Abstract
Steroid hormones are a group of hormones derived from cholesterol that act as chemical messengers in the body. The steroid hormones regulate many physiologic processes, including the development and function of the reproductive system. The actions of the steroid hormones are mediated by the steroid hormone receptors, intracellular proteins belonging to the nuclear family of transcription factors. These receptors mediate signal transduction through genomic and nongenomic actions in a context-specific manner. The complex molecular events orchestrated by the steroid hormones and their receptors reflect the diversity in signals activating the receptors and the dynamic mechanisms by which the steroid hormone receptors integrate these signals into a physiologic response.
Keywords
Mineralocorticoid, glucocorticoid, estrogen, progestin, androgen, nuclear receptor hormone, response element coactivators, corepressors
Steroid Hormone Receptors Act as Ligand-Dependent Transcription Factors or Repressors
- ◆
Steroid hormones are metabolites of cholesterol that bind to intracellular receptors with high specificity.
- ◆
Steroid hormones exert their functions by diffusing through the cell membrane, binding their receptors, inducing a conformational change in the receptor, associating with DNA, interacting with cofactors, and altering gene expression.
- ◆
Precise regulation of gene transcription is essential for development, physiology, and homeostasis.
Steroids are small, lipophilic hormones synthesized from a common precursor molecule, cholesterol, through a complex biosynthetic process in tissues and glands throughout the body (see Chapter 4 ). Essential for life, the adrenal gland is the most important steroidogenic organ in the human body. Within the adrenal cortex, mineralocorticoids and glucocorticoids are produced, while the sex steroids ( estrogens , progestins , and androgens ) are primarily generated by the gonads. Despite their shared molecular origin and basic structural similarities, mineralocorticoids, glucocorticoids, estrogens, progestins, and androgens are distinct classes of steroid hormones that interact with specific, high-affinity receptors to exert their biological effects (mineralocorticoid receptor [MR], glucocorticoid receptor [GR], estrogen receptor [ER], progestin receptor [PR], and androgen receptor [AR]). These hormones control diverse physiologic and cellular processes and affect almost all aspects of eukaryotic physiology, from sexual differentiation, growth, and reproduction to immunity, metabolism, and behavior. Consequently, a clear and complete understanding of the basic mechanisms of steroid hormone action is of critical importance for health and disease. Moreover, it is also necessary to define the unique mechanisms that lead to hormone-specific effects and functions, which also occur in a cell-specific manner.
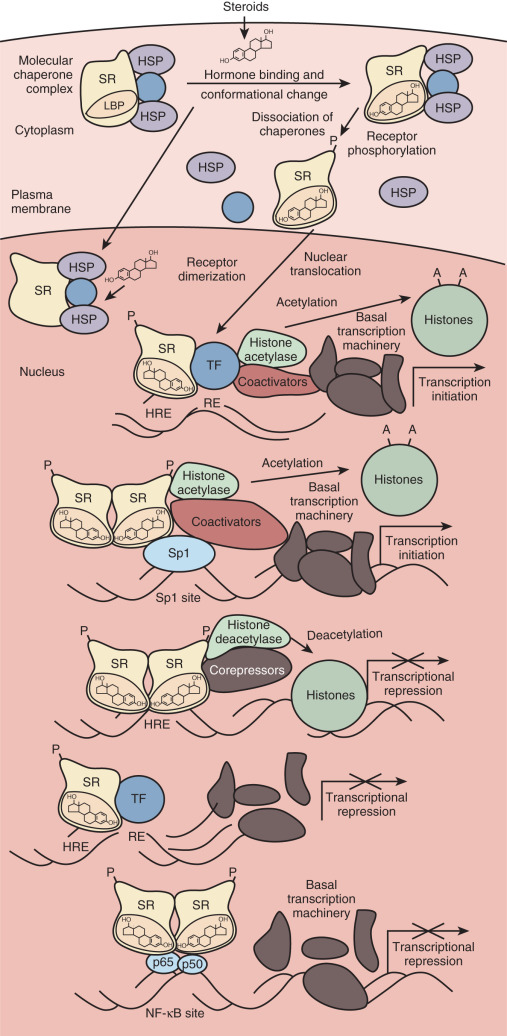
This chapter reviews what is known about the mechanisms of steroid action. The classic mode of action entails simple diffusion of steroid hormones into the cell, where they interact with cognate receptors and stimulate or inhibit transcription of target genes ( Fig. 5.1 ). The hormone-dependent changes in receptor conformation drive transactivation and transrepression of gene expression by altering interactions with molecular chaperones that keep the receptor in a ligand-independent state, inducing posttranslational modifications of the receptor that alter activity, promoting the formation of receptor dimers, enhancing interactions with specific DNA sequences (hormone response element) , and facilitating recruitment of coactivator or corepressor proteins that alter chromatin structure and contact the basal transcription machinery. Mechanistic studies have built on the classic mode of action to reveal a complex regulatory network of interacting factors and chromatin state.
To understand the plasticity of steroid hormone action and appreciate the specificity of signaling by different classes of steroids, it is important to understand the evolution of receptors from a common ancestral protein. Evolving with unique specificity, steroid receptors gained distinct functions in physiology; the processes governed by each steroid receptor are discussed in this chapter. General factors that influence steroid hormone action are also highlighted. Finally, the chapter examines studies that describe alternative modes of action for steroid hormones and their receptors, including interactions with other transcription factors and nongenomic effects mediated by second messenger signaling pathways.
