follicles. Follicle-stimulating hormone stimulation in granulosa cells induces CYP19A1 expression and LHCGR transcription and expression levels necessary for the final steps in the maturation process of the oocyte and for follicular rupture. These orphans, including TR4, also bind the human DR element in the testis LHCGR . Epigenetic regulation also has been involved in LHCGR expression, with acetylation and demethylation of histones playing important roles. Acetylation occurs through inhibition of HDAC1/2 (histone deacetylase) bound to the mSin3A/RbAP48 complex or the Sp1 factor at the Sp1(I) site, and recruitment of acetylated H3 and H4 to the promoter. DNA demethylation of the promoter seems necessary to overcome the inhibitory constraint imposed by the HDAC/mSin3A complex. Thus epigenetic silencing and activation of the LHCGR is achieved through the concerted action at both histone and DNA levels, with coactivators and corepressor factors playing important regulatory roles. Several signaling pathways are involved in the complex regulation of the LHCGR , including the PI3/PKCζ pathway, which phosphorylates Sp1 and de-recruits corepressor factors, and the PKC-α/ERK pathway, which also phosphorylates Sp1 leading to dissociation of inhibitor complexes such as HDAC1/mSin3A and recruitment of TFIIB.
Transcripts
Multiple transcripts of the gonadotropin receptors have been found, which apparently arise from alternative splicing, different transcriptional start sites, or the use of different polyadenylation sites (reviewed in Simoni ). The ontogeny of the multiple FSHR and LHCGR transcripts in female and male rats has been reviewed elsewhere. Although there is some controversial evidence regarding the number and nature of multiple FSHR transcripts, at least 4 and 2 FSHR mRNA transcripts, ranging in size from 7.7 to 1.8 kb, have been detected in rat ovaries and testis, respectively, with the 2.5-kb transcript being the most abundant. Several heterogeneous FSHR splice variants have been detected in various animal species, including humans. Four splicing products affecting the ECD of the hFSHR have been detected in cumulus cells surrounding oocytes isolated from follicular aspirates of women undergoing in vitro fertilization. When these variants were expressed in vitro, a reduced response to exogenous FSH stimulation was observed. There also are reports of transcripts in the sheep FSHR that arise from alternative splicing, and that are presumably translated into functional proteins with distinct signaling properties. Since many of the alternative transcripts detected are not translated into a protein, or translation yields severely misfolded proteins unable to traffic to the cell surface PM, the function and regulatory role of FSHR transcript variants has been scarcely studied. Two alternately spliced transcripts of the ovine FSHR gene with altered exon 10 are particularly interesting, because of their potential functional consequences. The first variant is similar to the full-length WT receptor, except that differential splicing leads to divergence in protein sequence in the Ctail and that the variant is 25 amino acids shorter; this FSHR variant is expressed at the PM, fails to signal via the cAMP/PKA pathway, and behaves as a dominant negative receptor when coexpressed with the WT receptor in HEK293 cells. The second FSHR variant consists of a transcript encoding only exons 1 to 8, along with a single putative TMD, unlike anything in FSHR, and a carboxyl-terminal extension, all more characteristic of cytokine/growth factor receptors. When expressed in granulosa cells, the growth promoting effects of this variant were independent of the cAMP/PKA pathway and were rather mediated by activation of Ca 2+ and ERK1/2-dependent pathways. Further studies demonstrated that this particular variant is also expressed in the mouse ovary, and that it is upregulated by gonadotropin stimulation. More recent studies in ewes found that the FSH-growth factorlike receptor was more highly expressed than the WT variant in all size follicles, particularly in medium-size follicles obtained during the beginning of estrus, suggesting that this receptor isoform may participate in follicle development in this particular animal species. Additional studies are still necessary to more precisely define the role of these and other FSHR transcript variants in mediating the pleiotropic effects of FSH (discussed later), particularly in humans.
The transcript sizes of the LHCGR gene varies depending on the species studied. The rat receptor gene expresses four transcripts, ranging in size from 6.7 to 1.8 kb, whereas in human granulosa cells two mRNA transcripts have been detected. Several LHCGR mRNA splice variants have been identified in different animal species, including humans. A variant lacking exon 9 isolated from a human corpus luteum is particularly interesting, since it exerts dominant negative effects on both hFSHR and hLHCGR, which may represent a mechanism that regulates the gonadotropic stimulus in this ovarian structure. A cryptic exon (exon 6A), localized between exons 6 and 7 identified in primates, produces transcripts leading to nonsense-mediated mRNA decay; although the physiological significance of these variants is unknown, mutations in this cryptic exon have been associated with human male pseudohermaphroditism secondary to Leydig cell hypoplasia.
Recent studies suggest that gonadotropin receptors may be expressed in a number of extragonadal tissues, where they could exert distinct roles in certain physiological and/or pathological conditions. This interesting aspect is further discussed later in this chapter.