Evolution of Steroid Hormone Receptor Structure and Function
- ◆
The steroid hormone receptors belong to a large family of transcription factors called nuclear receptors.
- ◆
The structure of the steroid receptors is modular, with distinct domains. The steroid receptors contain a highly conserved DNA-binding domain, a moderately conserved ligand-binding domain, and less well-conserved amino- and carboxy-terminal domains.
- ◆
Phylogenetic analysis determined that the steroid receptors clustered separately from other ligand-dependent transcription factors. The relatedness of the steroid hormone receptors reflects gene duplication and divergence.
Steroid receptors belong to a larger family of structurally and evolutionarily related proteins called nuclear receptors , which are encoded by 48 genes in the human genome. Through alternative splicing and alternative translation initiation, it is predicted that these 48 genes could result in many more receptors in the human proteome. All nuclear receptors, including steroid receptors, exhibit a modular structure composed of distinct domains ( Fig. 5.2 ). In general, these receptors contain a variable amino-terminal region (A/B), a highly conserved DNA-binding region (C), a highly variable hinge region (D), and a moderately conserved hormone or ligand-binding domain (E). Some receptors also contain a carboxy-terminal F domain. The primary structure of each human steroid receptor is shown, along with its physiologic ligand. Specific residues within the DNA-binding (C) and ligand-binding (E) domains play an important part in receptor dimerization, which is critical, because most nuclear receptors are only transcriptionally active as homodimers or heterodimers. Finally, nuclear receptors have one or two regions called activation function 1 and 2 (AF1 and AF2), which are required to transactivate gene expression. Whereas the activity of AF1 is usually ligand-independent and located in the A/B domain, AF2 is found in the ligand-binding domain and is predominantly regulated by hormone binding.
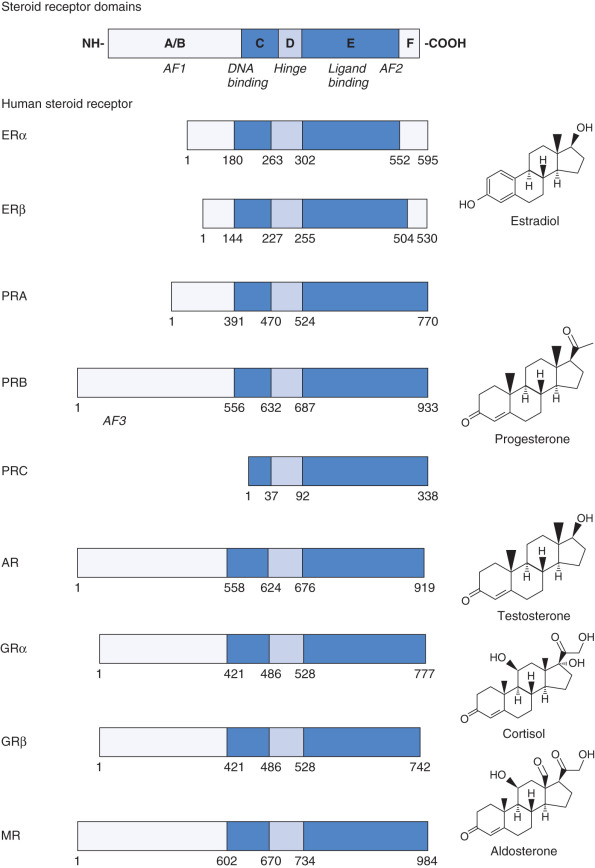
Some nuclear receptors have defined natural ligands—such as the steroid hormones, thyroid hormones, retinoids, or vitamin D—but the ligand for some nuclear receptors has not yet been identified. These receptors are therefore called orphan receptors. The finding that diverse compounds act as ligands for nuclear receptors and that some receptors have no apparent ligand led to the hypothesis that ancestral nuclear receptors were constitutive transcription factors that independently evolved the ability to bind ligand. However, a second hypothesis posits that ancestral receptors were ligand-dependent transcription factors that evolved specificity for different ligands by gene duplication, mutation, and functional divergence. There are several lines of evidence that favors the latter hypothesis for the evolution of ligand binding in the steroid receptor family. First, the primary, secondary, and tertiary structures of the ligand-binding domain of different steroid receptors are highly similar. Second, detailed sequence, structural, and functional analyses strongly support the hypothesis that the ancestral steroid receptor bound estrogens and specificity for other steroids evolved by serial and parallel duplications of the ancestral gene, mutation of nucleotides coding for specific amino acids, and structural and functional divergence of the paralogues. Finally, steroid hormone receptors are nuclear receptors unique to the chordate lineage, indicating that they originated when the first chordates evolved.
Gene Duplication of Ancestral Steroid Hormone Receptors
Phylogenetic analysis of the primary amino acid sequences of 73 steroid receptors from jawed vertebrates (e.g., fish, reptiles, birds, mammals) and a jawless fish (the sea lamprey) indicate that there were two serial duplications of an ancestral steroid receptor before the divergence of these lineages approximately 450 million years ago. Maximum likelihood reconstructions of the ancestral amino acid sequence indicate that the first steroid receptor was probably an ER-like molecule ( Fig. 5.3 ). After this gene was duplicated, one copy was constrained by natural selection and retained its function as an “estrogen receptor,” whereas the other copy evolved specificity for 3-ketosteroid–like ligands. Duplication of the latter gene then produced a corticoid receptor–like protein and a receptor for 3-ketogonadal steroid–like molecules (androgens, progestins, or both). Whereas these three steroid receptors (i.e., the ER, corticoid, and 3-ketogonadal steroid receptors) are present in the sea lamprey, six steroid receptors are present in fish and tetrapods, suggesting a genomewide duplication in the last common ancestor of jawed vertebrates ( blue boxes in Fig. 5.3 ).
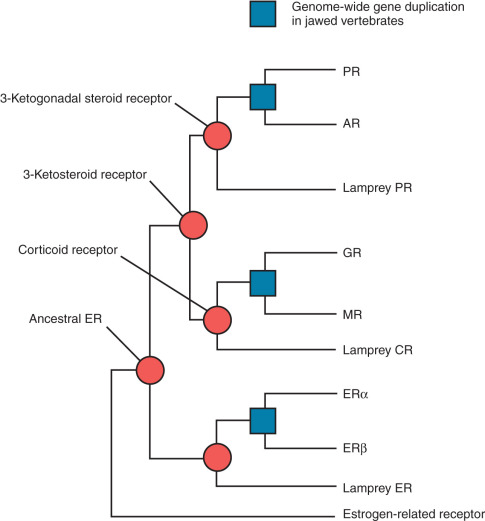
This final duplication then led to the evolution of the true androgen and progesterone receptors from the ancestral 3-ketogonadal steroid receptor, GR and MR from the ancestral corticoid receptor, and ERα and ERβ from the ancestral ER. The number of paralogues for other gene families, such as the homeobox (Hox) genes, supports the hypothesis that many genes were duplicated in parallel in the ancestor of jawed vertebrates.
Structural and Functional Divergence of Steroid Hormone Receptors
Experiments deleting large regions of different steroid receptors have clearly shown that domain C is responsible for DNA binding and that domain E is responsible for ligand binding. Furthermore, domain swapping of the entire ligand-binding domain between different receptors has shown that this region determines specificity for particular classes of steroid hormone. Nevertheless, crystal structures of various steroid hormone receptors show that all ligand-binding domains fold into a highly homologous three-layered structure with a small ligand-binding pocket in the center. This pocket is composed of roughly 30 amino acids that are in close proximity or make direct contact with hormones when bound to their cognate receptors. In agreement with structural studies, experiments using site-directed mutagenesis indicate that specific but minor changes of amino acids within the ligand-binding pocket can lead to dramatic changes in the hormone-binding specificity of steroid receptors. For example, the human PR, GR, and MR contain a conserved cysteine residue that appears to be critical for contacting the C20 keto group found in progestins, glucocorticoids, and mineralocorticoids. Mutation of the corresponding threonine to cysteine in the AR reduces its affinity for androgens and allows the receptor to transactivate gene expression in the presence of progesterone and corticoids. Based on structure-function studies of this sort and phylogenetic analyses, Thornton proposed a series of relatively minor amino acid changes that may account for broad changes in hormone specificity during the evolution of steroid receptors.
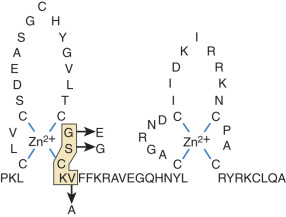
Similar studies of the DNA-binding domain have defined the molecular basis for interactions between steroid receptors and particular DNA sequences. The DNA-binding domain of nuclear receptors contains two zinc fingers. The first finger interacts with the major groove of DNA, whereas the second is involved in receptor dimerization. Mutation of three residues within a five-residue motif known as the Proximal box (P box) of the first zinc finger of the GR to the corresponding residues in ER changes the binding specificity from DNA sequences called glucocorticoid-responsive elements to estrogen-responsive elements, and vice versa ( Fig. 5.4 ). Although the DNA-binding domain is very highly conserved among nuclear receptors, these residues are variable among different receptors, which may in part account for receptor-specific regulation of distinct sets of genes. These examples illustrate that site-directed mutagenesis and fine-scale comparison of amino acid sequences among receptors, in the context of the tertiary structure of the DNA- and ligand-binding domains, can lead to testable hypotheses about the evolution of signaling and regulation of gene expression by different classes of steroids.
Steroid Hormone Receptor Function
- ◆
The steroid hormone receptors have well-described functions in the reproductive tract but also critically regulate general physiology.
- ◆
Transgenic animal models and mutations identified in humans have provided insight into the cell-specific activities of the steroid hormones.
- ◆
ER, PR, and AR are indispensable for reproductive function but also regulate nonreproductive aspects of physiology.
- ◆
GR signaling is essential for life after birth, maintaining general physiologic homeostasis, and integrating the hypothalamic-pituitary-adrenal (HPA) axis with reproductive function.
- ◆
MR is required for electrolyte balance and fluid transport, where insufficiency or deficiency can lead to mortality.
Steroid receptors have taken on distinct physiologic roles during evolution. Transgenic animal models and case reports in humans with genetic mutations/deletions in the steroid receptors have provided insight into these unique functions. ERα, ERβ, PR, and AR are essential for sexual differentiation and reproduction. Although androgens, estrogens, and progestins are sex-typical hormones, they are not sex-limited, and they play a physiologic role in both sexes. In contrast, GR and MR principally govern general physiology including, development, immune function, cognition and behavior, cardiovascular health, electrolyte balance, and metabolic homeostasis. In addition to mediating physiologic homeostasis, stress-induced and endogenous levels of glucocorticoids regulate fertility, whereas the role of mineralocorticoids in reproductive functions is not well defined.