Posttranscriptional Regulation
Posttranscriptional regulation is also an important aspect of the regulation of the LHCGR mRNA and the LHCGR levels during the preovulatory LH surge or in response to supra-physiological levels of the gonadotropin. This important level of regulation seems to be mediated by several factors, including an LHCGR mRNA binding protein (LRBP), which has been identified as mevalonate kinase, an enzyme that is involved in cholesterol metabolism and that is also an LRBP. LHCG-induced activation of steroidogenesis and subsequent depletion of cholesterol triggers an increase in the transcription of genes that participate in cholesterol biosynthesis, including mevalonate kinase, through a PKA/ERK1/2 signaling pathway. The increased levels of this protein may thus serve the dual role of enhancing cholesterol synthesis (an enzymatic function) and decreasing the levels of LHCGR mRNA by virtue of its ability to bind the LHCGR mRNA and promote its degradation, transiently downregulating receptor expression. This transient decrease in LHCGR mRNA levels is then followed by a full recovery and increased expression during corpus luteum development only to eventually fall again with the regression of this highly steroidogenic ovarian structure. More recent studies have shown that levels of LRBP are regulated by the microRNA miR-122, which activates the sterol regulatory element binding protein (SREBP) pathway leading to LHCG-stimulated, PKA/ERK1/2-mediated up regulation of mRNA and protein LRBP levels. Another miRNA involved in the posttranscriptional regulation of LHCGR mRNA is miR-513a-3p, which interacts with the mRNA through three binding sites located in the 3’UTR region, leading to downregulation of LHCGR mRNA levels in human granulosa cells, as well as MiR-136-3p, which has been found to be associated with downregulation of the LHCGR in rat granulosa cells.
Posttranscriptional regulation of FSHR expression has been less extensively studied, although it is well known that several factors, including TGFβ and activin, increase the stability and half-life of FSHR mRNA, upregulating FSHR expression. In female rats, FSH stimulation may up- and downregulate FSHR mRNA and FSHR protein expression, depending on the stage of follicular maturation. Apart from miR-126, which was recently shown to downregulate FSHR mRNA and to act synergistically with androgens to inhibit FSHR protein production in porcine granulosa cells, little is known about whether other miRNAs are involved in the posttranscriptional regulation of the FSHR mRNA.
Folding, Maturation, and Trafficking
The life cycle of GPCRs begins at the ER, where synthesis, folding, and assembly of proteins occur ( Fig. 2.12 ). Properly folded receptors are then targeted to the ER-Golgi intermediate complex, and thereafter to the Golgi apparatus and trans-Golgi network; here, processing is completed and the receptor proteins are ready to complete their outward trafficking to the cell surface PM, where they become accessible to their cognate ligands. Interaction between GPCRs and agonists at the PM triggers trafficking of the receptor through a series of distinct processes, including (1) phosphorylation (which terminates G protein-mediated signaling) and β-arrestin recruitment, which interacts with clathrin and the clathrin adaptor, AP2, to drive receptor internalization into endosomes; and (2) either recycling of the receptor back to the PM or targeting to the lysosomes and/or proteasomes for degradation. Thus the balance between trafficking from the ER to the PM and the endocytosis-recycling/degradation pathway determines the net amount of receptor protein available to interact with the agonist and elicit a measurable biological response. GPCRs are subjected to a strict quality control system (QCS) that monitors and corrects, when necessary, the folding of the nascent receptor into a three-dimensional structure. By monitoring the structural and conformational features of newly synthesized proteins, the QCS determines which proteins must be retained at the ER and eventually degraded or routed to the Golgi apparatus and thereafter to the PM. Thus the QCS prevents accumulation of misfolded proteins that may aggregate and interfere with cell function. GPCR export from the ER to the Golgi is modulated by interaction of the trafficking proteins with specialized folding factors, escort proteins, retention factors, enzymes, and members of the molecular chaperone families, which belong to the ER QCS and the so-called proteostasis network. In particular, molecular chaperones are essential components of the ER QCS that evaluate native receptor conformation and promote delivery from the ER to the Golgi, where the protein molecule is processed before final delivery of the mature form to the PM. Molecular chaperones are an important quality control mechanism that not only recognizes but also retains and targets misfolded, nonnative protein conformers for their eventual degradation via the polyubiquitination/proteasome pathway.

Similar to other GPCRs, the gonadotropin receptors have to be correctly folded into a conformation that will pass the QCS and be compatible with ER export ( Fig. 2.12 ). Glycosylation, which together with disulfide bonds is a frequent feature of GPCRs, occurs during biosynthesis and facilitates folding of protein precursors by increasing their solubility and stabilizing protein conformation. In fact, mutations that affect glycosylation at the ECD or formation of disulfide bonds hampers the folding process of these receptors leading to intracellular retention of the misfolded intermediates, and eventually to disease. Thus glycosylation plays an important role not only in folding, but also in the maturation and intracellular trafficking of the receptors from the ER to the cell surface PM. Studies in the rat have shown that at least one glycosylation site at the FSHR ECD is required for proper receptor folding and efficient trafficking to the PM. Lack of glycosylation of the mature rat FSHR does not affect binding or affinity, thus indicating that this structure does not participate in ligand interaction. Likewise, mutants of rat LHCGR that prevented glycosylation at the first three consensus sequences did not affect receptor synthesis or ligand binding but decreased the efficiency of receptor folding, leading to reduced maturation and increased degradation of the precursor protein. Thus carbohydrates in both gonadotropin receptors, although not involved in hormone binding, are important players in the maturation process of the newly synthesized receptors, promoting its folding and conformational stability and its trafficking to the PM.