Estrogen Receptor
The biological effects of estrogens were believed to be mediated by a single receptor (ERα) until the cloning of a second receptor in the late 20th century (ERβ). The ERs are products of different genes and demonstrate distinct expression patterns across tissues and cell types ( Table 5.1 ). Consequently, these two receptors regulate unique biological functions, which have been characterized in transgenic animal models. For example, both ERα and ERβ are localized to the mammalian ovary, but the ovarian phenotypes of ERα and ERβ knockout mice are dissimilar, likely due to the distinct cellular distribution of these two receptors in the ovary. The ERα knockout mouse is anovulatory and accumulates cystic and hemorrhagic follicles, whereas the ERβ knockout mouse contains histologically normal ovaries but still displays impaired ovulation. In the uterus, the absence of ERα results in a significant infertile phenotype due to a hypoplastic uterus and defects in implantation and decidualization; however, the loss of ERβ does not alter uterine development, function, or cellular responses to estradiol. Cell-specific deletion models have evaluated the biological effects of ERα in the uterine stroma and epithelial cells. Gene targeting technology has also led to the creation of mouse models with mutations in the functional domains of ERα, which have been characterized for uterine pathologies. The mammary gland is also sensitive to ERα loss. ERα knockout mice demonstrate impaired ductal growth and differentiation following the onset of endogenous estrogen production during puberty. The loss of ERβ does not alter ductal growth but appears to play a role in differentiation of the mammary gland during pregnancy. In males, loss of ERα results in infertility related to defects in fluid resorption in the efferent ducts and epididymis and progressive deterioration of testicular morphology. The ERβ knockout males are fertile, with phenotypically normal testis.
| Tissue | Receptor * | |
|---|---|---|
| ERα | ERβ | |
| Epididymis | +++ | + |
| Prostate | + | +++ |
| Testis | +++ | + |
| Pituitary | ++ | + |
| Ovary | +++ | +++ |
| Uterus | +++ | ++ |
| Bladder | + | ++ |
| Lung | 0 | + |
| Liver | + | 0 |
| Kidney | ++ | 0 |
| Thymus | + | + |
| Adrenal | ++ | 0 |
| Olfactory lobe | 0 | + |
| Cerebellum | 0 | + |
| Brain stem | 0 | + |
| Spinal cord | 0 | + |
| Heart | + | 0 |
* Relative levels of expression are indicated by the number of plus signs: 0, not detected; +, low; ++, medium; +++, high.
Despite the fact that both ERα and ERβ bind estradiol with similar affinities ( Table 5.2 ), in vitro studies and computational modeling have determined that the relative affinity for exogenous estrogenic compounds differs between the receptor subtypes. The differences in ligand binding have been exploited for the benefit of various therapies (selective ER modulators). However, exogenous estrogenic compounds are also able to interfere with endogenous ligand binding, resulting in aberrant activation or repression of ER function. One of the first examples of endocrine disruption by a synthetic estrogenic compound occurred when women were exposed to diethylstilbestrol (DES) during pregnancy. Prenatal exposure was linked to reproductive tract cancers in offspring. Subsequently, other estrogenic endocrine disruptors have been studied with reported impacts on ER signaling that vary in severity and duration.
| Ligand | Relative Binding Affinity * | |
|---|---|---|
| ERα | ERβ | |
| E2 | 100 | 100 |
| Diethylstilbestrol | 468 | 295 |
| Hexestrol | 302 | 234 |
| Dienestrol | 223 | 404 |
| Estrone | 60 | 37 |
| 17α-Estradiol | 58 | 11 |
| Moxestrol | 43 | 5 |
| Estriol | 14 | 21 |
| 4-OH-Estradiol | 13 | 7 |
| 2-OH-Estradiol | 7 | 11 |
| Estrone-3-sulfate | <1 | <1 |
| 4-OH-Tamoxifen | 178 | 339 |
| ICI-164384 | 85 | 166 |
| Nafoxidine | 44 | 16 |
| Clomifene | 25 | 12 |
| Tamoxifen | 7 | 6 |
| Coumestrol | 94 | 185 |
| Genestein | 5 | 36 |
| Bisphenol A | 0.05 | 0.33 |
| Methoxychlor | 0.01 | 0.13 |
* Relative binding affinity is the ratio of concentrations of E2 and competitor required to displace 50% of specific radioligand binding. Relative binding affinity was set to 100% for E2.
In addition to their physiological role in regulating reproductive tract function, estrogens are a risk factor for the initiation and progression of breast cancer. However, ER antagonists can slow or stop the growth of breast cancer by blocking estrogen-regulated gene expression. The carcinogenic effects of estrogen in breast tissue may, in part, be mediated by the induction of vascular endothelial growth factor (VEGF), as tumor growth is generally dependent on angiogenesis and a steady blood supply. Interestingly, the breast cancer susceptibility gene (BRCA1) can directly interact with ER and block the induction of VEGF. Mutations that inactivate BRCA1 and confer increased risk for breast cancer may be due to a loss of the ability to antagonize the actions of estrogen.
ER loss-of-function mutations are rare but have been reported in a small number of patients. The clinical phenotypes of these patients were comparable to the ER knockout mouse model. Females presented with amenorrhea, cystic ovaries, thin endometrium, and no breast development. The predominant phenotype of affected males was delayed bone maturation. Somatic mutations and genetic polymorphisms in the human ER are also associated with various disease states. For example, a number of ER mutations have been detected in breast cancer. A high-frequency somatic mutation between D and E region in the ER was identified in breast hyperplasias and was found to enhance estrogen sensitivity when evaluated in vitro. Breast cancer cell models of two other common ER mutations, Y537S and D538G, demonstrate that some mutations bestow ligand-independent gene regulation, growth, and resistance to ER modulators. Polymorphisms in ERα and ERβ have been associated with breast cancer severity. Moreover, polymorphisms identified in genomic ER binding sites have been predicted to alter global ER activity and contribute to breast cancer pathogenesis. These mutations and polymorphisms likely influence the progression of cancer as well as the response to treatment.