Co-immunoprecipitation studies have identified some interacting proteins that support folding of the gonadotropin receptors during their residency at the ER. These studies have shown that the folding process of the rat FSHR and LHCGR precursors (co-translationally glycosylated at the ER by the addition of the core sugars N-acetyl glucosamine, mannose, and glucose) involves interactions with the chaperones calnexin and calreticulin, which facilitate proper folding of intermediate glycoprotein molecules. The cycle of these chaperones predominantly centers on substrate N-glycans present on the glycoprotein receptor precursor, adding hydrophobicity to the folding protein. The calnexin/calreticulin cycle depends on the concerted action of carbohydrate-modifying enzymes (glycosidases I and II), which lead to formation of monoglycosylated oligosaccharide structures that interact with the chaperones, as well as to removal of the remaining glucose residue from the oligosaccharide, terminating the association with the chaperones. Then the GPHR (already in its native conformation) is exported to the Golgi complex to continue processing of the oligosaccharide chains. Another GPHR interacting chaperone is the protein disulfide isomerase, or PDI, which is an ER-resident enzyme involved in disulfide bond formation of folding intermediates, and that probably acts as a co-chaperone with calnexin and calreticulin during their association with the GPHR. No interaction of the GPHR with other molecular chaperones has been yet documented, with the exception of the TSHR, which apparently interacts with BiP—a chaperone that maintains proteins in a state competent for subsequent folding and oligomerization, and that mediates retrograde translocation of misfolded conformers for proteosomal degradation.
Several sequence motifs present in the GPCRs are involved in the exit of the receptors from the ER and the Golgi. Among these motifs is the previously mentioned Phe (X) 6 LeuLeu sequence identified in the Ctail of several GPCRs, including the gonadotropin receptors (see Figs. 2.5 and 2.6 ). In the hFSHR, this export motif is located between amino acid residues 633 and 641, whereas in the hLHCGR, this motif is located between residues 630 and 638. The Ctail peptide of the hFSHR also contains the minimal BBXXB motif reversed in its juxtamembrane region (residues 631 to 635) ; the last two residues of this motif (R634 and R635) and the preceding F633 constitute the NH 2 -terminal end of the highly conserved F (X) 6 LL motif, and thus mutations in these residues impairs receptor trafficking and PM localization of the receptor. The IL3 of the hFSH and hLHCG receptors also contains this BXXBB motif (residues 569 to 573 in the hFSHR and 566 to 570 in the hLHCGR), and either deletion or replacement of the basic residues of this motif with alanine impair PM expression of the modified receptors. Another motif that influences gonadotropin receptor folding is the AFNGT motif (amino acid residues 193 to 197 in the hLHCGR and 189 to 193 in the hFSHR), which bears a potential glycosylation site (N195GT and N191GT, in the hLHCGR and hFSHR, respectively). Thus mutations in this motif influence receptor folding and trafficking to the PM.
Mammalian cells expressing the recombinant hLHCGR display several distinct glycoprotein species with molecular masses (estimated from SDS gels) ranging from 65 to 240 kDa. The mature, PM-expressed hLHCGR has been identified as an 85- to 95-kDa protein, whereas a 65- to 75-kDa band has been identified as an immature, partially glycosylated precursor that is located in the ER. The higher molecular weight bands ranging from 165 to 240 kDa are oligomers of the immature or mature receptors. Precursor, mature, and oligomeric forms of the hFSHR can also be detected in transfected cells, but there is more variation on the reported molecular weights of these products. Estimates for the molecular mass of the mature cell surface FSHR range from 74 to 80 kDa, whereas estimates for the molecular mass of the immature intracellular precursor range from 67 to 75 kDa. Higher molecular weight forms of the FSHR (~170 kDa) have also been identified and appear to be oligomers of the immature intracellular precursor. It should be noted that many of the studies on the sizes and nature of the different forms of gonadotropin receptors have relied on immunological detection of epitope-tagged receptors expressed in heterologous cell lines. Immunological detection of the endogenous gonadotropin receptors expressed in the testes, ovaries, or extragonadal tissues has been much more difficult, because they are expressed at low densities and because of the nature of the available gonadotropin receptor antibodies. Using monoclonal or polyclonal antibodies (some of which have been rigorously validated), the mature and immature forms of the hLHCGR and hFSHR described previously can indeed be immunologically detected in target tissues and homologous cell lines.