Progesterone Receptor
Like estrogens, progestins are necessary for female fertility. In fact, the etymology of the word progesterone is “ progest ational ster oid horm one .” Progesterone signaling is mediated by PR, which has two isoforms, PR-A and PR-B, derived from the same gene. The necessity of progesterone signaling for female fertility was demonstrated in a mouse model devoid of both PR isoforms. The female PR knockout mice were infertile due to defects in mating behavior, ovarian follicle rupture, implantation, and uterine function. Females also displayed disrupted mammary gland development. In contrast to females, PR-deficient males appear normal, and fertility was comparable to that of wild-type males.
The isoforms PR-A and PR-B are alike except for the addition of 164 amino acids at the amino-terminal end of PR-B due to the utilization of an alternative translational initiation site. These 164 amino acids confer an ancillary activation function domain (AF3), which contributes to the functional differences between the two isoforms. The AF3 domain prevents the activity of a transcription inhibitory domain in the AF1 of PR-B, while the absence of the AF3 in PR-A allows the inhibitory domain to function and renders this isoform more “transcriptionally repressive.” Moreover, at sites of transcriptional activity, PR-B preferentially recruits coactivators, while PR-A recruits corepressors. Mice with targeted deletion of either PR-A or PR-B have allowed the functions of the PR isoforms to be characterized in different tissues in vivo. Deletion of the PR-A isoform phenocopied the fertility defects described in PR null mice, whereas selective ablation of PR-B only affected mammary gland development. Overexpression of PR-A in the uterus also resulted in infertility due to disrupted embryo attachment, indicating that precise regulation over the spatiotemporal expression of PR-A is required for the establishment of pregnancy. A third potential PR isoform, PR-C, has also been cloned and results from translation initiation 430 amino acids downstream of the PR-A start site. Although PR-C lacks the AF1 and DNA-binding domains, it has been shown to heterodimerize with PR-B and alter its transcriptional activity.
One of the primary functions of progesterone signaling in the uterus prior to implantation is to inhibit the early actions of estradiol. For example, estrogen signaling drives uterine epithelial cell proliferation. However, estrogen signaling also promotes the expression of PR in the uterus, which subsequently inhibits epithelial proliferation through paracrine signaling to allow embryo implantation. Likewise, mucin 1 (MUC1), a cell surface glycoprotein that provides a protective barrier to the uterine epithelium, is induced by estradiol in the uterus. However, the preimplantation rise in progesterone levels leads to the downregulation of MUC1, which is necessary for the embryo to adhere, implant, and invade. PR also critically coordinates paracrine signaling across the stroma and epithelium during early pregnancy. Progesterone target genes in the epithelium mediate downstream signaling events in the stroma, which are critical during embryo implantation and for postimplantation maintenance of pregnancy.
The antiproliferative properties of progesterone in the uterus have been targeted for the treatment of endometrial cancer. The standard treatment for endometrial carcinomas is often surgical. However, patient desire to preserve fertility has prompted the use of progestin hormonal therapy as an alternative treatment option with varied success. In uterine fibroids, benign tumors of the uterine smooth muscle, progesterone promotes growth by stimulating proliferation and hypertrophy. For this reason, selective progesterone receptor modulators with antagonistic activity have been evaluated as therapeutic agents for fibroids.
In accord with the finding that PR is necessary for development of the mammary ducts, progesterone signaling has also been shown to play a role in the pathogenesis of breast cancer. Progesterone regulates proliferation of the mammary epithelium through autocrine and paracrine pathways. One of the main paracrine factors mediating progesterone-induced proliferation is receptor activator of nuclear factor kappa-B ligand (RANKL). The induction of mitogenic factors by progesterone also contributes to the maintenance and expansion of the mammary gland stem cells. In the Women’s Health Initiative trial, combined estrogen plus progesterone therapy as part of a hormone replacement regimen in postmenopausal women increased the risk of breast cancer. However, studies have also demonstrated that PR activation in certain types of breast cancer improved progression-free survival. The differential actions of progesterone may reflect the distinct cellular context of healthy and malignant tissue or the dose and specificity of the progestogen used.
Androgen Receptor
Androgens, the main male sex steroids, drive the differentiation of the male reproductive tract during development and are crucial for reproductive functions in the adult. During development, the androgen-producing cells of the mammalian gonad arise from precursor cells in the interstitium to become Leydig cells in males and theca cells in females. Androgens produced by the fetal Leydig cells direct the development of the male external genitalia, vas deferens, and related structures from the wolffian ducts. In mouse models of disrupted androgen signaling, the wolffian ducts regress and female external genitalia develop. Likewise, mutations in the human AR, causing androgen insensitivity syndrome, result in a female phenotype in individuals who are genotypically male. Mutations in AR have also been identified that lead to partial androgen insensitivity by disrupting AR-cofactor interactions. Studies with the AR-deficient mouse model also demonstrated a role for androgen signaling in female reproduction. AR-null female mice undergo premature ovarian senescence due to follicle atresia. Subsequent analysis of the AR-null mice revealed defective luteinization of the preovulatory follicles. In agreement with the animal studies, AR gene mutations have been characterized in female patients with premature ovarian failure.