Another posttranslational modification important for GPCR trafficking is palmitoylation. Cysteine residues in the Ctail of several GPCR have been shown to be the target for S-acylation with palmitic acid; in some receptors, this posttranslational modification is often required for efficient delivery of the protein to the cell membrane, where it facilitates anchoring of the receptor Ctail to the PM. The hFSHR has in its Ctail two conserved cysteine residues (at positions 646 and 672) and one nonconserved cysteine residue at position 644. Although the hFSHR is palmitoylated at all cysteine residues, regardless of their location in the Ctail of the receptor, S-acylation at C627 and C655 is not essential for efficient hFSHR PM localization, whereas at C629 it is, as replacement of this residue with glycine or alanine reduced detection of the mature form of the receptor by approximately 40% to 70%. Further, when all palmitoylation sites are removed from the hFSHR, cell surface PM expression is reduced to approximately 10% to 30% of that shown by the WT receptor. The hLHCGR is palmitoylated at two conserved cysteine residues (643 and 644), but in contrast to the hFSHR, palmitoylation of this receptor is not important for trafficking to the PM, as abrogation of palmitoylation did not appear to affect PM expression and agonist binding. The importance of palmitoylation of gonadotropin receptors in internalization and postendocytic processing of the PM-expressed receptor following formation of the hormone-receptor complex depends on the receptor. While in the hFSHR, palmitoylation does not play any role in internalization of the receptor/hormone complex, in the hLHCGR prevention of palmitoylation increases the rate of agonist-stimulated internalization. In both receptors, abrogation of palmitoylation impairs receptor recycling to the PM and increases the fraction of receptor/hormone complex targeted to degradation via the proteasome/lysosome pathways. Most of the internalized hFSHR is recycled back to the PM, whereas in the hLHCGR, only 30% of the internalized receptor recycles back to the cell surface. In addition to palmitoylation, postendocytic sorting is also influenced by specific sequences in the Ctail of the receptors.
Agonist stimulation of GPCRs is followed by a series of structural modifications and associations with scaffold proteins that eventually lead to effector uncoupling, internalization, and either recycling back to the PM or degradation in lysosomes and/or proteasomes ( Fig. 2.12 ). The hFSHR has been reported to be phosphorylated by second messenger-dependent kinases PKA and PKC, but also by GRKs 2, 3, 5, and 6 in various models. PKA and PKC contribute to both agonist-dependent (homologous) and agonist-independent (heterologous) desensitization of the receptor. GRK-mediated phosphorylation leads to more complex effects, as they are centrally involved in homologous desensitization, while simultaneously regulating β-arrestin recruitment and subsequent β-arrestin effects on receptor internalization through clathrin-coated pits and G protein-independent signaling. As described previously, a cluster of five serine and threonine residues located in the Ctail of the hFSHR (see Fig. 2.5 ) has been shown to account for the bulk of FSH-induced phosphorylation as a result of GRK2 action. It has also been documented that β-arrestins are recruited to the GRK-phosphorylated and agonist-occupied FSHR. β-arrestins recruited to GRK2- or GRK5/6-phosphorylated FSHR have been suggested to exert distinct intracellular functions ; GRK2-phosphorylated hFSHR predominate in the β-arrestin-mediated desensitization process, whereas GRK5- and 6-induced phosphorylation of the activated FSHR is required for β-arrestin-dependent signaling pathway in the immortalized cell line HEK293. It is well established that β-arrestin 1 and 2 binding to GRK-phosphorylated FSHR leads to the internalization and recycling of the receptor. In contrast to the hFSHR, the hLHCGR does not recruit GRKs to promote its desensitization, which is instead mediated by the interaction of the receptor with ADP ribosylation factor nucleotide-binding site opener (ARNO), an exchange factor for ADP ribosylation factor 6 (ARF6), which then recruits β-arrestins when bound to GTP. Here it is important to mention that recruitment to β-arrestins bound by the hFSHR plays an important role for initiating distinct arrestin-mediated signaling pathways, mainly the MAPK ERK, as will be described later. In the case of the hLHCGR, β-arrestins are not apparently involved in MAPK-ERK signaling, and thus this receptor does not demonstrate biased signaling through this particular signaling cascade.
Extragonadal Expression
LHCGR and the FSHR localize to gonadal cells, which contain their targets, evidenced by early hormone ablation experiments and subsequent development of in vitro bioassays. In the testes, the canonical location of LHCGR and FSHR is restricted to Leydig and Sertoli cells, respectively. In the ovary, expression of the LHCGR occurs in theca, interstitial, and granulosa cells from preovulatory follicles and in the corpus luteum, whereas the FSHR is limited to the granulosa cells of the maturing follicles. In all cases, one can easily demonstrate readouts, following treatment with physiological levels of hormone and importantly the binding of radiolabeled hormone to these tissues with high affinity and capacity. Certainly, the main physiological roles of the gonadotropin receptors can be attributed to their actions in the ovaries and the testes, as demonstrated by the phenotypes of individuals harboring activating or inactivating mutations of these genes (discussed later). Moreover, the phenotypes of genetically engineered mice with exaggerated or absent gonadotropin actions, or mice with targeted inactivation or constitutively activated receptors, appear to be explained entirely by the classical actions of LH and FSH in gonadal tissues.
Nevertheless, in recent years there have been reports of low level FSHR expression in extragonadal tissues, where it has been proposed to have distinct physiological roles. The extragonadal sites where the FSHR has been detected include bone osteoclasts and monocytes, endothelial cells from umbilical vein, tumor vasculature and metastases, different sites of the female reproductive tract, the developing placenta, the endometrium, and the liver. Data on the expression (mRNA and protein) of either the FSHR WT or the splice variant originally identified in bovine granulosa cells in both osteoclasts and monocytes, and consequently on the role of FSH in bone function, are still quite controversial. Immunohistochemical studies employing monoclonal anti-FSHR antibodies have been the mainstay for documentation of the receptor, because ligand binding is not detectable. The expression of the hFSHR in blood vessels from a number of malignant tumors and tumor metastases has been reported. Although the authors posit that FSHR at these locations may contribute to the growth and expansion of the tumor tissue by promoting angiogenesis, further studies are still necessary to validate these findings, given that such claims can lead to unnecessary uncertainty in women undergoing hormone therapy for infertility or during menopause, when FSH levels are high. FSHR in several nonovarian reproductive tissues have been recently identified. Interestingly, fetoplacental haploinsufficiency of the Fshr in mice was associated with defects in placental growth and fetal loss in both heterozygous and homozygous null Fshr animals. More recently, a role for the FSHR in endometriotic lesions and in hepatic tissue has been proposed; the authors claimed that at these locations the FSHR might be involved in FSH-stimulated estrogen production and progression of endometriosis, as well as in the expression of the low-density lipoprotein receptor and regulation of low-density lipoprotein cholesterol clearance, respectively. Nevertheless, further studies are still warranted to confirm the existence and elucidate more strongly the expression and physiological significance of extragonadal FSHRs and, if so, whether they are structurally similar to that primarily expressed in the gonads or are splice variants of the WT receptor that may be expressed in the PM in sufficient quantities to provoke a biological effect upon exposure to physiological concentrations of FSH.
There is a large and equally controversial body of literature suggesting that functional LHCGR may also be expressed in a number of extragonadal tissues. The suggestion of extragonadal LHCGR expression has been based in many cases upon the detection of fragments of the LHCGR mRNA. Attempts to detect immunoreactive LHCGR protein often result in the identification of a protein (or proteins) that do not match the expected molecular weight of the authentic gonadal LHCGR (discussed previously). A comparison of the actions of LH/CG in isolated cells or tissues from WT and LHCGR null mice would go a long way in shedding light on this controversy, but curiously, this has not been done often. In a recent study on the potential angiogenic activity of CG, it was shown that the proangiogenic effect of the gonadotropin was clearly ameliorated in blood vessels isolated from the LHCGR null mice. This finding suggests that a functional LHCGR might be expressed in blood vessels. Further, the LHCGR has been detected in the secretory endometrium, particularly around the spiral arteries, as well as in the site of implantation in nonhuman primates. The authors claimed that the temporal expression of the LHCGR in this tissue might play an important role in inducing expression of specific genes important in modulating several processes, including decidualization, the immune system, cell survival, and the vascularization at the maternal-fetal interface.
Some of the clearest evidence for the extragonadal expression of a functional LHCGR comes from studies done on a woman who developed pregnancy-associated Cushing syndrome and later developed it again after menopause. After menopause, there is clear evidence that the cortisol levels in this woman were controlled by LH because suppression of the hypothalamic/pituitary axis with GnRH agonist controlled the hypercortisolism. Clinical findings in a number of other cases also seem to be explained by the inappropriate expression of the LHCGR in the adrenal cortex or in adrenocortical tumors. Ectopic expression of functional LHCGR in the adrenal cortex seems to be a common finding in transgenic or knock-out mouse models with elevated levels of gonadotropins, and expression of the mature LHCGR can be readily documented in the adrenal glands of pregnant rats.
Lastly, some reports indicate that during development other extragonadal rat tissues such as the kidneys express the immature form of the LHCGR, whereas nervous tissues express both the mature and immature forms. Direct actions of gonadotropins on these tissues are still controversial.