Following reproductive tract development, androgens, primarily testosterone and 5α-dihydrotestosterone, induce and maintain the secondary sex characteristics, stimulate male reproductive behavior, and support spermatogenesis. In the testis, the primary target of androgen action is the Sertoli cell. Mice with a targeted deletion of AR in Sertoli cells are infertile due to spermatogenic arrest at meiosis and impaired Sertoli cell barrier formation (blood–testis barrier). AR has also been conditionally deleted from the Leydig cells, which caused spermatogenesis arrested at the round spermatid stage, empty and atrophied epididymides, and infertility. The Leydig cell–specific knockout mice also exhibited higher serum gonadotropin levels and lower serum testosterone levels. These observations indicate that testicular AR signaling is required to maintain spermatogenesis. During normal spermatogenesis, germ cell apoptosis is a constant feature of the adult testis and is required to maintain homeostasis within the seminiferous tubule. Exposure to excess and insufficient levels of testosterone can cause testicular cell death. In vitro culture of testicular tissue samples with testosterone improved measures of apoptosis. The mechanism by which testosterone acts as a prosurvival factor has been partially attributed to the inhibition of prodeath signals. Conversely, excess testosterone can cause cell death through the induction of prodeath signals, namely Fas cell surface death receptor (Fas) and Fas ligand (FasL).
The prostate epithelium is also a target of androgen action during development, and androgen signaling is required to maintain cellular homeostasis in the adult prostate. Castration results in prostate involution due to epithelial cell apoptosis, which is reversible by androgen supplementation. Cell-specific knockout models of AR in the prostate indicate that paracrine signaling by stromal AR regulates epithelial cell proliferation. The prosurvival actions of AR in the prostate play a fundamental role in the pathogenesis of prostate cancer. In fact, over 75 years ago, it was shown that androgen deprivation by castration caused regression of prostate cancer. Androgen-initiated prostate cancer growth is believed to occur through cross-talk with several signaling pathways, namely pathways involved in metabolic regulation, cell-cycle progression, and growth-factor signaling. Moreover, AR signaling in the epithelium, independent of paracrine AR signaling in the stroma, has been implicated in the “switch” from nonmalignant to a cancerous prostate. AR also contributes to prostate cancer progression by promoting angiogenesis through the induction of VEGF. Therefore AR is the primary therapeutic target in prostate cancer. It is now clear that AR signaling contributes to cancers in organs other than the prostate, including the breast, bladder, pancreas, ovary, and endometrium.
Glucocorticoid Receptor
Glucocorticoids are often referred to as stress hormones because they are synthesized and secreted in response to physical pain, emotional trauma, caloric restriction, or general anxiety. However, glucocorticoids are also released in a diurnal pattern to mediate the general activity of the HPA axis. The essential nature of glucocorticoid signaling was demonstrated in mice homozygous for a null GR mutation, which were nonviable shortly after birth due to developmental defects. Subsequently, tissue- and cell-specific knockout models have demonstrated the importance of glucocorticoid signaling in postnatal physiology and the pathogenesis of disease. The diversity in GR signaling stems in part from several transcriptional and translational isoforms produced from a single gene. Alternative splicing in the ligand-binding domain generates two receptor isoforms: GRα and GRβ. GRβ functions as a dominant negative regulator of GRα activity but also has inherent transcriptional activity. Using alternative AUG start sites, at least eight translational isoforms of GR with unique tissue distribution and properties have been characterized. Interestingly, the human placenta expresses both GRα and GRβ in addition to several of the translational isoforms, and expression of the isoforms has been associated with sex of the offspring and pregnancy complications.
Glucocorticoids alter the physiology of numerous tissues throughout the body during periods of acute stress. For example, the inhibitory effects of stress on reproduction are mediated by glucocorticoid signaling at all levels of the hypothalamic-pituitary-gonadal axis. In the hypothalamus, stress and exogenous glucocorticoid exposure inhibit the production of the gonadotropin-releasing hormone (GnRH). The inhibitory effects of glucocorticoids on GnRH represent both direct and indirect regulation. Downstream of GnRH, secretion of the gonadotropins luteinizing hormone (LH) and follicle-stimulating hormone (FSH) is also regulated by glucocorticoids. In addition to the effects on the central nervous system, high levels of glucocorticoids suppress the functions of the ovaries, uterus, placenta, and testis. In the ovary, glucocorticoid exposure alters steroid biosynthesis. The effects on steroidogenesis are partially mediated by induced apoptosis of mural granulosa cells. In the uterus, exogenous glucocorticoids antagonize estrogen-induced uterine growth, proliferation, and embryo implantation in mice and estrogen-regulated gene expression in human uterine cell lines. Glucocorticoid exposure during early pregnancy inhibits placentation and results in fetal growth restriction. Both stress-induced levels and exogenously administered glucocorticoids suppress testicular function by inhibiting steroidogenesis and inducing programmed cell death in the testis.
In addition to mediating the stress response, glucocorticoids play an essential role in normal reproductive physiology, which has been demonstrated in animal models of adrenalectomy and tissue-specific knockouts. Uterine-specific deletion of GR resulted in subfertility as a consequence of impaired implantation and decidualization, indicating an obligate role for GR in maintaining normal uterine function. Deletion of GR in Sertoli cells led to fewer meiotic spermatocytes, postmeiotic spermatids, and Sertoli cells, indicating that GR expression in Sertoli cells is required to maintain normal testicular function. Adrenalectomy in male rats demonstrated that glucocorticoid signaling is required to support steroidogenesis, spermatogenesis, and sperm maturation.