Oligomerization
A number of biochemical, pharmacological, and biophysical studies support the concept of receptor self-association or oligomerization as a fundamental process enabling GPCR activity. It is currently accepted that multimerization of GPCRs is involved in fine-tuning regulation of several physiological processes, such as ligand binding, receptor trafficking, and regulation of PM expression levels, as well as activation of signaling pathways. As with many other GPCRs, GPHR also associate to form homodimers/oligomers and even heterodimers, in which two closely related receptors simultaneously expressed in the same cell associate (e.g., the LHCGR and FSHR in ovarian mature granulosa cells ). In the case of the gonadotropin receptors and the TSHR, a number of studies have shown that these receptors self-associate. Although the structure of the FSH-FSHR HB revealed that the FSHR ECD may form weakly associated dimers, with each molecule bearing one FSH molecule, later studies employing combined biochemical and biophysical approaches directly demonstrated that the FSHR self-associates early during receptor biosynthesis, and that it can be identified as FSHR/FSHR homodimers or FSHR/LHCGR heterodimers in the surface membrane of HEK293 cells. The mechanism and extent of FSHR self-association are not known, but it seems reasonable to assume that contacts occurring via the TMDs play an important role. The more recent crystal structure of the ligand-bound ECD of the FSHR demonstrated an additional mode of association of FSH with the entire FSHR ECD and a quaternary structure bearing three individual ECDs (i.e., a trimeric receptor structure; see Fig. 2.9A ); this structure predicts hosting of one fully glycosylated FSH molecule or three deglycosylated molecules (see Fig. 2.9B ). In the former scenario, the FSHR may become activated, whereas in the latter the structure remains inactive, supporting early observations on the lack of effect of deglycosylated FSH for triggering signal transduction at the FSHR. The trimeric model also explains the reported differences in receptor binding activity between fully glycosylated and hypoglycosylated FSH isoforms, with the former exhibiting delayed and lower binding activities, and provides a mechanism for FSHR association with multiple effector proteins and drawing on several signaling pathways.
The LHCGR also forms oligomers in the ER and at the PM, in a process that is unrelated to receptor activation. Importantly, co-expression of a misfolded LHCGR with its WT counterpart impairs the cell surface expression of the wild-type receptor and attenuates signaling, a dominant-negative effect. Co-expression of splice variants of the LHCGR may also regulate the expression of the LHCGR and FSHR by forming intracellular oligomers that prevent the proper processing of the intracellular LHCGR precursor. For example, LHCGR transcripts lacking exon 9 are prevalent in normal human ovaries, but the resulting protein is not able to bind hCG or to be adequately processed to be expressed at the cell surface. When co-expressed with the WT hLHCGR or hFSHR, the mutant lacking exon 9 associated with the immature forms of these receptors and exerted dominant-negative effects by decreasing their cell surface expression.
Formation of FSHR/FSHR complexes explains intermolecular complementarity between mutant FSHR dimers, in which the function of a hormone binding-deficient FSHR protomer may be rescued by a signal deficient counterpart following agonist exposure, as well as the negative cooperativity between FSHR protomers (i.e., when upon ligand binding one protomer blocks or decreases affinity of the second protomer for its ligand or intracellular signaling partners leading to a right shift of the dose–response curve). Functional complementarity in the LHCGR has also been documented to occur both in vitro and in vivo. Complementarity between gonadotropin receptor homo- or heterocomplexes conveys important physiological implications for the gonadotropin receptors, as temporal association and alternating negative cooperativity between the FSHR and the LHCGR in the ovarian granulosa cells might protect the follicle from premature luteinization or conversely from ovarian hyperstimulation, depending on the expression level of each receptor. Heterozygous mutations that inactivate receptors may remain silent by virtue of rescue with WT receptor, or may conversely express as clinically adverse, by virtue of the inactive receptor (i.e., with a trafficking defect), preventing the native WT receptor from trafficking to the cell surface. The occurrence of homo- and heterodimerization as a mechanism for negative cooperativity or intermolecular complementarity between gonadotropin receptors represents a unique opportunity for therapeutic interventions.
Signaling Pathways Mediated by Gonadotropin Receptors
- ◆
Gonadotropin receptors as GPCRs represent as canonical G-protein signaling receptors.
- ◆
Emerging concepts indicate that gonadotropin receptor signaling is more complex.
- ◆
Recently it has been discovered that gonadotropin receptors exhibit biased signaling.
Canonical and New Pathways
Although most investigators agree that the effects of the gonadotropins on the differentiated function of their target cells are mainly mediated by the activation of the canonical Gs/adenylyl cyclase/cAMP/PKA pathway, which subsequently activates CREB thereby modulating gene transcription, it is now clear that this is not the only signaling cascade activated by these receptors. Additional pathways are activated, as detailed later, and these may be involved in several gonadotropin-dependent events, such as proliferation and/or differentiation of target cells and, at the molecular level, functional selectivity and differential gene expression.
In recent years it has become evident that the FSHR is connected via conformational selectivity to a nonlinear and complex signaling network mediated either by several G protein subtypes, including the Gs, Gi, Gq/11, and Gh proteins ; other types of receptors (e.g . , the insulin-like growth factor 1 receptor and the epidermal growth factor [EGF] receptor [EGFR] ) ; and/or other proteins associated with the receptor (e.g., β-arrestins and adapter proteins containing pleckstrin homology domain, phosphotyrosine binding domain, and leucine zipper motif [APPL] ) to promote activation of a number of signaling pathways, including those mediated by distinct kinases (such as PKA, PKC, PI3K, PKB/Akt, and ERK1/2). This complex network of pathways allows for a fine-tuning regulation of the gonadotropic stimulus, where the activation/inhibition of its multiple components may vary depending on the cell context, developmental stage of the host cells, and concentration of receptors and ligands. Of particular interest are FSHR-mediated pathways triggered by cAMP accumulation but independent from PKA activation, such as those activated by the exchange protein directly activated by cAMP (EPAC) . In addition to CREB, cAMP-activated PKA activates p38 mitogen-activated protein kinases, ERK 1/2, p70S6 kinase (p70S6K), and PI3K via insulin receptor substrate 1 (IRS-1), leading to PKB/Akt and FOXO1 transactivation and thereby to activation/repression of FSH-regulated genes. Meanwhile, EPAC promotes p38 MAPK and PKB/Akt phosphorylation, and upregulation of the EGFR. In addition to G proteins, FSHR also associates with other cytoplasmic interacting proteins, including the APPL1,2 and 14-3-3τ adapters and the scaffolds β-arrestin 1,2. Linking of the FSHR with APPL1 occurs at the IL1 of the receptor, specifically at Lys393 (see Fig. 2.5 ), whereas association with 14-3-3τ maps to the IL2, overlapping with the canonical G protein binding sites. Agonist stimulation rapidly induces FOXO1a phosphorylation in HEK293 cells leading to inhibition of apoptosis via either serum and glucocorticoid-induced kinase (Sgk) or APPL1 interaction with the upstream PI3K/Akt pathway. In addition, APPL1 also participates in the FSH-induced inositol 1,4,5 triphosphate (IP3) pathway and implicates it in intracellular calcium signaling in granulosa cells. The coexistence of APPL1 and 14-3-3τ, associated with the FSHR, suggests that FSH causes phosphorylated FOXO1a to be sequestered by 14-3-3τ, with APPL1 facilitating this antiapoptotic process.
The FSHR activates ERK1/2 phosphorylation and signaling, which have been shown to play an important role in immature granulosa cells on the induction of a subset of FSH gene targets that define the mature preovulatory granulosa cells, including the genes encoding the α-subunit of inhibin, the LHCGR, the EGFR, and the enzyme aromatase. ERK1/2 activation occurs through two distinct pathways, one mediated by Gsα and the other by β-arrestins, which are primarily involved in agonist-stimulated desensitization and internalization of the FSH/FSHR complex (noted previously). As discussed earlier, phosphorylation by GRK5 and 6 is required for β-arrestin-dependent, G s α-independent signaling. When activated by the G s α-PKA pathway, ERK1/2 phosphorylation occurs via PKA-promoted dissociation of ERK from a 100-kDa phosphotyrosine phosphatase that inhibits ERK activation and its translocation to the nucleus. PKA-stimulated ERK activation occurs early and is transient, peaking at 5 to 10 minutes after FSH exposure, whereas in the G s α-independent pathway mediated by β-arrestins, activation is slower and sustained, and mainly occurs during the ensuing 10 to 30 minutes of agonist exposure.
LH is essential to stimulate androgen production by the Leydig and theca cells, as well as for ending the FSH-regulated program for steroidogenesis and granulosa cells growth, and for promoting the luteinization of cells into luteal cells. LH also induces genes necessary to promote oocyte maturation, follicle rupture, and ovulation. To do this, the LH-activated LHCGR must turn on a variety of signaling pathways. The LHCGR was one of the first GPCRs shown to independently activate distinct G protein-dependent signaling pathways, adenylyl cyclase (through G s α), and phospholipase C (through Gαq/11). Although this observation was initially made in heterologous cells expressing the recombinant mouse LHCGR, it has now been extensively reproduced in a variety of cell lines transfected with either the rodent or human LHCGR. LHCGR-mediated activation of the G s α/PKA/cAMP pathway leads to activation of several downstream signaling cascades, such as the EGF network, which includes expression of the EGF-like growth factors amphiregulin (AREG) , epiregulin (EREG) , and betacellulin (BTC) that bind and transactivate the EGFR in mural and cumulus cells in an autocrine/paracrine fashion, leading to activation of the ERK1/2 cascade and the expression of genes important for cumulus expansion. Activation of the EGFR-regulated network is also one important factor involved in resumption of oocyte meiosis, as it promotes the decline in cGMP levels, whose concentrations negatively regulate the activity of the phosphodiesterases necessary to reduce intraoocyte cAMP and promote meiotic resumption. The ERK1/2 cascade, activated through EGFR-Ras and also probably by cAMP and/or PKC, is critical for several LH stimulated processes, including androgen production, and those occurring around ovulation, such as cumulus expansion, oocyte resumption of meiosis, ovulation, and luteinization. Further, it has been shown that most of the ovarian genes that are up- or downregulated by LH are targets of ERK1/2.