Mineralocorticoid Receptor
Aldosterone, the major circulating mineralocorticoid, specifically binds to and activates the MR. In contrast, cortisol (or corticosterone in rodents) binds to and activates both the MR and GR. To prevent inappropriate MR activation by glucocorticoids, 11β-hydroxysteroid dehydrogenase (11β-HSD) converts cortisol (or corticosterone) to inactive metabolites in cells containing MR (see Chapter 4 ). One critical function of aldosterone is to regulate electrolyte balance and blood pressure. Aldosterone’s primary target tissue is the distal tubule of the kidney, where it promotes the retention of Na + and the elimination of K + during urine formation. Retention of Na + leads to absorption of water from the distal tubules, an increase in blood volume, and maintenance of blood pressure. Mineralocorticoid insufficiency causes death by Na + and water loss, a fall in blood volume, and circulatory shock. Accordingly, MR knockout mice also die of Na + and water loss. On the other hand, mineralocorticoid excess contributes to hypertension and pathologies associated with high blood pressure. Additional targets of aldosterone include the heart, brain, smooth muscle, endothelial cells, and adipose tissue, although less is known regarding the cellular and molecular mechanisms of mineralocorticoids in these tissues.
Work has identified several mineralocorticoid-induced genes involved in Na + resorption in tight epithelia. Aldosterone, for example, increases both the mRNA and protein of serum- and glucocorticoid-regulated kinase (Sgk), activating the phosphatidylinositol-3-OH kinase (PI3K) pathway, stabilizing the epithelial Na + channel (ENaC) in an open state as well as increasing the number of ENaCs in the apical membrane. It is also important to note that aldosterone induces the α subunit of ENaC and the α 1 subunit of the sodium/potassium ATPase pump (Na + /K + -ATPase) by the classic transcriptional mechanism attributed to steroid hormones, which is critical for generating the electrochemical gradient necessary for Na + resorption at the apical membrane. Aldosterone also induces the expression of the Na + /H + transporter and numerous other genes that may play a role in electrolyte balance. The role of genes repressed by aldosterone, however, remains unclear.
General Factors That Influence Steroid Hormone Action
- ◆
Relative abundance of the receptor and ligand alters steroid hormone action.
- ◆
Steroid hormones can be intermetabolized from inactive to active forms and vice versa.
- ◆
Some steroid hormones circulate bound to proteins, which keeps them in an inactive state.
- ◆
Steroid hormone receptor expression can be regulated through transcriptional or translational mechanisms and posttranscriptional and posttranslational mechanisms, including modifications that alter receptor stability.
- ◆
Posttranslational phosphorylation can alter receptor activity.
- ◆
Interactions with cofactors in the nucleus can act as a molecular switch, inducing or repressing transcription.
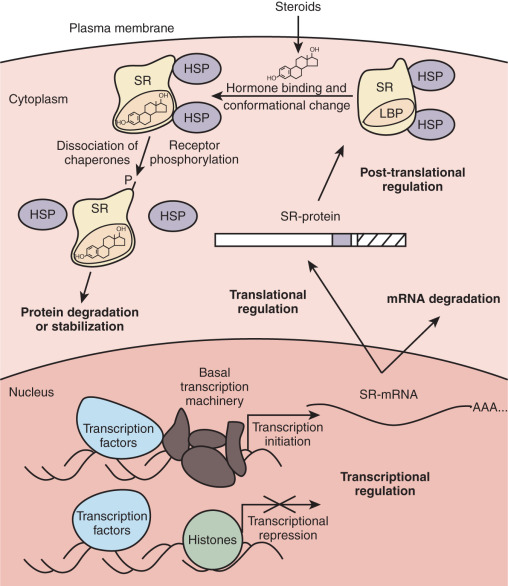
Although numerous factors affect signaling by steroid hormones in vivo, this chapter focuses on some of the more important and well-characterized regulatory mechanisms. Foremost among the factors determining whether a cell responds to circulating steroid hormones is the intracellular availability of a given steroid and its receptor. Intracellular enzymes are able to interconvert steroids to active and inactive forms, providing preemptory control over steroid hormone action. The number of receptors within a cell also determines its responsiveness to hormone, with regulation of receptor expression at all levels from transcription to protein degradation. When sufficient hormone and receptor are present in a cell, the ligand binds and activates the monomeric receptor. The ligand-bound receptor undergoes a conformational change, dissociates from regulatory heat-shock proteins, and is phosphorylated. Cytoplasmic receptors translocate to the nucleus, where they bind as homodimers to specific DNA sequences called hormone-responsive elements. The MR, GR, PR, and AR are predominantly cytoplasmic in the absence of ligand. In contrast, ERα and ERβ are localized in the nucleus in the absence of ligand and do not require nuclear translocation for their transcriptional activity. Within the nucleus, the ligand–receptor complex interacts with coactivator and corepressor proteins that remodel chromatin or contact the basal transcription machinery, thereby increasing or decreasing the probability of initiation of transcription. Although we discuss basic regulatory mechanisms at each step in the dynamic process just outlined, the reader is referred to primary research and review articles that treat each mechanism in greater detail.