Studies in several experimental models have found that LHCGR activates members of other families of G proteins, including the Gαq/11 protein, which via the progesterone receptor contributes to the ovulatory process, as has been demonstrated in mice with a granulosa cell-specific deletion of Gαq/11. Although these Gαq/11-deficient mice resumed meiosis and showed cumulus expansion upon hCG exposure, they failed to increase inositol phosphate accumulation and express the progesterone receptor, and exhibited marked subfertility because of trapping of the oocytes destined for ovulation in preovulatory follicles or corpora lutea. LHCG-activated Gαq/11 and phospholipase C is also involved in Ca 2+ mobilization from intracellular stores; although Ca 2+ is not necessary for oocyte maturation, it is apparently involved in luteinization.
In addition, LHCGR activates PI3K and PKB/Akt in a cAMP-independent fashion. It is thought that this pathway is involved in oocyte meiotic resumption, albeit in a dispensable manner, given that mice lacking PTEN (phosphatase and tensin homolog deleted on chromosome 10, which functions as a negative regulator of PI3K action) or PDK (phosphoinositide-dependent kinase, which phosphorylates PKB/Akt) did not exhibit alterations in meiosis II progression or germinal vesicle breakdown. The LHCGR also activates the mammalian target of rapamycin complex 1 (mTORC1) signaling through the cAMP/PI3K/Akt signaling pathway to regulate cell proliferation and expression of several key enzymes involved in androgen biosynthesis, including P450 side-chain cleavage enzyme (P450scc), 3β-hydroxysteroid dehydrogenase type 1, and 17α-hydroxylase/17,20lyase (P450c17), without modifying expression of the steroidogenic acute regulatory protein (StAR) in theca-interstitial cells.
In summary, a number of studies in experimental animals and humans indicate that several gonadotropin-regulated intracellular signaling pathways must coalesce to coordinately regulate distinctly different cellular processes involved in steroidogenesis, follicular growth and maturation, cell differentiation, and ovulation. These signaling cascades are activated in a time-regulated fashion, depending on the differential expression of gonadotropin receptors during the different stages of the ovarian cycle. Thus granulosa cells from small antral follicles express the FSHR, with a progressive increase during follicular maturation. LHCGR expression initially confined to the theca-interstitial cells is also induced to express in antral follicles, and expression increases during follicular growth in response to FSH, FSH-stimulated estrogen production, and other paracrine factors. In fact, both gonadotropin receptors are simultaneously expressed in granulosa cells from mature, preovulatory follicles, in which activation of the LHCGR by its cognate ligand eventually suppresses the effects of FSH on steroidogenesis and follicle growth and promotes the ovulatory process. After ovulation and differentiation of granulosa cells into luteal cells, LHCGR expression regulates progesterone and estradiol production by the luteal cells until corpus luteum regression occurs.
Biased Agonism
It is currently accepted that preferential activation of distinct GPCR-mediated signaling cascades may occur through biased signaling that results from stabilization of distinct receptor conformations in response to particular ligands or receptor mutations. In the case of the FSHR, functional selectivity or biased agonism toward Gi-mediated signaling has been documented for some naturally occurring glycosylated variants of human FSH and recombinant FSH expressed in insect cells, as well as for selective allosteric compounds with FSH agonist or antagonist properties. Bias toward β-arrestin-mediated ERK1/2 MAPK signaling also has been observed when FSHR is treated with a modified agonist (truncated equine LHβ [Δ121 to 149], combined with Asn56-deglycosylated equine LHβ) ; this molecule is particularly interesting, because while preferentially activating the β-arrestin-ERK1/2 phosphorylation module, it concomitantly behaves as an antagonist of FSH-stimulated cAMP-PKA signaling when tested at equimolar concentrations. At the receptor level, biased agonism has been documented when hFSHR PM expression is severely impaired as a result of a loss-of-function mutation (Ala189Val, discussed later ; see Fig. 2.5 ). In this case, β-arrestins recruited to the agonist-bound receptor assembled a MAPK module, whereas G protein-dependent signaling remained impaired. Also interesting is the missense Met512Ile hFSHR mutation, which led to an impaired FSH response in terms of cAMP accumulation and PI3K activation but not ERK1/2 phosphorylation. Conditional signaling (biased signaling dependent on the cell context) at the LHCGR has been recently reported for two negative allosteric modulators of the FSHR (discussed later), in which they differentially inhibited hCG-mediated steroidogenesis when tested in the murine Leydig cell tumor-derived mLTC-1 cell line and rat primary Leydig cells.
Although the physiological role of biased signaling at the gonadotropin receptors remains to be defined, this phenomenon opens the door for the design of drugs that may positively or negatively regulate selective signaling pathways on the follicle and that may be potentially useful in the clinical arena to treat estrogen- and/or androgen-dependent diseases and infertility, as well as in contraception.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