Hormone Availability
Intracellular enzymes can activate or inactivate steroid hormones to produce tissue-specific or even cell-specific patterns of hormone responsiveness. Aromatase P450, which converts androgens to estrogens, is expressed in the gonads, brain, adipose tissue, skin, and placenta. In most female mammals, the circulating level of estradiol varies during the menstrual or estrous cycle. At the nadir of the cycle, circulating estrogen levels are usually below the concentration necessary to activate ER. In contrast, estrogen levels at the peak of the cycle are generally high enough to activate the ER. Consequently, the synthesis and secretion of estrogens from the ovary into general circulation is the main mechanism controlling estrogen action in females. Whereas males have plasma levels of estrogens that are below the concentration necessary to activate ER, they still require estrogen for normal reproductive function and behavior, as demonstrated by ERα knockout mice. Therefore, it would seem that the expression of male copulatory behavior in a variety of vertebrates depends on the local conversion of testosterone to 17β-estradiol by aromatase and subsequent activation of ER within the medial preoptic area of the brain.
Similarly, localized expression of 5α-reductase, which converts testosterone to dihydrotestosterone, plays a crucial role in regulating androgen action. This reaction is particularly important because dihydrotestosterone binds to the AR with higher affinity than testosterone and is essential for full virilization of the external genitalia during male development. Interestingly, female knockout mice for type 1 5α-reductase show significant impairment of parturition, suggesting that androgens also play an important role in female reproductive physiology. In general accord with this finding, female knockout mice for AR have smaller litter sizes than wild-type littermates.
In another example of steroid-metabolizing enzymes regulating steroid hormone action, two isoforms of 11β-HSD oppose each other’s action and regulate local levels of active and inactive glucocorticoids. Whereas 11β-HSD type 1 is a reductase that converts weak glucocorticoids (cortisone and 11-deoxycorticosterone) into potent glucocorticoids (cortisol and corticosterone), 11β-HSD type 2 is a dehydrogenase that catalyzes the reverse reaction to inactivate the more potent glucocorticoids. Clinical and experimental evidence suggests that the relative activity of these enzymes in particular tissues may play an important role in normal glucocorticoid physiology and pathophysiology. These examples illustrate how local biosynthesis or inactivation of steroid hormones can facilitate or inhibit steroid hormone action in a tissue- or cell-specific manner.
Receptor Expression
All other factors being equal, the level of expression of steroid receptors is a major factor regulating steroid hormone action. Whereas certain cells or tissues are insensitive to steroid hormones because they lack the appropriate receptors, other cells and tissues express large numbers of receptors and are exquisitely sensitive to low concentrations of hormones. In addition, hormone responsiveness is often modulated by up- or downregulation of steroid receptors. One of the best-characterized mechanisms of sensitization is induction of PR gene expression by estrogens. During the ovarian cycle in the rat, 17β-estradiol concentrations slowly rise as ovulation approaches, and 4 hours before ovulation, progesterone concentrations surge. These changes in hormone levels lead to biochemical changes in neurons of the ventromedial hypothalamus that facilitate the display of female sexual behavior. The surge in 17β-estradiol activates the ER and increases transcription and subsequent translation of the PR gene, thereby sensitizing the ventromedial hypothalamus to the subsequent surge in progesterone. Essentially the same pattern and mechanism of PR induction by estrogens occurs in stromal cells of the uterus, priming these cells for decidualization in response to progesterone and the implanting blastocyst. Another uterine cell type, the luminal epithelium, displays the opposite pattern of PR expression during the reproductive cycle. Uterine epithelial cells express PR constitutively and rapidly downregulate PR in response to estrogens. However, the mechanism responsible for this decrease in PR protein is currently unknown. It is also unclear whether PR downregulation leads to hormone desensitization in these cells.
Changes in mRNA stability provide a second mechanism for regulating the levels of receptor expression in tissues. The preovulatory surge in LH decreases levels of ERβ mRNA in granulosa cells of rat preovulatory follicles. This decrease in ERβ mRNA is due to a decrease in the half-life of the message from approximately 18 to 5 hours, rather than a decrease in transcription of the ERβ gene. Moreover, the LH-induced decline in steady-state levels of ERβ mRNA is mimicked by forskolin, indicating that LH signaling occurs through the activation of adenylyl cyclase and the generation of cyclic adenosine monophosphate (cAMP). Interestingly, a similar effect was also seen with the protein kinase C (PKC) activator phorbol myristoyl acetate (PMA). The stability of ERα mRNA is also regulated in the Michigan Cancer Foundation-7 (MCF-7) breast cancer cell immortalized line. In these cells, 17β-estradiol decreases the half-life of ERα mRNA from 4 hours to 40 minutes by increasing the ribonuclease activity associated with polyribosomes. In contrast, 17β-estradiol upregulates ERα mRNA in the endometrium by selectively increasing message half-life from 9 to 24 hours. These studies demonstrate hormone- and tissue-specific effects on the stability of steroid receptor mRNA.
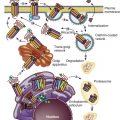
Another well-established mechanism for steroid receptor regulation occurs at the protein level. Treatment with a given steroid generally results in a time-dependent decrease in the level of its cognate receptor in a process called homologous downregulation. Accordingly, a decrease in receptor levels usually results in desensitization to subsequent treatment with the same steroid. Receptor degradation occurs by targeting of the receptor into the ubiquitin–proteasome pathway. The effects of proteasome inhibition on steroid hormone action clearly need further study because there are paradoxical effects on receptor levels and receptor function. Whereas GR accumulation by proteasome inhibition leads to increased transcriptional activity of the receptor, proteasome inhibition of ER and PR degradation results in a decrease in hormone-induced transcription. In sum, regulation of steroid receptor expression occurs at multiple levels from gene transcription to mRNA stability to degradation of the mature protein ( Fig. 5.5 ).